

Abstract
Ribosome biogenesis – the complex and highly coordinated cellular process leading to the production of ribosomes – is strictly dependent on the activity of RNA polymerase I (Pol I) transcriptional machinery. Pol I activity is continually increased in proliferating cells to sustain the increased demand for ribosome production and protein synthesis, which are necessary for appropriate cell growth and division. The integrity of the process of ribosome biogenesis represents an important sensor of cellular stress: when this process is altered, a tumor suppressor response is triggered, which leads to proliferative arrest. The present review focuses on the possible implications of Pol I targeting in the treatment of human malignancies.
Keywords: RNA polymerase I inhibitors, ribosome biogenesis, cell cycle control, cancer, Pol I
Introduction
The genome of eukaryotic cells is transcribed by means of three different enzymes, which carry out the synthesis of different types of RNA: RNA polymerase I (Pol I) synthesizes the precursor rRNA 45S, which, when mature, forms the major RNA part of the ribosome;1 RNA polymerase II (Pol II) synthesizes the precursors of mRNAs and most small nucleolar RNA, small nuclear RNA, and microRNAs;2,3 and RNA polymerase III (Pol III), which synthesizes transfer RNAs, rRNA 5S, and other small RNAs.4
Of the three polymerases, RNA Pol I is considered the most actively engaged (or, the “busiest”), since it accounts for at least 60% of cell transcriptional activity.5 RNA Pol I activity is further increased in proliferating cells and/or neoplastic cells to sustain increased production of the ribosomes required for the protein synthesis necessary for unbridled cell growth.
The present review focuses on both the role of RNA Pol I in human malignancies and the possible implications of its targeting in cancer treatment. To introduce the topic, a brief summary of both the structure of the Pol I machinery and the functional importance of its molecular components is provided.
The Pol I transcriptional machinery
The RNA Pol I holoenzyme is a complex formed by 14 subunits. In eukaryotes, all three nuclear polymerases share some components, but the subunits located in the core of the complex, which retain the catalytic activity, are specific for each polymerase.
The initiation of ribosomal DNA (rDNA) transcription requires the assembly of a multiprotein complex including Pol I and a number of basal transcription initiation factors at the rDNA promoter. In mammalian cells, the action of two of these basal factors, upstream binding factor (UBF) and selectivity factor 1 (SL1), synergistically regulates Pol I recruitment to the rDNA gene promoter and transcriptional initiation.6 UBF binds both to a specific region upstream of the rDNA core promoter, termed the “upstream core element,” and to a guanine-cytosine-rich element in the core promoter, thus allowing the subsequent binding of SL1, which confers promoter specificity, and the engagement of Pol I. After initiation, the transcript may be elongated by Pol I. The elongation phase of rDNA gene transcription appears to be less finely regulated than initiation; however, there is evidence indicating that rRNA synthesis might also be regulated during this step.7,8 The transcription ends at specific sequence signals – termed “terminator elements” – with the aid of specific termination factors, such as both DNA-bound transcription terminator factor I (TTF-I) and Pol I transcript release factor (PTRF).9 TTF-I also affects transcription initiation by Pol I and binds several sites upstream of the Pol I promoter.10
Figure 1 summarizes the organization of the ribosomal gene unit and shows the major basal factors binding to the promoter region that are required for the transcription of ribosomal genes transcription. Mammalian cells contain hundreds of tandemly repeated rDNA genes. The transcription of ribosomal genes by Pol I produces 45S RNA precursor molecules, which are then specifically site-modified and processed to generate the mature 18S, 5.8S, and 28S rRNAs. Mature rRNAs are then assembled with 5S rRNAs (which are transcribed in the nucleoplasm by RNA Pol III) and with ribosomal proteins to form the large 60S and the small 40S subunits of mature ribosomes. The large 60S subunit contains one each of the 28S, 5.8S, and 5S RNAs, together with 47 ribosomal proteins, whereas the small 40S subunit contains one 18S RNA and 32 ribosomal proteins. Since the rate of ribosome production in the cell is strictly dependent on RNA Pol I activity, rRNA transcription is considered the limiting step in ribosome biogenesis.11
Figure 1.
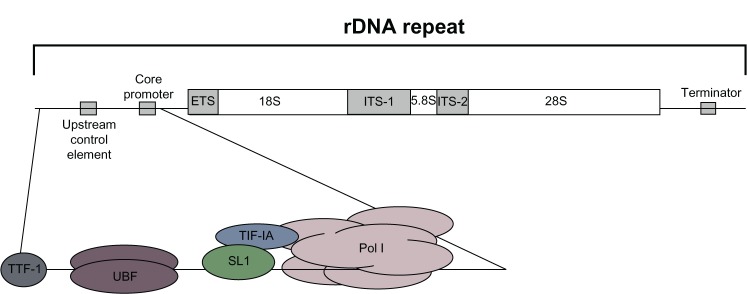
Schematic representation of the organization of the ribosomal gene unit. The scheme illustrates the organization of the ribosomal gene unit, which is found tandemly repeated approximately 400 times in the human genome. One unit includes the promoter, the transcribed region, and the terminator. The major basal factors binding to the promoter region and required for ribosomal gene transcription are also shown.
Abbreviations: ETS, external transcribed region; ITS, internal transcribed region; rDNA, ribosomal DNA; SL1, selectivity factor 1; TIF-1A, transcription initiation factor 1A; TTF-I, transcription terminator factor I; UBF, upstream binding factor; Pol I, RNA polymerase l.
