

Abstract
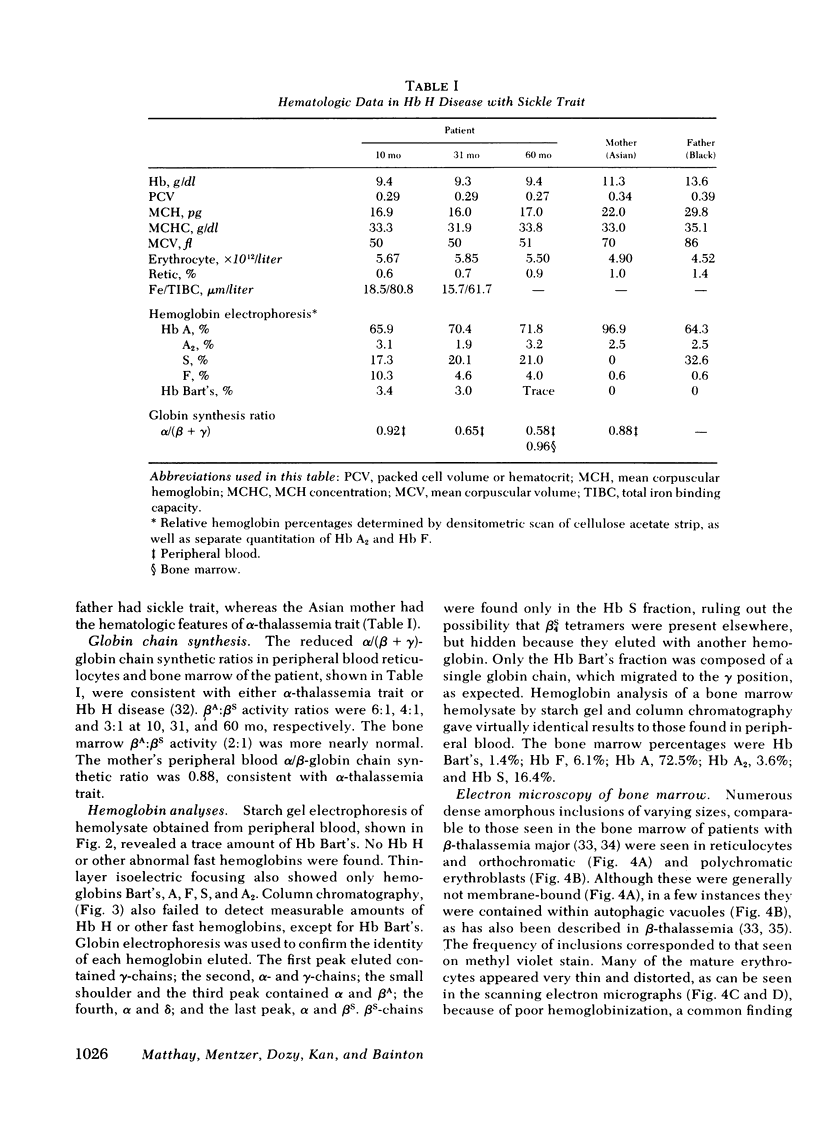
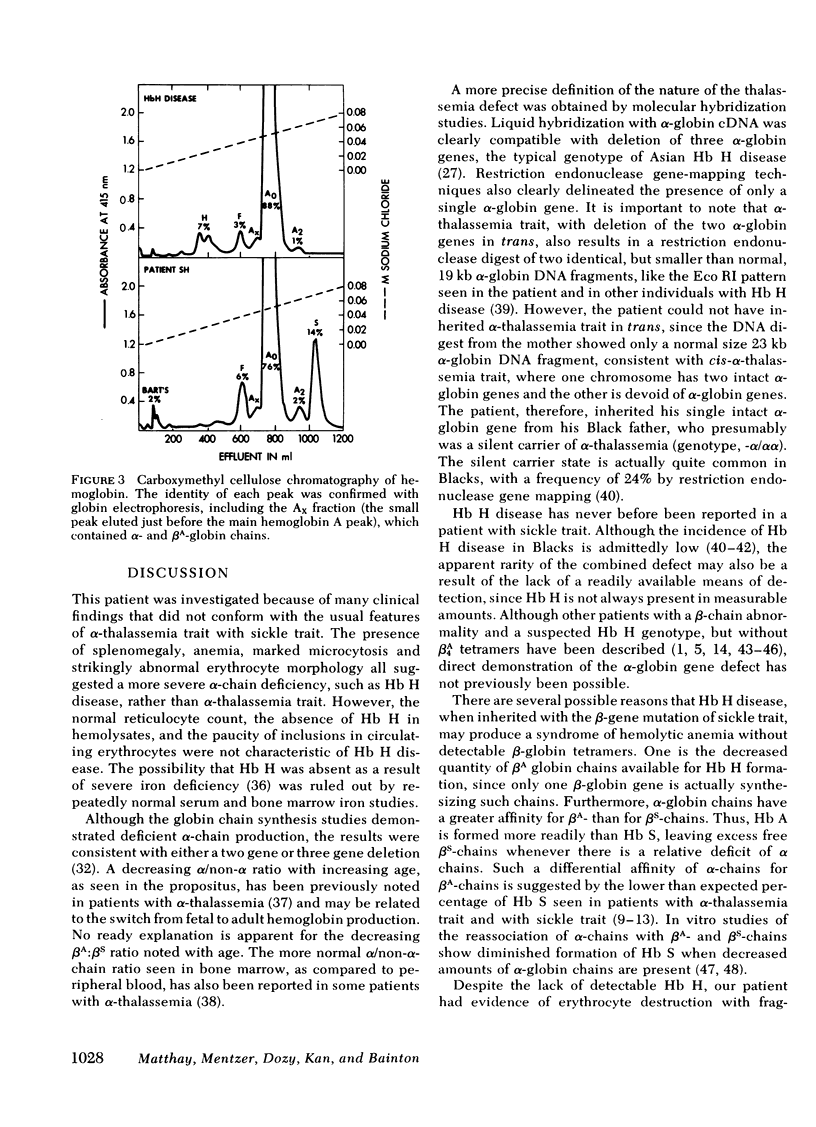
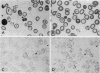
The rarity of hemoglobin (Hb) H disease in combination with sickle trait may be due in part to the absence of actual Hb H in individuals who, nonetheless, have inherited the deletion of three α-globin genes. We describe here a boy with persistent microcytic, hypochromic anemia despite adequate iron stores, who exhibited splenomegaly with a normal reticulocyte count and only rare inclusions in circulating erythrocytes. Starch gel electrophoresis and isoelectric focusing at age 5 yr showed 21% Hb S, persistent Hb Bart's, but no Hb H. Recticulocyte α/non-α globin chain synthesis ratio was 0.58 at age 5. The mother (Asian) had laboratory evidence of α-thalassemia trait and the father (Black) had sickle trait. The nature of α-thalassemia in this patient was investigated both by liquid hybridization and by the Southern method of gene mapping, in which DNA is digested with restriction endonucleases and the DNA fragments that contained the α-globin structural gene identified by hybridization with complementary DNA. The patient had only one α-globin structural gene, located in a DNA fragment shorter than that found in normal or α-thalassemia trait individuals, but similar to that present in other patients with Hb H disease. Morphologic studies of bone marrow by light and electron microscopy revealed erythroid hyperplasia with inclusions in polychromatic and orthochromatic erythroblasts, suggesting early precipitation of an unstable hemoglobin. The lack of demonstrable Hb H may be the result of both diminished amounts of βA available for Hb H formation (since one β-globin gene is βS) and the greater affinity of α-chains for βA than βS-globin chains leading to the formation of relatively more Hb A than Hb S. The presence of a βS gene may thus modify the usual clinical expression of Hb H disease.
Full text
PDF







Images in this article




Selected References
These references are in PubMed. This may not be the complete list of references from this article.
