

Abstract
Methylation at position 5 of cytosine in DNA is being intensively studied in many areas of biological sciences, as the methylation is intimately associated with the control of gene functions. The principal analytical method for determining the sites of 5-methylcytosine in genome at the sequence level involves bisulfite modification of DNA. The utility of this chemical treatment is based on the property of bisulfite to selectively deaminate cytosine residues. The bisulfite-mediated cytosine deamination was discovered in 1970 by us in the University of Tokyo. At the same time, Shapiro and his coworkers in New York University found the same reaction independently. We also reported that 5-methylcytosine was deaminated by bisulfite only very slowly. These findings were later utilized by a group of Australian scientists to devise a means to analyze 5-methylcytosine in DNA; thus, a method called ‘bisulfite genomic sequencing’ was invented by these researchers in 1992. This review describes the author’s reflection of the discovery of bisulfite reactions with pyrimidine bases. The author’s recent work that has resulted in an improvement of the procedure of analysis by use of a newly devised high concentration bisulfite solution is also described.
Keywords: bisulfite, 5-methylcytosine, deamination of cytosine, concentration dependency, chemical modification
1. Introduction
The presence of 5-methylcytosine in DNA is a current focus of attention in studies of the genome. A textbook on DNA, for example, has a newly added chapter ‘Cytosine Methylation and DNA Epigenetics’ in its 2004 edition.1) In animal and plant DNAs, a portion of cytosine is methylated at position 5 (Fig. 1). The methylation occurs by a post-replication enzymatic modification, and is a heritable event. The methylated cytosine is present primarily at 5′-CpG-3′ sites in the genome. The DNA methylation is associated with gene function control, and is known to intimately related to human diseases, including cancer.
Fig. 1.
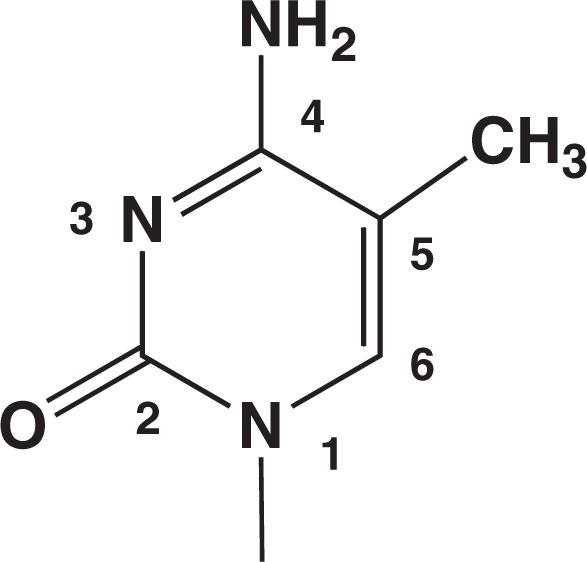
Structure of 5-methylcytosine residue.
A principal method for determining the 5-methylcytosine sites in genes at the sequence level is the bisulfite genomic sequencing. The principle of this methodology resides in the selectivity of bisulfite actions, attacking cytosine rapidly, but only very sluggishly 5-methylcytosine. The bisulfite-mediated specific deamination of cytosine was discovered in 1970 by Shapiro and coworkers in New York City and by my research group in Tokyo independently.2),3) The use of this chemical treatment for analyzing 5-methylcytosine in DNA was initiated by a group of Australian scientists in 1992.4)
This article is a short historical account of our discovery of bisulfite reactions with pyrimidine bases in DNA.
2. Background of the research
The major training I received in the undergraduate- and the 2-year graduate-courses, 1955–1958, in the University of Tokyo, was organic chemistry, under the guidance of the great chemist, Professor E. Ochiai (1898–1974), whose expertise was synthesis and reactions of aromatic amine oxides. He dictated to students that a chemist may read literatures but should not be led by them. I happily complied with this instruction and spent most of my time doing experimental bench-work. This life-style of mine has persisted many decades until today. My doctoral work under the supervision of Professor T. Ukita (1915–1972) was chemical synthesis of nucleotides, and then I did post-doctoral work in Professor H. G. Khorana’s laboratory in the University of Wisconsin, 1964–1967, synthesizing oligonucleotides. Coming back to Tokyo, I began searching for reagents that can modify DNA bases in aqueous solutions under mild conditions. Soon I found that potassium permanganate, a commonly used oxidizing agent, is capable of modifying thymine selectively among major bases.5) Along with that work, a finding was made; i.e., a fast reaction of 4-thiouridine, a minor nucleoside in transfer RNA, with permanganate, forming its 4-sulfonate6) (Fig. 2). It became necessary to measure the progress of this oxidation, and for that purpose, the permanganate in the reaction mixture had to be degraded by adding a reducing agent. The reducing agent I chose was sodium bisulfite. Then, an unexpected phenomenon was observed.
Fig. 2.