The role of ribosomal biogenesis in malignancy
Long before a series of studies conducted from the late 1950s through the early 1960s made it possible to establish that the nucleolus is the site of ribosome production, investigation into the relationship between ribosome biogenesis and cancer had been begun unknowingly. In fact, in the late nineteenth century, “nucleolar hypertrophy” – which we now know is the morphological counterpart of an upregulated ribosome biogenesis – had started to attract the attention of tumor pathologists as a peculiar morphological aspect of cancer cells.12–15 Since then, the relationship between nucleolar function and cancer has been studied in great depth and, more recently, studies have been devoted mainly to clarifying the cause of the upregulated ribosome production in cancer cells, and to ascertaining whether an upregulated ribosome biogenesis of normal cells might be responsible for their neoplastic transformation. The major results obtained by these two research topics will be reported separately.
Why ribosome biogenesis is upregulated in cancer cells
In proliferating cells, there is a higher biosynthetic demand for increasing the production of cell constituents, which should ensure that daughter cells have the necessary complement for survival and normal functioning.16,17 This result is accomplished by the increased synthesis of proteins which, in turn, is induced by an upregulation of the rate of ribosome production. The generally high ribosome biogenesis in cancer cells is a consequence of the fact that ribosome production is mainly controlled by the same products of proto-oncogene and tumor suppressor genes that also control cell proliferation. In fact, in cancer cells, the changes in either tumor suppressors or proto-oncogenes, which are responsible for uncontrolled cell proliferation, also upregulate ribosome biogenesis.18,19
Regarding the relationship between oncogenes and ribosome biogenesis, it is worth noting that cell proliferation is stimulated by mitogens and growth factors that activate the c-myc proto-oncogene product (MYC),20 which is the major factor responsible for variations in the ribosome biogenesis rate. In fact, all the steps of ribosome biogenesis are controlled by MYC: the product of this oncogene increases Pol I activity by recruiting SL1 to promoters, stimulates ribosomal protein synthesis by enhancing Pol II transcription, and stimulates Pol III transcription by activating transcription factor for polymerase III B (TFIIIB).21–23
Mitogens and growth factors also trigger the extracellular signal-regulated kinase (mitogen-activated protein kinases/extracellular signal-regulated kinases) pathway. This leads to the activation of both Pol I transcription, through the phosphorylation of UBF,7,24 and Pol III transcription, through the phosphorylation TFIIIB.25 Also, mammalian target of rapamycin (mTOR) is stimulated by mitogens and growth factors. The activated mTOR induces Pol I transcription by activating UBF and transcription initiation factor 1A, and Pol III transcription by facilitating the association of TFIIIB and transcription factor for polymerase III C with 5S rRNA genes.26
In this context, two other factors, nuclear ErbB227 and the proto-oncogene PELP1,28 have been described to stimulate rRNA transcription even though their mechanism of action on the Pol I transcription complex has not yet been clearly clarified.
Regarding the relationship between tumor suppressors and ribosome biogenesis, it should be remembered that the tumor suppressors which modulate cell cycle progression also control ribosome biogenesis. Retinoblastoma protein (pRB) negatively controls the passage through the G1/S phase restriction point by binding to E2F1, a transcription factor whose target gene products are necessary for the entry and progression through the S phase. pRB inhibits both rRNA synthesis by binding to UBF29–32 and Pol III transcription by binding to TFIIIB.33,34 During the G1 phase, a phosphorylation of pRB occurs which functionally inactivates the tumor suppressor, thus allowing the cell to progress to the S phase. At the same time, phosphorylation of pRB, in addition to facilitating cell cycle progression, also induces a progressive increase in the rRNA transcription from the G1 phase to G2 phase by removing its inhibitory effects on Pol I and Pol III function. p53, the other major tumor suppressor which negatively controls cell cycle progression, directly hinders ribosome biogenesis. p53 binds to the selectivity factor SL1, which is necessary for Pol I recruitment to the rRNA gene promoter,35 and to TFIIIB,34 thus inhibiting Pol I and Pol III transcription, respectively. This inhibitory effect is reduced in cycling cells as a consequence of a downregulation of p53 expression. p14ARF contributes to the p53-mediated control of ribosome biogenesis. The Arf gene is induced by a series of stress signals such as hyperproliferative signals emanating from oncogenic Ras and overexpressed MYC,36,37 and p14Arf helps the stabilization of p53 by binding to Hdm2, which is the factor responsible for p53 degradation. This tumor suppressor, in addition to activating the p53 pathway, reduces the ribosome biogenesis rate both by hindering UBF recruitment on the Pol I transcription complex38 and by downregulating the activity of “nucleophosmin,” a multifunctional protein involved in rRNA processing.39 Lastly, another important tumor suppressor involved in the control of ribosome biogenesis is phosphatase and tensin homolog deleted in chromosome 10 (PTEN), which represses Pol I transcription by disrupting the SL1 complex.40
Since the neoplastic transformation is characterized by either the uncontrolled activity of oncogenes or the inactivation of tumor suppressors, all the data just reported indicate that some very frequent changes in proto-oncogenes and tumor suppressor genes in a variety of human cancers, which are responsible for the loss of the normal control mechanisms of cell proliferation and cell cycle progression, are also responsible for an enhanced ribosome biogenesis. In fact, MYC overexpression and the aberrant activation of the mitogen-activated protein kinases/extracellular signal-regulated kinases pathway, which are very frequently observed in human cancers,41 both result in increased rRNA synthesis; pRB inactivation due to genetic changes42 strongly reduces its braking power on rRNA transcription, as well as TP53 mutations, resulting in p53 inactivation, which characterizes about 50% of all human tumors.43,44 Also, the Arf gene may be mutated or silenced in cancers,36,37 thus it may enhance ribosome biogenesis both directly and through action on p53 stabilization. Lastly, the repressive action on Pol I transcription by PTEN may be lost in human cancers in which the tumor suppressor is deleted or mutated.45
We may conclude that both the nucleolar hypertrophy and the upregulated ribosome biogenesis that frequently characterize cancer cells are the consequences of the changes in proto-oncogene and tumor suppressor protein expression that control cell proliferation: the highly variable severity of these changes explains the highly variable nucleolar size and function in cancer.46–48
From the teleological point of view, the upregulation of ribosome biogenesis in cancer cells appears to be an advantage for cancer growth. In fact, the acquired upregulated ribosome biogenesis may allow the complement of constituents necessary to always achieve the appropriate division in dividing cells, independently of the loss of cell cycle progression checkpoints.