- AKSOY M. THE FIRST OBSERVATION OF HOMOZYGOUS HEMOGLOBIN S-ALPHA THALASSEMIA DISEASE AND TWO TYPES OF SICKLE CELL THALASSEMIA DISEASE: (A) SICKLE CELL-ALPHA THALASSEMIA DISEASE, (B) SICKLE CELL-BETA THALASSEMIA DISEASE. Blood. 1963 Dec;22:757–769. [PubMed] [Google Scholar]
- BETKE K., MARTI H. R., SCHLICHT I. Estimation of small percentages of foetal haemoglobin. Nature. 1959 Dec 12;184(Suppl 24):1877–1878. doi: 10.1038/1841877a0. [DOI] [PubMed] [Google Scholar]
- Bainton D. F., Ullyot J. L., Farquhar M. G. The development of neutrophilic polymorphonuclear leukocytes in human bone marrow. J Exp Med. 1971 Oct 1;134(4):907–934. doi: 10.1084/jem.134.4.907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bank A., Mears G., Weiss R., O'Donnell J. V., Natta C. Preferential binding of beta s globin chains associated with stroma in sickle cell disorders. J Clin Invest. 1974 Oct;54(4):805–809. doi: 10.1172/JCI107820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basset P., Beuzard Y., Garel M. C., Rosa J. Isoelectric focusing of human hemoglobin: its application to screening, to the characterization of 70 variants, and to the study of modified fractions of normal hemoglobins. Blood. 1978 May;51(5):971–982. [PubMed] [Google Scholar]
- Bate C. M., Humphries G. Alpha-beta thalassaemia. Lancet. 1977 May 14;1(8020):1031–1034. doi: 10.1016/s0140-6736(77)91261-2. [DOI] [PubMed] [Google Scholar]
- Bellevue R., Dosik H., Rieder R. F. Alpha thalassaemia in American blacks: a study of a family with five cases of haemoglobin H disease. Br J Haematol. 1979 Feb;41(2):193–202. doi: 10.1111/j.1365-2141.1979.tb05848.x. [DOI] [PubMed] [Google Scholar]
- Beuzard Y., Tulliez M., Breton-Gorius J., Griscelli C., Cosson A., Schaison G. Beta thalassemia with reticulocytopenia: clinical, biochemical, and ultrastructural studies. Blood Cells. 1978;4(1-2):269–289. [PubMed] [Google Scholar]
- Brittenham G. Genetic model for observed distributions of proportions of haemoglobin in sickle-cell trait. Nature. 1977 Aug 18;268(5621):635–636. doi: 10.1038/268635a0. [DOI] [PubMed] [Google Scholar]
- DeSimone J., Kleve L., Longley M. A., Shaeffer J. Unbalanced globin chain synthesis in reticulocytes of sickle cell trait individuals with low concentrations of hemoglobin S. Biochem Biophys Res Commun. 1974 Jul 24;59(2):564–569. doi: 10.1016/s0006-291x(74)80017-3. [DOI] [PubMed] [Google Scholar]
- Dozy A. M., Kabisch H., Baker J., Koenig H. M., Kurachi S., Stamatoyannopoulos G., Todd D., Kan Y. W. The molecular defects of alpha-thalassemia in the Filipino. Hemoglobin. 1977;1(6):539–546. doi: 10.3109/03630267709003418. [DOI] [PubMed] [Google Scholar]
- Embury S. H., Lebo R. V., Dozy A. M., Kan Y. W. Organization of the alpha-globin genes in the Chinese alpha-thalassemia syndromes. J Clin Invest. 1979 Jun;63(6):1307–1310. doi: 10.1172/JCI109426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FESSAS P. Inclusions of hemoglobin erythroblasts and erythrocytes of thalassemia. Blood. 1963 Jan;21:21–32. [PubMed] [Google Scholar]
- Fessas P., Yatachanas X. Intraerythroblastic instability of hemoglobin beta-4 (Hgb H). Blood. 1968 Mar;31(3):323–331. [PubMed] [Google Scholar]