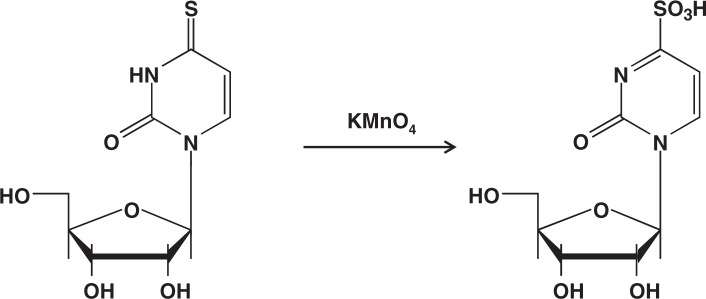
Permanganate oxidation of 4-thiouridine to form its sulfonate derivative.
3. Power of an old-fashioned spectrophotometer — Encounter with an unanticipated reaction mediator, oxygen
In 1969, the UV-visible spectrophotometer we used was a Beckman apparatus having a window with a moving arrow-headed needle. Another window showing absorbance values had a dial to be moved by hand. The experimenter adjusted the dial to make the needle arrow-head to come to the central ‘0’ point and then recorded the absorbance value.
4-Thiouridine was treated with permanganate, and at a desired period, the reaction was stopped by addition of an excess sodium bisulfite (NaHSO3). 4-Thiouridine has a unique absorption peak at 330 nm, so the use of A330 allows quantification of unreacted 4-thiouridine after the treatment. With this setting, the progress of the permanganate oxidation of 4-thiouridine was determined, and I soon became aware that something peculiar was going on, because the results were not reproducible. I noticed after decomposing permanganate with bisulfite, that the A330 value still continued to decrease. I was excited by the possibility that bisulfite itself might be reacting with 4-thiouridine. I thought “Now, no permanganate anymore, let’s try to see if bisulfite can react with 4-thiouridine”. So, I mixed this nucleoside with bisulfite in buffers and tried to see if it may undergo changes. Indeed, the A330 decreased gradually. However, the results were erratic. I tried various combinations of reaction conditions; pH, temperature, and concentrations of components. At times, the decrease in A330 was large, and at others, it was not so large. But the most annoying was non-reproducibility of each experiment. I never understood why.
One day, a few-ml solution containing 4-thiouridine and NaHSO3 was placed in a small flask, the flask was tightly closed, and the solution was incubated for several hours. For determining the progress of the transformation, the A330 value was measured: only a small decrease from the original value was observed. I transferred the solution from the Beckman cuvette back to the flask, with some gloomy feeling. This time, I wanted to reconfirm the recorded value. So, I poured the solution again into the cuvette and started the measurement. Now, I found the value somewhat smaller than the first record. Suddenly, I saw that the needle arrow-head in the window was moving to the downside, indicating that the absorbance of the sample was rapidly decreasing. I then understood what was going on. The action of transfer of the solution to and from the flask and cuvette must have caused this sudden acceleration of reaction: the key factor must have been AERATION.
Thus, the puzzle was solved. By bubbling air through a reaction mixture, the conversion of 4-thiouridine into a product was completed within 20 min (Table 1).7) The non-reproducible nature of the trial runs was due to inefficient and variable supply of air. Moreover, to my surprise the product turned out to be the sulfonate. Bisulfite is a reducing agent and yet the product was the same as the one produced in permanganate oxidation.
Table 1.
Air-dependent reaction of 4-thiouridine with bisulfite7)
| Conditions | Reaction extent at room temperature | ||
|---|---|---|---|
|
| |||
| 5 min | 60 min | 120 min | |
| Air-bubbling | 70% | 100% | 100% |
| N2-bubbling | 0% | 0% | |
| With mechanical stirring in an open flask, but without air-bubbling | 50% | 65% | |
| Air-bubbling in the absence of bisulfite | 0% | 0% | |
The compositions of reaction mixture (5 ml) were 0.1 mM 4-thiouridine, 10 mM sodium bisulfite, and 20 mM sodium phosphate buffer pH 6.9. The container used was a 20-ml Erlenmeyer flask.
It took almost a year to fully elucidate the mechanism of this reaction. Figure 3 shows the sequence of the events.8) The aerobic oxidation of bisulfite was known to be a free-radical-generating chain reaction:
The •SO3− radical thus formed can react with 4-thiouridine to form an –SSO3− derivative. This intermediate product can undergo nucleophilic substitution with SO32− to produce the sulfonate.
Fig. 3.
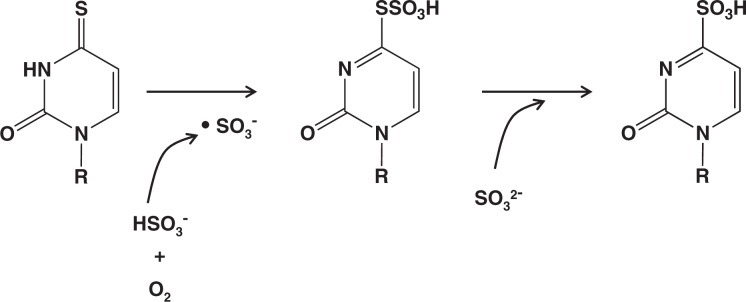
Conversion of 4-thiouridine mediated by aerobic oxidation of bisulfite.