At the same time, the enhanced ribosome biogenesis accelerates the cell cycle progression and consequently, the cell proliferation rate.49
Upregulated ribosome biogenesis and neoplastic transformations
Can an upregulation of ribosome biogenesis be responsible for a neoplastic transformation? Many experimental data are consistent with an affirmative answer to this question. The depletion of TTF-1-interacting-protein-5 – a component of the nucleolar remodeling complex which keeps a portion of ribosomal genes in a silent heterochromatin organization – not only increases rRNA transcription but also induces a transformed phenotype in NIH3T3 cells.50
The loss of MTG16a, a ribosomal gene repressor, increases ribosome biogenesis and induces morphological and molecular changes which are typical of breast cancer initiation in breast epithelial cells.51 Further, the importance of an enhanced ribosome biogenesis in tumor development was demonstrated by the fact that the lymphomagenesis occurring in Eμ–MYC+/+ transgenic mice – in which MYC is overexpressed in the B-cell compartment – was reduced by their intercrossing with RPL24+/−, as well as with RPL38+/− mice.52,53 In these mice, the level of ribosome biogenesis and protein synthesis was lowered remarkably, thus restoring the increased protein synthesis in Eμ–MYC/+ transgenic mice to normal levels and suppressing the oncogenic potential of MYC.
With regard to human pathology, a series of epidemiological studies has shown that many diseases with different etiologies, associated with an increased risk of developing cancer, are also characterized by the focal and systemic release of factors which stimulate ribosome biogenesis.
There is evidence that chronic inflammation,54,55 obesity,56,57 and type 2 diabetes58 represent important risk factors for the onset of malignancies. Interestingly, all these conditions exhibit an increased plasma level of the inflammatory cytokine, interleukin 6 (IL-6),59,60 which has been proved to stimulate rRNA transcription (Brighenti et al, unpublished data, 2013). Further, in obese and early stage type 2 diabetes patients, a higher plasma level of insulin is also present,60 enhancing per se ribosome biogenesis.61 What is the cause that lets an enhanced ribosome biogenesis increase the risk of cancer onset? The answer lies in the mechanism by which ribosome biogenesis controls the function of p53. A large body of data has shown that a perturbation in any of the several steps of ribosome biogenesis causes p53 stabilization and activation,62–65 whereas an enhancement of rRNA transcription is responsible for a downregulation of p53 expression and activity.61 p53 is regulated by murine double minute 2 (MDM2; HDM2 [ie, human double minute 2] in humans), which adversely controls p53 activity in two ways: by binding to the protein, thus interfering with its transactivation activity, and by facilitating p53 proteasomal degradation, acting as an E3 ubiquitin ligase.66–68 After the inhibition of rRNA transcription, the ribosomal proteins, no longer used for ribosome building, bind to and inactivate MDM2, thus allowing p53 stabilization.68,69 Conversely, when rRNA transcription is stimulated, the ribosomal protein that binds and inactivates MDM2 is reduced, thus resulting in a greater degradation of p53.61 This was clearly demonstrated in human cell lines exposed to either IL-6 or insulin, in which both the amount of p53 and the p53-mediated response to cytotoxic stresses decreased.61 Therefore, cells with upregulated ribosome biogenesis would have greater difficulty in activating their tumor suppressor mechanisms, thus be more subject to neoplastic transformation. This may be the root of the increased risk of developing cancer in those human pathological conditions in which high plasma levels of IL-6 and insulin in responsive tissues induce an upregulation of ribosome biogenesis with the consequent downregulation of p53-related tumor suppressor potential.70
Targeting Pol I for the treatment of human cancer
Currently, chemotherapy is a widely chosen option for treating cancer, either before or after the surgical removal of tumor masses. Chemotherapeutic agents may be broken down into several families according to their mechanism of action: alkylating drugs, anthracyclines, antimetabolites, DNA damaging agents, spindle poisons, topoisomerase inhibitors, oncoprotein-targeting agents, and antihormone drugs. With the exception of the last two classes, the other chemotherapeutic agents exert their toxic action on cancer cells by damaging DNA, hindering DNA synthesis, and disrupting the mitotic process. Therefore, the targets of these drugs are mainly proliferating cells, which are abundant in cancer tissues. Unfortunately, this toxic action is also exerted on the proliferating cells of normal tissues, thus establishing strict pharmacological limits to the use of these anticancer drugs. This has led to the need for continuous efforts to find new therapeutic strategies that more specifically address cancer cells.