- HUISMAN T. H. Properties and inheritance of the new fast hemoglobin type found in umbilical cord blood samples of Negro babies. Clin Chim Acta. 1960 Sep;5:709–718. doi: 10.1016/0009-8981(60)90013-9. [DOI] [PubMed] [Google Scholar]
- Honig G. R., Gunay U., Mason R. G., Vida L. N., Ferenc C. Sickle cell syndromes. I. Hemoglobin SC-alpha-thalassemia. Pediatr Res. 1976 Jun;10(6):613–620. doi: 10.1203/00006450-197606000-00010. [DOI] [PubMed] [Google Scholar]
- Honig G. R., Koshy M., Mason R. G., Vida L. N. Sickle cell syndromes. II. The sickle cell anemia-alpha-thalassemia syndrome. J Pediatr. 1978 Apr;92(4):556–561. doi: 10.1016/s0022-3476(78)80287-x. [DOI] [PubMed] [Google Scholar]
- Huisman T. H., Schroeder W. A., Brodie A. N., Mayson S. M., Jakway J. Microchromatography of hemoglobins. II. A simplified procedure for the determination of hemoglobin A2. J Lab Clin Med. 1975 Oct;86(4):700–702. [PubMed] [Google Scholar]
- Huisman T. H. Trimodality in the percentages of beta chain variants in heterozygotes: the effect of the number of active Hbalpha structural loci. Hemoglobin. 1977;1(4):349–382. doi: 10.3109/03630267708996895. [DOI] [PubMed] [Google Scholar]
- Kan Y. W., Dozy A. M. Polymorphism of DNA sequence adjacent to human beta-globin structural gene: relationship to sickle mutation. Proc Natl Acad Sci U S A. 1978 Nov;75(11):5631–5635. doi: 10.1073/pnas.75.11.5631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kan Y. W., Golbus M. S., Dozy A. M. Prenatal diagnosis of alpha-thalassemia. Clinical application of molecular hybridization. N Engl J Med. 1976 Nov 18;295(21):1165–1167. doi: 10.1056/NEJM197611182952104. [DOI] [PubMed] [Google Scholar]
- Kan Y. W., Nathan D. G. Mild thalassemia: the result of interactions of alpha and beta thalassemia genes. J Clin Invest. 1970 Apr;49(4):635–642. doi: 10.1172/JCI106274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kan Y. W., Schwartz E., Nathan D. G. Globin chain synthesis in the alpha thalassemia syndromes. J Clin Invest. 1969 Nov;47(11):2512–2522. doi: 10.1172/JCI105933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H. C., Weierbach R. G., Friedman S., Schwartz E. Globin biosynthesis in sickle cell, Hb SC, and Hb C diseases. J Pediatr. 1977 Jul;91(1):13–20. doi: 10.1016/s0022-3476(77)80434-4. [DOI] [PubMed] [Google Scholar]
- Nathan D. G., Gunn R. B. Thalassemia: the consequences of unbalanced hemoglobin synthesis. Am J Med. 1966 Nov;41(5):815–830. doi: 10.1016/0002-9343(66)90039-8. [DOI] [PubMed] [Google Scholar]
- Natta C. Failure of the alpha-thalassemia gene to decrease the severity of sickle cell anemia. Blood. 1978 Jun;51(6):1163–1168. [PubMed] [Google Scholar]
- O'Brien R. T. The effect of iron deficiency on the expression of hemoglobin H. Blood. 1973 Jun;41(6):853–856. [PubMed] [Google Scholar]
- Polliack A., Rachmilewitz E. A. Ultrastructural studies in -thalassaemia major. Br J Haematol. 1973 Mar;24(3):319–326. doi: 10.1111/j.1365-2141.1973.tb01656.x. [DOI] [PubMed] [Google Scholar]