If I had used a modern, digitalized spectrophotometer instead of the old-fashioned Beckman at that time, the sudden rapid fall of the absorbance could have escaped attention. The visual recognition of the quickly occurring decline in absorbance through the downward needle movement was the key for the discovery.
4. Unique usefulness of paper chromatography — Discovery of uracil-bisulfite adduct formation
The excitement of solving the puzzle of the bisulfite action toward 4-thiouridine led me to wonder whether bisulfite may act on major nucleosides as well. In the spring of 1969, Mr. Y. Wataya, then a student, started to collaborate with me (he is at present supervising the laboratory of Okayama University where I am currently visiting for work). I asked him to explore the possible reaction of bisulfite with uridine. He treated uridine with 1.5 molar equivalent NaHSO3 at pH 7, and performed paper chromatography for analysis. The solvent used for the development of the paper in an ascending manner was a mixture of n-butanol and aqueous acetic acid. After 7 hrs of patient waiting for the solvent to run up the sheet of paper to a distance of ∼20 cm, we took out the paper from the chromatography-tank and wasted no time to take a view against UV-light in the dark. The smell of the wet paper-sheet was terrible. The smell notwithstanding, we did careful viewing with great enthusiasm. Nothing, however, other than the starting material, uridine (Rf about 0.5), was detectable. We were disappointed, and the paper was hung in a draft chamber for drying (this was a necessary step before its eventual disposal).
Then, an important instant came. Two days later, Wataya told me that a very faint UV-absorbing zone showed up near the origin of the chromatographic paper. I viewed the paper, which still bore some acidic odor, and saw the faint zone. It had not been there two days before. I thought that a product was there, and that during the course of 2 day-standing it underwent a change into a UV-absorbing material. Moved by intuition, I said, “Let’s spray alkali to the paper and see what will happen”. We applied a mist of dilute NaOH to the paper and took a look against UV-light. There, the zone in question had intensified its absorption and the intensity rapidly grew stronger under our eyes. We exclaimed, “That’s it!”. The zone must have contained a product that was convertible to a UV-absorbing material on treatment with alkali. It turned out that the compound formed after alkali-treatment was uridine, the starting material.
The reaction between bisulfite and uridine produced an adduct, which can be reverted to uridine by alkali, but only slowly by acid. We were very excited and immediately started hard working to elucidate the whole picture of bisulfite reactivity with nucleosides. Within a week, we found that bisulfite can form a similar adduct with cytidine, and furthermore, that the cytidine-bisulfite can undergo deamination to form uridine-bisulfite.
A feature of paper chromatographic analysis is that some specific treatments can be performed on the paper-sheet, and, as in this case, a change occurring on the paper may be detected by direct viewing. In this way, we were luckily serendipitous again.
As my background was organic synthesis, my core expertise was isolation of pure products. I took solid uracil, suspended it in a small amount of water, heated the mixture at ∼70°C, and an excess of Na2SO3-NaHSO3 (3:1, w/w) was added to it. Within 1 min, a homogeneous solution of pH about 7 resulted, and after several minutes, precipitates began to appear. After 30 min of reaction, the precipitates were collected and recrystallized from dilute acetic acid. The crystalline material thus prepared proved to be the uracil-bisulfite adduct. By a full range of chemical and physical analyses, the structure was established as 5,6-dihydrouracil-6-sulfonate Na salt (Fig. 4).3),9)
Fig. 4.
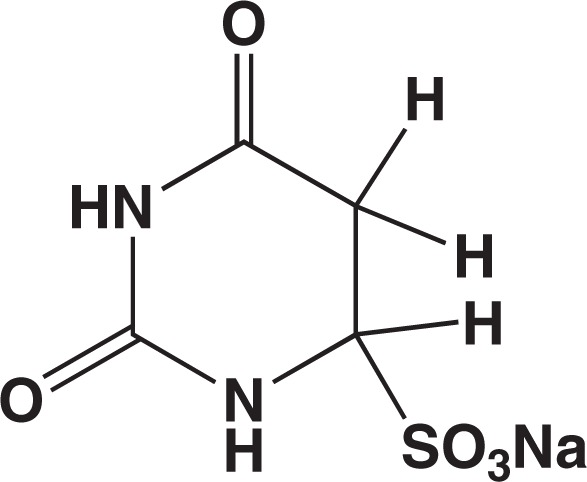
Uracil-bisulfite adduct.