Considering that an ongoing, appropriate ribosome production is necessary both for cell cycle progression and for the generation of viable cells, a new therapeutic approach to killing cancer cells based on the specific inhibition of Pol I activity has been recently proposed.71–75
At first glance, a similar approach based on the inhibition of ribosome biogenesis would appear to be even less specific for cancer cell elimination than the above-reported classic chemotherapeutic procedures. In fact, unlike the DNA synthesis, the synthesis of rRNA occurs in both proliferating and resting cells. What is more, the specific inhibition of the Pol I activity might sound less efficient than a large series of chemotherapeutics which, in addition to the inhibition of DNA synthesis, also hinder the rRNA production at either the transcription or processing level.71 However, a series of data dealing with the relationship between ribosome biogenesis and cell proliferation has indicated that inhibition of the Pol I activity may result in specific damage to neoplastic cells, at least in some instances. Indeed, there is evidence that the inhibition of rDNA transcription may have specific lethal effects on cancer cells in the following two conditions: (1) when cancer cells have lost checkpoint mechanisms which control the cell cycle progression and (2) when cancer cells are characterized by an upregulated ribosome biogenesis. In fact, in both cases, the inhibition of rRNA synthesis has been demonstrated to have deleterious effects only on cancer cells, while sparing normal tissues.71–76
Regarding the first condition, it has been shown that a transient, selective inhibition of the Pol I-dependent transcription induced by actinomycin D exposure caused cell cycle arrest in pRB and p53 proficient cells but not in pRB- and p53-deficient cells.76 The cell cycle arrest observed in pRB-and p53-profcient cells, due to the activation of the p53-pRB checkpoint pathway, was reversible: after recovery of ribosome biogenesis, cell cycle progression started up again, and cells could achieve a ribosomal complement sufficient for giving rise to normal daughter cells (Figure 2A). This was not the case for pRB- and p53-deficient cells in which rRNA inhibition did not alter the cell cycle progression and cells underwent a progressive ribosome depletion that led to apoptotic death (Figure 2B).76 Therefore, these data indicate that the selective inhibition of Pol I-dependent transcription may be very useful in killing cancer cells lacking the two tumor suppressors, without having significant effects on the proliferation of noncancerous cells in which the two tumor suppressors are functioning normally.
Figure 2.

Different effects of RNA polymerase I (Pol I) inhibition according to the function of the p53/pRb pathway. (A) Pol I inhibition induces G1/S cell cycle arrest through activation of the p53/pRb pathway. (B) In cells lacking the functional p53/pRb pathway, the inhibition of Pol I does not induce cell cycle arrest; rather, without a sufficient ribosomal complement, the cell divides and dies.
Abbreviation: pRb, retinoblastoma protein.
The other pathological cancer condition in which the inhibition of rDNA transcription may selectively destroy cancer, but not normal cells, is that of tumors characterized by highly upregulated ribosome biogenesis. This has been clearly demonstrated in a study conducted using the CX-5461 inhibitor of rRNA synthesis in a transgenic murine model of spontaneous lymphoma (Eμ-MYC), in which MYC is overexpressed in lymphocytes of the B lineage.73 CX-5461 (Cylene Pharmaceuticals Inc, San Diego, CA, USA), is a powerful small molecule that selectively inhibits Pol I-driven transcription.74 It was demonstrated that CX-5461 disrupts the binding of the SL1 transcription factor to the rDNA promoter, thus preventing the initiation of rRNA synthesis. The drug inhibits Pol I transcription without having almost any effect on DNA replication, protein translation, and general cellular transcription.74
MYC overexpression was responsible for the high elevation of ribosome biogenesis in malignant B cells from Eμ-MYC transgenic mice. Administration of CX-5461 to mice caused p53-dependent apoptotic death in neoplastic but not normal B cells.75 This different sensitivity to CX-5461 exposure may be explained by considering the mechanism involved in the MDM2 inactivation by the ribosomal protein binding, with the consequent p53 stabilization, after the inhibition of rDNA transcription (Figure 3). Since the synthesis of rRNA requires the stoichiometric production of ribosomal proteins, in cells with low rRNA synthesis activity, and therefore with low ribosomal protein production, even a marked reduction of rDNA transcription should not make ribosomal proteins available in an amount sufficient to inactivate MDM2 (Figure 3A). In contrast, in the case of cells with highly upregulated ribosome biogenesis, a slight reduction in rRNA molecules would make ribosomal proteins free to an extent sufficient for inactivating MDM2 (Figure 3B).
Figure 3.

Different effects of RNA polymerase I (Pol I) inhibition according to the rate of ribosome biogenesis. A hypothesis explaining the different sensitivity to ribosomal RNA (rRNA) transcription inhibition in cells with different rate of ribosomal biogenesis. rRNA synthesis requires the stoichiometric production of ribosomal proteins. (A) In cells with a low ribosomal biogenesis rate, inhibition of rRNA transcription produces a limited amount of ribosomal proteins (RPs) free to bind and inactivate murine double minute 2 (MDM2) protein. Some MDM2 molecules can still bind p53 for degradation. (B) In cells with a high ribosomal biogenesis rate, inhibition of rRNA transcription produces a large amount of RPs free to bind and inactivate the majority of MDM2 molecules, thus leading to p53 accumulation.