- Salmon J. E., Nudel U., Schiliro G., Natta C. L., Bank A. Quantitation of human globin chain synthesis by cellulose acetate electrophoresis. Anal Biochem. 1978 Nov;91(1):146–157. doi: 10.1016/0003-2697(78)90825-4. [DOI] [PubMed] [Google Scholar]
- Schroeder W. A., Pace L. A. Chromatography of hemoglobins on CM-cellulose with bis-tris and sodium chloride developers. J Chromatogr. 1976 Apr 7;118(3):295–302. doi: 10.1016/s0021-9673(00)82166-4. [DOI] [PubMed] [Google Scholar]
- Schwartz E. Abnormal globin synthesis in thalassemic red cells. Semin Hematol. 1974 Oct;11(4):549–567. [PubMed] [Google Scholar]
- Schwartz E., Atwater J. -thalassemia in the American Negro. J Clin Invest. 1972 Feb;51(2):412–418. doi: 10.1172/JCI106827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaeffer J. R., DeSimone J., Kleve L. J. Hemoglobin synthesis studies of a family with alpha-thalassemia trait and sickle cell trait. Biochem Genet. 1975 Dec;13(11-12):783–788. doi: 10.1007/BF00484410. [DOI] [PubMed] [Google Scholar]
- Shaeffer J. R., Kingston R. E., McDonald M. J., Bunn H. F. Competition of normal beta chains and sickle haemoglobin beta chains for alpha chains as a post-translational control mechanism. Nature. 1978 Dec 7;276(5688):631–633. doi: 10.1038/276631a0. [DOI] [PubMed] [Google Scholar]
- Southern E. M. Detection of specific sequences among DNA fragments separated by gel electrophoresis. J Mol Biol. 1975 Nov 5;98(3):503–517. doi: 10.1016/s0022-2836(75)80083-0. [DOI] [PubMed] [Google Scholar]
- Steinberg M. H., Adams J. G., 3rd, Dreiling B. J. Alpha thalassaemia in adults with sickle-cell trait. Br J Haematol. 1975 May;30(1):31–37. doi: 10.1111/j.1365-2141.1975.tb00514.x. [DOI] [PubMed] [Google Scholar]
- TUCHINDA S., RUCKNAGEL D. L., MINNICH V., BOONYAPRAKOB U., BALANKURA K., SUVATEE V. THE COEXISTENCE OF THE GENES FOR HEMOGLOBIN E AND ALPHA-THALASSEMIA IN THAIS, WITH RESULTANT SUPPRESSION OF HEMOGLOBIN E SYNTHESIS. Am J Hum Genet. 1964 Sep;16:311–335. [PMC free article] [PubMed] [Google Scholar]
- Taylor J. M., Dozy A., Kan Y. W., Varmus H. E., Lie-Injo L. E., Ganesan J., Todd D. Genetic lesion in homozygous alpha thalassaemia (hydrops fetalis). Nature. 1974 Oct 4;251(5474):392–393. doi: 10.1038/251392a0. [DOI] [PubMed] [Google Scholar]
- Wasi P., Sookanek M., Pootrakul S., Na-Nakorn S., Suingdumrong A. Haemoglobin E and alpha-Thalassaemia. Br Med J. 1967 Oct 7;4(5570):29–32. doi: 10.1136/bmj.4.5570.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weatherall D. J., Clegg J. B., Blankson J., McNeil J. R. A new sickling disorder resulting from interaction of the genes for haemoglobin S and alpha-thalassaemia. Br J Haematol. 1969 Dec;17(6):517–526. doi: 10.1111/j.1365-2141.1969.tb01402.x. [DOI] [PubMed] [Google Scholar]
- Yataganas X., Fessas P. The pattern of hemoglobin precipitation in thalassemia and its significance. Ann N Y Acad Sci. 1969 Nov 20;165(1):270–287. doi: 10.1111/j.1749-6632.1969.tb27797.x. [DOI] [PubMed] [Google Scholar]
- van Enk A., Lang A., White J. M., Lehmann H. Benign obstetric history in women with sickle-cell anaemia associated with -thalassaemia. Br Med J. 1972 Dec 2;4(5839):524–526. doi: 10.1136/bmj.4.5839.524. [DOI] [PMC free article] [PubMed] [Google Scholar]