5. Bisulfite-mediated deamination of cytosine
These observations were all surprising. Sodium bisulfite is a common laboratory reagent. It was unbelievable that nobody had noticed these reactions in the past. We completed piling up necessary data within several months, wrote a short report, and submitted it in November, 1969, to J. Am. Chem. Soc. as a Communication. It was smoothly accepted, and in the end of that year, a 3-page proof-prints came in. Attached there was a message, saying that a similar study from another laboratory was presently in press and that the corrected proof should be returned as quickly as possible. Thus, we sent it back immediately, and kept watching the journal. A Communication by Shapiro and coworkers appeared in the January 28th issue of the journal and ours in the next issue February 11, 1970.2),3) Both had a diagram of the reaction scheme as shown here in Fig. 5. Their submission was done in October, a month earlier than ours. We were lucky that the journal did not turn down our paper. It was in fact quite fair to publish these almost identical contents from two independent groups in a journal virtually simultaneously.
Fig. 5.
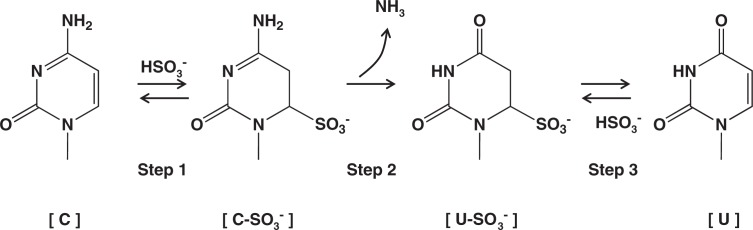
Bisulfite-mediated deamination of cytosine.
During these periods, we had accumulated a large volume of data about this new reaction. We wrote a full paper on these details and sent it to Biochemistry, a journal also of the American Chemical Society, in March, 1970. The paper was published in that journal in the fall of that year.9)
As shown in Fig. 5, the bisulfite-mediated deamination of cytosine proceeds by three steps. Step 1 is a reversible addition of HSO3− to cytosine. Step 2 is liberation of NH3 by hydrolysis, and Step 3 is release of HSO3− to regenerate the 5,6-double bond, forming uracil.
The [C-SO3−] formed in Step 1 is unstable in neutral solutions, regenerating [C] rapidly. On the other hand, [C-SO3−] is stable in acid: in fact, I found it easy to isolate crystalline cytidine-bisulfite as an inner salt from an acidic mixture of cytidine and sodium bisulfite. The equilibrium between [C] and [C-SO3−] is reached rapidly at pH 7 when bisulfite concentration is 0.5 M or higher. Step 2 is the key process of the overall procedure. It is the rate-limiting step for the conversion of [C] to [U-SO3−]. [U-SO3−] is stable in neutral conditions, but can be easily converted to [U] by alkali (Step 3). With respect to pH, the [C] to [U-SO3−] conversion is optimal at pH 5–6.
The competitive research of Shapiro’s group and ours had since continued for about a decade. Obviously, it accelerated the identification of factors affecting the bisulfite modification of DNA and RNA. Several of these factors are worthy of description here, as they are relevant in discussing, later in this article, the methodology used today in the analysis of epigenetic states of DNA.
6. Slow and complicated reaction with 5-methylcytosine
In early 1970s, 5-methylcytosine was known to constitute minor portions of cytosine in the genome of various species. According to a table produced by Hall and published in 1971,10) the 5-methylcytosine contents in organisms were 0.5–10% in animals, 10–37% in plants, 0–2% in bacteria, and 0.1–0.5% in phages. No biological roles of this minor constituent, however, were known. Mr. S. Iida, then a student collaborating with me, insisted to explore its reaction with bisulfite, perhaps because he instinctively felt its possible importance (Dr. Iida is presently running a laboratory in the National Institute of Basic Biological Sciences, Okazaki, studying plant epigenetics). It should be noted that the Hall table10) mentioned an outstanding, exceptional case for a phage: bacteriophage XP12 from Xanthomonas oryzae has DNA in which 100% of its cytosine residues are replaced by 5-methylcytosine.11) This phenomenon poses now an interesting issue as to why and how this replacement exists in this organism.
We observed that 5-methylcytosine can be deaminated by bisulfite, but much more slowly than cytosine.9) Further studies showed that the rate of the deamination is nearly two orders of magnitude lower than that for cytosine.12) Moreover, it was revealed that the deaminated products, thymine-bisulfite adducts are a mixture of two diastereomers.13) The structures of these isomers are shown in Fig. 6. They were produced in approximately 1 to 1 ratio. The trans isomer is the same adduct as that formed in the treatment of thymine with bisulfite (trans, regarding the –SO3− at position 6 and the –H at position 5). This trans isomer readily generates thymine on treatment with alkali. In contrast, the other one, cis adduct, is quite stable. It can be converted to thymine only with a harsh alkaline treatment. It had been common knowledge that cis–elimination is difficult to occur.14) Ehrlich and coworkers reported in 1980 that bisulfite-mediated deamination of 5-methylcytosine in DNA in fact produced such alkali-resistant products.15)
Fig. 6.