Abbreviation: rDNA, ribosomal DNA.
Conclusion
Up to now, research efforts in the chemotherapeutic treatment of cancer have mainly addressed the development of drugs that hinder the neoplastic cell division by either inhibiting DNA synthesis or damaging the mitotic process, or by blocking abnormally upregulated pathways which stimulate cell proliferation. The results achieved over the past few years indicate that an appropriate ribosome biogenesis is necessary for adequate cell growth, which, in turn, is necessary for both normal cell cycle progression and cell division. Since the selective disruption of ribosome biogenesis by hindering rRNA synthesis has been proven to arrest cell cycle progression and/or cause apoptotic cell death, the targeting factors of the rDNA transcription complex may represent a promising new strategy for the chemotherapeutic treatment of cancers.76 Indeed, available data suggest that the specific inhibition of rRNA transcription could be used for the selective killing of cancer cells characterized by either a highly enhanced ribosome biogenesis or a deranged function of the p53 and pRB tumor suppressors.74,76
Acknowledgment
This work was supported by grants from the Associazione Italiana per la Ricerca sul Cancro (AIRC) (Grant number IG-11416 to LM and IG13480 to MD).
Footnotes
Disclosure
The authors declare no conflicts of interest in this work.
References
- 1.Grummt I. Regulation of mammalian ribosomal gene transcription by RNA polymerase I. Prog Nucleic Acid Res Mol Biol. 1999;62:109–154. doi: 10.1016/s0079-6603(08)60506-1. [DOI] [PubMed] [Google Scholar]
- 2.Hahn S. Structure and mechanism of the RNA polymerase II transcription machinery. Nat Struct Mol Biol. 2004;11(5):394–403. doi: 10.1038/nsmb763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lee Y, Kim M, Han J, et al. MicroRNA genes are transcribed by RNA polymerase II. EMBO J. 2004;23(20):4051–4060. doi: 10.1038/sj.emboj.7600385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Willis IM. RNA polymerase III. Genes, factors and transcriptional specificity. Eur J Biochem. 1993;212(1):1–11. doi: 10.1111/j.1432-1033.1993.tb17626.x. [DOI] [PubMed] [Google Scholar]
- 5.Warner JR. The economics of ribosome biosynthesis in yeast. Trends Biochem Sci. 1999;24(11):437–440. doi: 10.1016/s0968-0004(99)01460-7. [DOI] [PubMed] [Google Scholar]
- 6.Grummt I. Life on a planet of its own: regulation of RNA polymerase I transcription in the nucleolus. Genes Dev. 2003;17(4):1691–1702. doi: 10.1101/gad.1098503R. [DOI] [PubMed] [Google Scholar]
- 7.Stefanovsky V, Langlois F, Gagnon-Kugler T, Rothblum LI, Moss T. Growth factor signaling regulates elongation of RNA polymerase I transcription in mammals via UBF phosphorylation and r-chromatin remodeling. Mol Cell. 2006;21(5):629–639. doi: 10.1016/j.molcel.2006.01.023. [DOI] [PubMed] [Google Scholar]
- 8.Zhang Y, Smith AD, IV, Renfrow MB, Schneider DA. The RNA polymerase-associated factor 1 complex (Paf1C) directly increases the elongation rate of RNA polymerase I and is required for efficient regulation of rRNA synthesis. J Biol Chem. 2010;285(19):14152–14159. doi: 10.1074/jbc.M110.115220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jansa P, Grummt I. Mechanism of transcription termination: PTRF interacts with the largest subunit of RNA polymerase I and dissociates paused transcription complexes from yeast and mouse. Mol Gen Genet. 1999;262(3):508–514. doi: 10.1007/s004380051112. [DOI] [PubMed] [Google Scholar]
- 10.Längst G, Blank TA, Becker PB, Grummt I. RNA polymerase I transcription on nucleosomal templates: the transcription termination factor TTF-I induces chromatin remodeling and relieves transcriptional repression. EMBO J. 1997;16(4):760–768. doi: 10.1093/emboj/16.4.760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kopp K, Gasiorowski JZ, Chen D, et al. Pol I transcription and pre-rRNA processing are coordinated in a transcription-dependent manner in mammalian cells. Mol. Biol. Cell. 2007;18:394–403. doi: 10.1091/mbc.E06-03-0249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pianese G. Beitrag zur Histologie und Aetiologie der Carcinoma. Histologische und experimentelle Untersuchungen [Contribution to the histology and etiology of carcinoma. Histological and experimental studies] Beitr Pathol Anat Allgem Pathol. 1896;142:1–193. German. [Google Scholar]
- 13.MacCarty WC. The value of the macronucleolus in the cancer problem. Am J Cancer. 1936;26:529–532. [Google Scholar]
- 14.Koller PC. The nucleus of the cancer cell: a historical review. Exp Cell Res. 1963;24(Suppl 9):3–14. doi: 10.1016/0014-4827(63)90241-6. [DOI] [PubMed] [Google Scholar]
- 15.Busch H, Smetana K. Nucleoli of tumor cells. In: Busch H, Smetana K, editors. The Nucleolus. New York, NY: Academic Press; 1970. pp. 448–471. [Google Scholar]
- 16.Conlon I, Raff M. Size control in animal development. Cell. 1999;96(2):235–244. doi: 10.1016/s0092-8674(00)80563-2. [DOI] [PubMed] [Google Scholar]
- 17.Thomas G. An encore for ribosome biogenesis in the control of cell proliferation. Nat Cell Biol. 2000;2(5):E71–E72. doi: 10.1038/35010581. [DOI] [PubMed] [Google Scholar]
- 18.David-Pfeuty T. The flexible evolutionary anchorage-dependent Pardee’s restriction point of mammalian cells: how its deregulation may lead to cancer. Biochim Biophys Acta. 2006;1765(1):38–66. doi: 10.1016/j.bbcan.2005.08.008. [DOI] [PubMed] [Google Scholar]
- 19.Sulić S, Panić L, Dikić I, Volarević S. Deregulation of cell growth and malignant transformation. Croat Med J. 2005;46(4):622–638. [PubMed] [Google Scholar]
- 20.Zhu J, Blenis J, Yuan J. Activation of PI3K/Akt and MAPK pathways regulates Myc-mediated transcription by phosphorylating and promoting the degradation of Mad1. Proc Natl Acad Sci USA. 2008;105(18):6584–6589. doi: 10.1073/pnas.0802785105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.White RJ. RNA polymerases I and III, growth control and cancer. Nat Rev Mol Cell Biol. 2005;6(1):69–78. doi: 10.1038/nrm1551. [DOI] [PubMed] [Google Scholar]
- 22.Gomez-Roman N, Felton-Edkins ZA, Kenneth NS, et al. Activation by c-Myc of transcription by RNA polymerases I, II and III. Biochem Soc Symp. 2006;(73):141–154. doi: 10.1042/bss0730141. [DOI] [PubMed] [Google Scholar]
- 23.van Riggelen J, Yetil A, Felsher DW. MYC as a regulator of ribosome biogenesis and protein synthesis. Nat Rev Cancer. 2010;10(4):301–309. doi: 10.1038/nrc2819. [DOI] [PubMed] [Google Scholar]
- 24.Stefanovsky VY, Pelletier G, Hannan R, Gagnon-Kugler T, Rothblum LI, Moss T. An immediate response of ribosomal transcription to growth factor stimulation in mammals is mediated by ERK phosphorylation of UBF. Mol Cell. 2001;8(5):1063–1073. doi: 10.1016/s1097-2765(01)00384-7. [DOI] [PubMed] [Google Scholar]
- 25.Grummt I. Wisely chosen paths – regulation of rRNA synthesis: delivered on 30 June 2010 at the 35th FEBS Congress in Gothenburg, Sweden. FEBS J. 2010;277(22):4626–4639. doi: 10.1111/j.1742-4658.2010.07892.x. [DOI] [PubMed] [Google Scholar]
- 26.Mayer C, Grummt I. Ribosome biogenesis and cell growth: mTOR coordinates transcription by all three classes of nuclear RNA polymerases. Oncogene. 2006;25(48):6384–6391. doi: 10.1038/sj.onc.1209883. [DOI] [PubMed] [Google Scholar]
- 27.Li LY, Chen H, Hsieh YH, et al. Nuclear ErbB2 enhances translation and cell growth by activating transcription of ribosomal RNA genes. Cancer Res. 2011;71(12):4269–4279. doi: 10.1158/0008-5472.CAN-10-3504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gonugunta VK, Nair BC, Rajhans R, Sareddy GR, Nair SS, Vadlamudi RK. Regulation of rDNA transcription by proto-oncogene PELP1. PLoS One. 2011;6:e21095. doi: 10.1371/journal.pone.0021095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cavanaugh AH, Hempel WM, Taylor LJ, Rogalsky V, Todorov G, Rothblum LI. Activity of RNA polymerase I transcription factor UBF blocked by Rb gene product. Nature. 1995;374(6518):177–180. doi: 10.1038/374177a0. [DOI] [PubMed] [Google Scholar]
- 30.Voit R, Schäfer K, Grummt I. Mechanism of repression of RNA polymerase I transcription by the retinoblastoma protein. Mol Cell Biol. 1997;17(8):4230–4237. doi: 10.1128/mcb.17.8.4230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hannan KM, Hannan RD, Smith SD, Jefferson LS, Lun M, Rothblum LI. Rb and p130 regulate RNA polymerase I transcription: Rb disrupts the interaction between UBF and SL-1. Oncogene. 2000;19(43):4988–4999. doi: 10.1038/sj.onc.1203875. [DOI] [PubMed] [Google Scholar]
- 32.Ciarmatori S, Scott PH, Sutcliffe JE, et al. Overlapping functions of the pRb family in the regulation of rRNA synthesis. Mol Cell Biol. 2001;21(17):5806–5814. doi: 10.1128/MCB.21.17.5806-5814.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.White RJ, Trouche D, Martin K, Jackson SP, Kouzarides T. Repression of RNA polymerase III transcription by the retinoblastoma protein. Nature. 1996;382(6586):88–90. doi: 10.1038/382088a0. [DOI] [PubMed] [Google Scholar]