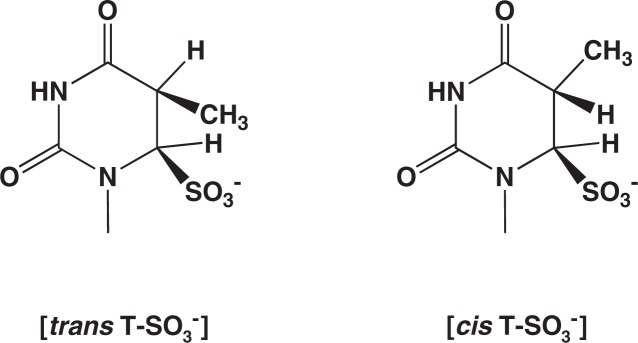
Two diastereomeric thymine-bisulfite adducts formed in the deamination of 5-methylcytosine.
This complicated situation for bisulfite modification of 5-methylcytosine could create an issue in the genomic sequencing presently practiced in epigenetic studies (see below).
7. Peculiar behavior of bisulfite — Low, but not high, concentration of bisulfite induces DNA degradation
Bisulfite is a fascinating agent. To me, the agent appears sometimes even mysterious. The 4-thiouridine-bisulfite interaction described above occurs very rapidly with 10 mM bisulfite, but only slowly with 1 M bisulfite. Why is it so? The explanation I settled on was that the reacting species is not bisulfite itself but the •SO3− radical generated by oxygen-bisulfite interaction. If the bisulfite concentration is high, the radical is short-lived, being decomposed by the sulfite ion surrounding the radical, so that no conversion of 4-thiouridine occurs.
The same low-concentration specific phenomenon was observed for treatments of DNA with bisulfite. Thus, the activity of B. subtilis DNA to transform the cell from Met− to Met+ was almost completely destroyed by treatment of the DNA with 20 mM bisulfite for 1 hr at pH 7 and 37 °C but only to a small extent by 1 M bisulfite.16) The ‘reverse’ dose dependence can be seen in Fig. 7. Hydroquinone, a radical scavenger, was very effective to prevent this bisulfite-mediated DNA degradation, and thus we concluded that •SO3−radical must be the agent responsible for this DNA degradation. DNA chain breaks caused by 10 mM bisulfite were directly detected by alkaline sucrose density gradient centrifugation.17) Again, hydroquinone was an effective inhibitor. The mechanism of this radical-mediated DNA degradation has remained to be investigated.
Fig. 7.

‘Reverse’ dose dependence in bisulfite-mediated inactivation of transforming DNA.16) It can been seen that the activity loss is smaller at higher concentrations of bisulfite.
While these studies were undergoing, I became aware that a similar low-concentration specific free-radical-mediated DNA damaging ability had been reported for hydroxylamine, an agent which can react with DNA bases forming covalently bonded adducts by ionic interactions.18) So, after all, the peculiar reversed dose-dependence is not a unique property of bisulfite. Still, it should be important to be aware of this phenomenon when one treats DNA with bisulfite, such as in genomic sequencing.
8. An unsolved problem in the mechanism
The mechanism of bisulfite-mediated deamination of cytosine was then shown to involve a mysterious action of bisulfite. The key process, [C-SO3−] to [U-SO3−] (Step 2 in Fig. 5), was revealed to be not a simple hydrolysis. This hydrolysis seemed to involve an additional role of bisulfite. If the hydrolysis is a simple attack of OH− to position 4 of [C-SO3−] to replace –NH2 with –OH, a theoretical consideration indicated that the relationship between the rate of the conversion [C] to [U-SO3−] and the concentration of bisulfite should be represented by a dotted line as shown in Fig. 8. However, the fact is that the observed rates of the reaction, as shown by the filled circles in the Figure, were far apart from the expected line. We found that by assuming an active participation of bisulfite in the process of this hydrolysis, a dose-response curve can be drawn fitting with those data points (the solid line in Fig. 8). How bisulfite catalyzes the hydrolysis, however, remains unknown until today.19) Shapiro’s team also made the same observation and considered various possibilities for the role of bisulfite in the hydrolysis, but they too encountered difficulty to uncover it.20)
Fig. 8.

Relationship between bisulfite concentration and rate of deamination of cytidine 5′-phosphate at pH 5.0 and 37 °C. The dotted line represents rates with no participation of bisulfite in hydrolyzing [C-SO3−], and the solid line those predicted in case of participation of bisulfite in the process of hydrolysis.
Nevertheless, the important fact is there; i.e., an increase in bisulfite concentration results in a more-than-linear increase in the rate of [C]-to-[U] conversion. This awareness is the basis of my recent effort for improving the method of bisulfite genomic sequencing (see below).
9. Bisulfite genomic sequencing, a major tool used for analyzing epigenetic states
Bisulfite modification of various bases, including minor constituents in RNA and DNA had been largely elucidated by the middle of 1970s, and these reactions and their use in biochemical studies were summarily presented in a review article.21) My contribution to science during the ensuing 1980s and 90s was mostly in the field of environmental mutagen research. As mutagens are DNA modification agents, I continued to enjoy solving puzzling phenomena associated with mutagenesis. During these periods, technologies for studying DNA advanced tremendously: e.g., base sequence determination, polymerase chain reaction (PCR) for amplification, and cloning for proliferation of individual DNA molecules. Also, biological significance of 5-methylcytosine had been revealed. In 1992, a group of Australian scientists invented an ingenious way of determining the sites of 5-methylcytosine in DNA.4) They treated denatured DNA with bisulfite to convert cytosine to uracil. Amplification by PCR of thus modified DNA produced a group of DNA molecules in which the original cytosines were replaced by thymines. Cloning into a vector, followed by sequencing gave DNA sequences in which all cytosines in the original had been changed into thymines. In these processes, 5-methylcytosines in the original DNA molecules stayed unchanged and the descendant DNA molecules possessed cytosines at these positions. In this way, one can determine the sites of 5-methylcytosines, and the whole procedure is called ‘bisulfite genomic sequencing’.
A large amount of data is now available with respect to the 5-methylcytosine sites in DNA, and the database is growing rapidly. These data are useful in diagnosing cancer in humans, and their use in basic biological sciences is expanding.
10. Device for preparing a high concentration bisulfite solution and its use in the DNA methylation analysis
It was my pleasure to learn and observe the recent situation of utilizing the bisulfite-chemistry that originated from my own work. Several years ago, I began to realize that it might be possible for me to contribute to the improvement of this chemical procedure: for one thing, the whole treatment may be speeded up. In the widely practiced procedure, the most time-consuming step is the incubation with bisulfite; 16–20 hr at 50–60 °C.22) I knew, as described above, that this slow step can be substantially quickened if the bisulfite concentration in the reaction solution can be increased (Fig. 8). In the practiced protocols, a sodium bisulfite solution of ∼5 M is used, 22),23) which is close to the saturation level. According to my experience, the ammonium salt is more soluble in water than the sodium salt of bisulfite. I began checking commercial reagent-catalogs, and soon found that there is a ‘50% ammonium bisulfite solution’ available from Wako Chemicals. A quick mental calculation made me realize that it contains bisulfite at 6.5 M! I obtained the reagent, which showed a pH of ∼4.5, and used it to dissolve additional bisulfite salts to produce a pH 5-solution containing bisulfite as much as possible. After several days of bench work, I succeeded to prepare a 10 M bisulfite solution (containing mostly ammonium, with a small portion of sodium, as cations).24)
Collaboration with DNA–chemistry experts Drs. K. Negishi and M. Shiraishi made it possible to explore the usefulness of this high concentration bisulfite in genomic sequencing. We showed that the incubation time can be significantly shortened by use of this agent, coupled with a high temperature for incubation.25),26) A 1-to-10 mixture of a DNA sample solution and 10 M bisulfite was incubated for 40 min at 70 °C. The DNA used was from a human cancer cell line, bearing 5-methyl-cytosines at known CpG sites. The results showed 100% [C]-to-[U] conversion, while 5-methylcytosines were completely preserved intact.25) Review articles that are intended to disseminate the usefulness of this methodology have been published.26)–28)
11. Issues and concluding remarks
From the beginning of the bisulfite genomic sequencing, an important issue inherent in the bisulfite treatment has been ‘DNA degradation’.4) The degradation has been believed to cause inefficient PCR performance of the treated DNA. In reality, however, there have been no systematic studies performed for elucidating the nature of the ‘degradation’ inflicted to DNA by the treatment. It is generally assumed that the treatment under acidic conditions would cause depurination, leading to DNA chain cleavage.
In addition, the possible effect of alkali-resistant adducts, occasionally formed from 5-methylcytosine during the treatment, must be investigated. Probably, such an adduct in the DNA chain would be a block against DNA polymerase in the polynucleotide synthesis. It is known that the presence of uracil-bisulfite adduct in the template DNA strongly inhibits the polynucleotide synthesis by E. coli DNA polymerase in vitro.29)
It should be pointed out that bisulfite treatment of DNA in general should be monitored more carefully. For example, the concentration of bisulfite in the incubation mixture should be quantified from time to time. A simple spectrophotometric method has been described in my previous publications.24),26)–28) Another recommended practice for the monitoring would be pH-measurement. For an extensive decay of bisulfite during treatments, a change in pH is a sensitive indicator.
In conclusion, I now feel that the presently available data for DNA methylation need carefully be looked at, and that a scientifically sound, assured methodology free from any false positive or false negative assignments be established as soon as possible.