- 34.Felton-Edkins ZA, Kenneth NS, Brown TR, et al. Direct regulation of RNA polymerase III transcription by RB, p53 and c-Myc. Cell Cycle. 2003;2(3):181–184. [PubMed] [Google Scholar]
- 35.Zhai W, Comai L. Repression of RNA polymerase I transcription by the tumor suppressor p53. Mol Cell Biol. 2000;20(16):5930–5938. doi: 10.1128/mcb.20.16.5930-5938.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sherr CJ. The INK4a/ARF network in tumour suppression. Nat Rev Mol Cell Biol. 2001;2(10):731–737. doi: 10.1038/35096061. [DOI] [PubMed] [Google Scholar]
- 37.Lowe SW, Sherr CJ. Tumor suppression by Ink4a-Arf: progress and puzzles. Curr Opin Genet Dev. 2003;13(1):77–83. doi: 10.1016/s0959-437x(02)00013-8. [DOI] [PubMed] [Google Scholar]
- 38.Ayrault O, Andrique L, Fauvin D, Eymin B, Gazzeri S, Séité P. Human tumor suppressor p14 ARF negatively regulates rRNA transcription and inhibits UBF1 transcription factor phosphorylation. Oncogene. 2006;25(58):7577–7586. doi: 10.1038/sj.onc.1209743. [DOI] [PubMed] [Google Scholar]
- 39.Itahana K, Bhat KP, Jin A, et al. Tumor suppressor ARF degrades B23, a nucleolar protein involved in ribosome biogenesis and cell proliferation. Mol Cell. 2003;12(5):1151–1164. doi: 10.1016/s1097-2765(03)00431-3. [DOI] [PubMed] [Google Scholar]
- 40.Zhang C, Comai L, Johnson DL. PTEN represses RNA Polymerase I transcription by disrupting the SL1 complex. Mol Cell Biol. 2005;25(16):6899–6911. doi: 10.1128/MCB.25.16.6899-6911.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100(1):57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 42.Knudsen ES, Knudsen KE. Tailoring to RB: tumour suppressor status and therapeutic response. Nat Rev Cancer. 2008;8(9):714–724. doi: 10.1038/nrc2401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vogelstein B, Lane B, Levine AJ. Surfing the p53 network. Nature. 2000;408(6810):307–310. doi: 10.1038/35042675. [DOI] [PubMed] [Google Scholar]
- 44.Vousden KH, Lane DP. p53 in health and disease. Nat Rev Mol Cell Biol. 2007;8(4):275–283. doi: 10.1038/nrm2147. [DOI] [PubMed] [Google Scholar]
- 45.Yin Y, Shen WH. PTEN: a new guardian of the genome. Oncogene. 2008;27(41):5443–5453. doi: 10.1038/onc.2008.241. [DOI] [PubMed] [Google Scholar]
- 46.Treré D, Ceccarelli C, Montanaro L, Tosti E, Derenzini M. Nucleolar size and activity are related to pRb and p53 status in human breast cancer. J Histochem Cytochem. 2004;52(12):1601–1607. doi: 10.1369/jhc.4A6454.2004. [DOI] [PubMed] [Google Scholar]
- 47.Derenzini M, Montanaro L, Treré D. What the nucleolus says to a tumour pathologist. Histopathology. 2009;54(6):753–762. doi: 10.1111/j.1365-2559.2008.03168.x. [DOI] [PubMed] [Google Scholar]
- 48.Montanaro L, Treré D, Derenzini M. Nucleolus, ribosomes, and cancer. Am J Pathol. 2008;173(2):301–310. doi: 10.2353/ajpath.2008.070752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Derenzini M, Montanaro L, Chillà A, et al. Key role of the achievement of an appropriate ribosomal RNA complement for G1-S phase transition in H4-II-E-C3 rat hepatoma cells. J Cell Physiol. 2005;202(2):483–491. doi: 10.1002/jcp.20144. [DOI] [PubMed] [Google Scholar]
- 50.Guetg C, Lienemann P, Sirri V, et al. The NoRC complex mediates the heterochromatin formation and stability of silent rRNA genes and centromeric repeats. EMBO J. 2010;29(13):2135–2146. doi: 10.1038/emboj.2010.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rossetti S, Hoogeveen AT, Esposito J, Sacchi N. Loss of MTG16a (CBFA2T3), a novel rDNA repressor, leads to increased ribogenesis and disruption of breast acinar morphogenesis. J Cell Mol Med. 2010;14(6A):1358–1370. doi: 10.1111/j.1582-4934.2009.00982.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Barna M, Pusic A, Zollo O, et al. Suppression of Myc oncogenic activity by ribosomal protein haploinsufficiency. Nature. 2008;456(7224):971–975. doi: 10.1038/nature07449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ruggero D. The role of Myc-induced protein synthesis in cancer. Cancer Res. 2009;69(23):8839–8843. doi: 10.1158/0008-5472.CAN-09-1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Coussens LM, Werb Z. Inflammation and cancer. Nature. 2002;420:860–867. doi: 10.1038/nature01322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mantovani A, Allavena P, Sica A, Balkwill F. Cancer-related inflammation. Nature. 2008;454(7203):436–444. doi: 10.1038/nature07205. [DOI] [PubMed] [Google Scholar]
- 56.Roberts DL, Dive C, Renehan AG. Biological mechanisms linking obesity and cancer risk: new perspectives. Annu Rev Med. 2010;61:301–316. doi: 10.1146/annurev.med.080708.082713. [DOI] [PubMed] [Google Scholar]