12. Acknowledgments
The late Prof. T. Ukita of the University of Tokyo is deeply thanked for his generosity and encouragement during the time of my research conducted in his department. I thank all the collaborators who contributed to the studies on bisulfite modification of nucleosides and nucleotides. The recent development in the use of high concentration bisulfite has been performed in collaboration with Dr. K. Negishi of Nihon Pharmaceutical University and Dr. M. Shiraishi of International University of Health and Welfare, to both of whom I am grateful. Dr. Y. Wataya of Okayama University is particularly thanked for his help in my writing here about the critical experiments he did in 1969.
Profile
Hikoya Hayatsu, born in 1934, studied organic chemistry and performed experimental work on the synthesis and reaction kinetics of heterocyclic aromatics during his undergraduate- and graduate-schooling courses at the University of Tokyo, Faculty of Pharmaceutical Sciences, 1956–1961. He was given Ph.D. in 1963 for research in nucleotide synthesis. He did his post-doc work with Prof. H. G. Khorana at the Univerisity of Wisconsin, 1964–1967, on the genetic code and on the polymer-supported oligonucleotide synthesis. He worked as Associate Professor at the University of Tokyo, in the laboratory of Professor T. Ukita, Department of Hygienic Chemistry, Faculty of Pharmaceutical Sciences, with his research focused on chemical modifications of nucleic acid constituents. He was Professor at Okayama University, Faculty of Pharmaceutical Sciences, during 1978–2000. His main research interest in that period was environmental mutagenesis, particularly on its inhibition; e.g., elucidation of antimutagenic mechanisms of porphyrin compounds such as chlorophyll and hemin. That study led him to the invention of a mutagen-trapping agent, blue cotton, a cellulose-supported copper phthalocyanine derivative useful for adsorbing polycyclic aromatic carcinogens and mutagens that are present in tiny amounts in environmental materials such as food and surface waters (the river-, lake- and sea-waters). In 2001, he organized the 8th International Conference on Environmental Mutagens, in Shizuoka, October 21–26, which was a success attended by 800 scientists worldwide in spite of the grave disaster of September 11th in the US. He served as Editor for Mutation Research, Genetic Toxicology and Environmental Mutagenesis, 1997–2006. In 1998, he was bestowed Purple Ribbon Award from the Japanese Government for his studies of nucleic acid chemistry. He worked as Dean at the School of Pharmacy, Shujitsu University, 2003–2007, and is presently Visiting Emeritus Professor at Okayama University, Department of Medicinal Chemistry.

References
- 1).Calladine, C.R., Drew, H.R., Luisi, B.F. and Travers, A.A. (2004) Understanding DNA, the Molecule and How it Works, 3rd ed., Elsevier, Amsterdam, pp. 270–294 [Google Scholar]
- 2).Shapiro, R., Servis, R.E. and Welcher, M. (1970) Reactions of uracil and cytosine derivatives with sodium bisulfite. A specific deamination method. J. Am. Chem. Soc. 92, 422–424 [Google Scholar]
- 3).Hayatsu, H., Wataya, Y. and Kai, K. (1970) The addition of sodium bisulfite to uracil and cytosine. J. Am. Chem. Soc. 92, 724–726 [DOI] [PubMed] [Google Scholar]
- 4).Frommer, M., McDonald, L.E., Millar, D.S., Collis, C.M., Watt, F., Grigg, G.W., Molloy, P.L. and Paul, C.L. (1992) A genomic sequencing protocol that yields a positive display of 5-methylcytosine residues in individual DNA strands. Proc. Natl. Acad. Sci. USA 80, 1579–1583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5).Hayatsu, H. and Ukita, T. (1967) The selective degradation of pyrimidines in nucleic acids by permanganate oxidation. Biochem. Biophys. Res. Commun. 29, 556–561 [DOI] [PubMed] [Google Scholar]
- 6).Hayatsu, H. and Yano, M. (1969) Permanganate oxidation of 4-thiouracil derivatives. Isolation and properties of 1-substituted-2-pyrimidone-4-sulfonates. Tetrahedron Lett. 9, 755–758 [DOI] [PubMed] [Google Scholar]
- 7).Hayatsu, H. (1969) The oxygen-catalyzed reaction between 4-thiouridine and sodium sulfite. J. Am. Chem. Soc. 91, 5693–5694 [DOI] [PubMed] [Google Scholar]