- 57.Gabay C. Interleukin-6 and chronic inflammation. Arthritis Res Ther. 2006;8( Suppl 2):S3. doi: 10.1186/ar1917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Roytblat L, Rachinsky M, Fisher A, et al. Raised interleukin-6 levels in obese patients. Obes Res. 2000;8(9):673–675. doi: 10.1038/oby.2000.86. [DOI] [PubMed] [Google Scholar]
- 59.Pickup JC, Chusney GD, Thomas SM, Burt D. Plasma interleukin-6, tumour necrosis factor alpha and blood cytokine production in type 2 diabetes. Life Sci. 2000;67(3):291–300. doi: 10.1016/s0024-3205(00)00622-6. [DOI] [PubMed] [Google Scholar]
- 60.Weyer C, Funahashi T, Tanaka S, et al. Hypoadiponectinemia in obesity and type 2 diabetes: close association with insulin resistance and hyperinsulinemia. J Clin Endocrinol Metab. 2001;86(5):1930–1935. doi: 10.1210/jcem.86.5.7463. [DOI] [PubMed] [Google Scholar]
- 61.Donati G, Bertoni S, Brighenti E, et al. The balance between rRNA and ribosomal protein synthesis up-and downregulates the tumour suppressor p53 in mammalian cells. Oncogene. 2011;30(29):3274–3288. doi: 10.1038/onc.2011.48. [DOI] [PubMed] [Google Scholar]
- 62.Mayer C, Grummt I. Cellular stress and nucleolar function. Cell Cycle. 2005;4(8):1036–1038. doi: 10.4161/cc.4.8.1925. [DOI] [PubMed] [Google Scholar]
- 63.Opferman JT, Zambetti GP. Translational research? Ribosome integrity and a new p53 tumor suppressor checkpoint. Cell Death Differ. 2006;13(6):898–901. doi: 10.1038/sj.cdd.4401923. [DOI] [PubMed] [Google Scholar]
- 64.Panić L, Montagne J, Cokarić M, Volarević S. S6-haploinsufficiency activates the p53 tumor suppressor. Cell Cycle. 2007;6(1):20–24. doi: 10.4161/cc.6.1.3666. [DOI] [PubMed] [Google Scholar]
- 65.Deisenroth C, Zhang Y. Ribosome biogenesis surveillance: probing the ribosomal protein-Mdm2-p53 pathway. Oncogene. 2010;29(30):4253–4260. doi: 10.1038/onc.2010.189. [DOI] [PubMed] [Google Scholar]
- 66.Momand J, Zambetti GP, Olson DC, George D, Levine AJ. The mdm-2 oncogene product forms a complex with the p53 protein and inhibits p53-mediated transactivation. Cell. 1992;69(7):1237–1245. doi: 10.1016/0092-8674(92)90644-r. [DOI] [PubMed] [Google Scholar]
- 67.Haupt Y, Maya R, Kazaz A, Oren M. Mdm2 promotes the rapid degradation of p53. Nature. 1997;387:296–299. doi: 10.1038/387296a0. [DOI] [PubMed] [Google Scholar]
- 68.Kubbutat MH, Jones SN, Vousden KH. Regulation of p53 stability by Mdm2. Nature. 1997;387(6630):299–303. doi: 10.1038/387299a0. [DOI] [PubMed] [Google Scholar]
- 69.Zhang Y, Lu H. Signaling to p53: ribosomal proteins find their way. Cancer Cell. 2009;16(5):369–377. doi: 10.1016/j.ccr.2009.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Montanaro L, Treré D, Derenzini M. Changes in ribosome biogenesis may induce cancer by down-regulating the cell tumor suppressor potential. Biochim Biophys Acta. 2012;1825(1):101–110. doi: 10.1016/j.bbcan.2011.10.006. [DOI] [PubMed] [Google Scholar]
- 71.Drygin D, Siddiqui-Jain A, O’Brien S, et al. Anticancer activity of CX-3543: a direct inhibitor of rRNA biogenesis. Cancer Res. 2009;69(19):7653–7661. doi: 10.1158/0008-5472.CAN-09-1304. [DOI] [PubMed] [Google Scholar]
- 72.Drygin D, Rice WG, Grummt I. The RNA polymerase I transcription machinery: an emerging target for the treatment of cancer. Annu Rev Pharmacol Toxicol. 2010;50:131–156. doi: 10.1146/annurev.pharmtox.010909.105844. [DOI] [PubMed] [Google Scholar]
- 73.Drygin D, Lin A, Bliesath J, et al. Targeting RNA polymerase I with an oral small molecule CX-5461 inhibits ribosomal RNA synthesis and solid tumor growth. Cancer Res. 2011;71(4):1418–1430. doi: 10.1158/0008-5472.CAN-10-1728. [DOI] [PubMed] [Google Scholar]
- 74.Bywater MJ, Poortinga G, Sanij E, et al. Inhibition of RNA polymerase I as a therapeutic strategy to promote cancer-specific activation of p53. Cancer Cell. 2012;22(1):51–65. doi: 10.1016/j.ccr.2012.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Burger K, Mühl B, Harasim T, et al. Chemotherapeutic drugs inhibit ribosome biogenesis at various levels. J Biol Chem. 2010;285(16):12416–12425. doi: 10.1074/jbc.M109.074211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Montanaro L, Mazzini G, Barbieri S, et al. Different effects of ribosome biogenesis inhibition on cell proliferation in retinoblastoma protein- and p53-deficient and proficient human osteosarcoma cell lines. Cell Prolif. 2007;40(4):532–549. doi: 10.1111/j.1365-2184.2007.00448.x. [DOI] [PMC free article] [PubMed] [Google Scholar]