- 8).Hayatsu, H. and Inoue, M. (1971) The oxygen-mediated reaction between 4-thiouracil derivatives and bisulfite. Isolation and characterization of 1-methyluracil-4-thiosulfonate as an intermediate in the formation of 1-methyluracil-4-sulfonate. J. Am. Chem. Soc. 93, 2301–2306 [DOI] [PubMed] [Google Scholar]
- 9).Hayatsu, H., Wataya, Y., Kai, K. and Iida, S. (1970) Reaction of sodium bisulfite with uracil, cytosine and their derivatives. Biochemistry 9, 2858–2865 [DOI] [PubMed] [Google Scholar]
- 10).Hall, R. (1971) The Modified Nucleosides in Nucleic Acids. Columbia University Press, New York, pp. 282–286 [Google Scholar]
- 11).Kuo, T., Huang, T. and Teng, M. (1968) 5-Methylcytosine replacing cytosine in the deoxyribonucleic acid of a bacteriophage for Xanthomonas oryzae. J. Mol. Biol. 34, 373–375 [DOI] [PubMed] [Google Scholar]
- 12).Hayatsu, H. and Shiragami, M. (1979) Reaction of bisulfite with 5-hydroxymethyl group in pyrimidines and in phage DNAs. Biochemistry 18, 632–637 [DOI] [PubMed] [Google Scholar]
- 13).Shiragami, M., Iida, S., Kudo, I. and Hayatsu, H. (1975) Formation of diastereomers of 5,6-dihydrothymine-6-sulfonate by deamination of 5-methylcytosine with bisulfite. Chem. Pharm. Bull. 23, 3027–3029 [Google Scholar]
- 14).Newman, M.S. (1956) Steric Effects in Organic Chemistry. Wiley, New York [Google Scholar]
- 15).Wang, R.Y., Gehrke, C.W. and Ehrlich, M. (1980) Comparison of bisulfite modification of 5-methyldeoxycytidine and deoxycytidine residues. Nucleic Acids Res. 8, 4777–4790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16).Inoue, M., Hayatsu, H. and Tanooka, H. (1972) Concentration effect of bisulfite on the inactivation of transforming activity of DNA. Chem. Biol. Interactions 5, 85–95 [DOI] [PubMed] [Google Scholar]
- 17).Hayatsu, H. and Miller, R.C. (1972) The cleavage of DNA by the oxygen-dependent reaction of bisulfite. Biochem. Biophys. Res. Commun. 46, 120–124 [DOI] [PubMed] [Google Scholar]
- 18).Freese, E. and Freese, E.B. (1965) The oxygen effect on deoxyribonucleic acid inactivation by hydroxylamine. Biochemistry 4, 2419–2433 [Google Scholar]
- 19).Sono, M., Wataya, Y. and Hayatsu, H. (1973) Role of bisulfite in the deamination and the hydrogen isotope exchange of cytidylic acid. J. Am. Chem. Soc. 95, 4745–4749 [DOI] [PubMed] [Google Scholar]
- 20).Shapiro, R., DiFate, V. and Welcher, M. (1974) Deamination of cytosine derivatives by bisulfite. Mechanism of the reaction. J. Am. Chem. Soc. 96, 906–912 [DOI] [PubMed] [Google Scholar]
- 21).Hayatsu, H. (1976) Bisulfite modification of nucleic acids and their constituents. Progress in Nucleic Acid Res. and Mol. Biol. 16, 75–124 [DOI] [PubMed] [Google Scholar]
- 22).Eads, C.A. and Laird, P.W. (2002) InDNA Methylation Protocols. Methods in Molecular Biology (eds. Millis, K.I. and Ramsahoye, B.H.). vol. 200, Humana Press, NJ, USA, pp. 71–85 [Google Scholar]
- 23).Clark, S.J., Stathem, A., Stirzaker, C., Molloy, P.L. and Frommer, M. (2006) DNA methylation: Bisulfite modification and analysis. Nature Protocols 1, 2353–2364 [DOI] [PubMed] [Google Scholar]
- 24).Hayatsu, H., Negishi, K. and Shiraishi, M. (2004) DNA methylation analysis: speedup of bisulfite-mediated deamination of cytosine in the genomic sequencing procedure. Proc. Jpn. Acad., Ser B 80, 189–194 [DOI] [PubMed] [Google Scholar]
- 25).Shiraishi, M. and Hayatsu, H. (2004) High-speed conversion of cytosine to uracil in bisulfite genomic sequencing analysis of DNA methylation. DNA Res. 11, 409–415 [DOI] [PubMed] [Google Scholar]
- 26).Hayatsu, H. (2006) Bisufite modification of cytosine and 5-methylcytosine as used in epigenetic studies. Genes and Environment 28, 1–8 [Google Scholar]
- 27).Hayatsu, H. (2008) The bisulfite genomic sequencing used in the analysis of epigenetic states, a technique in the emerging environmental geno-toxicology research. Mutat. Res. 659, 77–82 [DOI] [PubMed] [Google Scholar]
- 28).Hayatsu, H., Shiraishi, M., Negishi, K. (2008) Bisulfite modification of DNA for analyzing 5-methylcytosine and cytosine. Current Protocols in Nucleic Acid Chemistry 33, 6.10.1–6.10.15, Wiley, NC, USA [Google Scholar]
- 29).Kai, K., Tsuruo, T., Hayatsu, H. (1974) The effect of bisulfite modification on the template activity of DNA for DNA polymerase I. Nucleic Acids Res. 1, 889–899 [DOI] [PMC free article] [PubMed] [Google Scholar]