

Abstract
Host-specificity is an intrinsic feature of many bacterial pathogens, resulting from a long history of co-adaptation between bacteria and their hosts. Alpha-proteobacteria belonging to the genus Bartonella infect the erythrocytes of a wide range of mammal orders, including rodents. In this study, we performed genetic analysis of Bartonella colonizing a rodent community dominated by bank voles (Myodes glareolus) and wood mice (Apodemus sylvaticus) in a French suburban forest to evaluate their diversity, their capacity to recombine and their level of host specificity. Following the analysis of 550 rodents, we detected 63 distinct genotypes related to B. taylorii, B. grahamii, B. doshiae and a new B. rochalimae-like species. Investigating the most highly represented species, we showed that B. taylorii strain diversity was markedly higher than that of B. grahamii, suggesting a possible severe bottleneck for the latter species. The majority of recovered genotypes presented a strong association with either bank voles or wood mice, with the exception of three B. taylorii genotypes which had a broader host range. Despite the physical barriers created by host specificity, we observed lateral gene transfer between Bartonella genotypes associated with wood mice and Bartonella adapted to bank voles, suggesting that those genotypes might co-habit during their life cycle.
Introduction
Bacterial strain diversification within their natural host populations can restrict the potential future range of hosts that are susceptible to infection. As a result of adaptive evolution in their principal host, bacteria have a typically limited host range that they can successfully infect. At the molecular level, such host-parasite adaptation is reflected by specific receptor-ligand interactions between bacterial factors and their target host factors [1]. The resulting host specificity is an inherent feature of many bacterial pathogens, including species of the genus Bartonella. Although largely unexplored, the Bartonella species represents an interesting model for investigating the patterns and processes of host specificity [2].
Bartonella species infect mammalian reservoirs, including rodents, in which they induce an asymptomatic long lasting intra-erythrocytic bacteremia [3]. To date, more than 30 species and subspecies of Bartonella have been partially or completely characterized. Thirteen of which are recognized as emerging zoonotic pathogens, causing life-threatening infections in both animal and human populations. Bartonella species are mainly transmitted to their mammalian hosts through feces of ectoparasites after superficial scratching of their skin (or occasionally directly by the bite of blood feeding arthropods). They then colonize a primary niche from where they are seeded into the bloodstream before finally adhering to and invading erythrocytes [3], [4]. Infection is driven by two main pathogenic factors belonging to the type IV secretion systems (T4SS): the VirB/VirD4, involved in primary niche invasion and adaptation to the mammalian host, and the Trw T4SS, involved in host-specific erythrocyte invasion.
As a result of hypothesized adaptive radiation [5], each of the 30 Bartonella species or subspecies only infects one, or a few closely related mammalian host reservoirs [3], [6]. We have recently demonstrated that bartonellae host specificity is driven by their unique ability to adhere and infect erythrocytes from their natural host(s) via the Trw T4SS [7]. Equally important for Bartonella species host adaptation is the VirB T4SS machinery, which translocates a cocktail of related effector proteins into primary niche host cells, where they modulate various processes, resulting in the ability to adapt to a wide range of different hosts [5], [8].
While some species are highly specific for their host (i.e., B. henselae for the cat or B. bacilliformis for humans), other specific associations do not seem so clear-cut, especially for rodent-associated species. In addition the association between some Bartonella species and their rodent hosts remains a matter of controversy. Indeed, while B. vinsonii subsp. arupensis and B. washoensis have only been found to infect Peromyscus mice and ground squirrels respectively [9]–[12], B. elizabethae has been found to infect both Rattus spp. rats in Southern China [13] and Bandicota spp. rats in Thailand. In Europe, B. taylorii, B. grahamii and B. doshiae infect several sympatric woodland rodents at a single site [14]–[17], while a longitudinal study realized in Poland reported a strong host association between the majority of Bartonella species and their rodents (Apodemus mice and Myodes voles) [18]. Recently, the complete genome sequences of two rodent-carried Bartonella species (i.e. B. tribocorum and B. grahamii) were compared to Bartonella species from humans and cats, revealing that the rodent-associated species carried a higher number of T4SS host-adaptability factor encoding genes [19]. Interestingly, these host-adaptability genes are packaged into bacteriophage particles, resulting in an original method of gene exchange between B. grahamii strains promoting rapid diversification. This could therefore facilitate host shifting, which may explain the observed lack of host specificity of some species or strains [19].
In order to improve our understanding of how Bartonella species are associated with their mammalian hosts, it is important to accurately describe Bartonella species diversity within their natural host. Previous multi-locus sequence typing methods (MLST) of B. henselae strains isolated from humans and cats sampled across several continents revealed 14 genotypes, but did not disclose an obvious host association pattern, except for one genotype restricted to European cats [20]. However, a more recent MLST scheme based on eight loci indicated that the majority of human isolates are rare genotypes differing from those of cats [21]. Moreover, a variable number tandem repeat analysis performed on 178 B. henselae strains resulted in 99 profiles separated into two groups, where all human isolates were clustered within the same group [22]. All typing schemes applied to B. quintana strains, strictly human specific, indicated very low levels of diversity [23], [24]. In relation to rodent-adapted Bartonella species, the only published study utilized multi-locus sequence analysis (MLSA) of B. grahamii strains sampled worldwide, and demonstrated strict host specificity in Asia and North America, and low host specificity in Europe [25], [26].
Typically, intra-cellular bacteria have lower recombination rates, probably due to their relative niche isolation [27] and indeed, the Bartonella genus has one of the lowest recombination rates among bacteria [27]. This might be due to its intracellular location but also to its restricted host range. Interestingly, within the Bartonella genus, strains circulating within rodents showed more frequent recombination events compared to human and cat adapted species, suggesting a broader host range for rodent adapted species [26], [28].
To learn more about the adaptation of Bartonella species to their wild animal reservoirs, we conducted a field study in which Bartonella genotypes were identified from a rodent community living within a suburban forest. To determine the level of genotype specificity for rodent species, we performed robust genetic diversity analysis on the recovered Bartonella strains, based on a Multi-locus Sequence Analysis (MLSA) using six housekeeping genes. Recombination events between strains were analyzed to evaluate whether these strains have had the opportunity to co-habit. Finally, due to the important role of the T4SS VirB/D4 in Bartonella species host adaptability, we also investigated whether the pilus component VirB5 diversity revealed a possible host association.
Materials and Methods
Study site and animal processing
The study was conducted in 2007 and 2008 in the Forest of Sénart (3,200 ha, 02°29′E, 48°40′N), located in a dense urbanized area 22 km south-east of Paris [17]. Small mammals were collected from 5 sites in communities dominated by oaks (Quercus petraea, Q. robur) associated with hornbeam (Carpinus betulus) (sites 1, 3 and 5), or associated with the chestnut (Castanea sativa) (sites 2 and 4). The trappings were conducted in April 2008 at sites 1 to 4 and from March to October 2007 and 2008 on the site 5 [29], [30].
A set of small rodents were sacrificed by cervical dislocation: 68 bank voles (Myodes glareolus) and 70 wood mice (Apodemus sylvaticus) in April 2008 on sites 1–4, and 402 bank voles and 9 field voles (Microtus agrestis) on site 5 between 2007 and 2008 (Table 1). No wood mouse, strictly nocturnal, has been collected on the site 5 where traps were closed during the night, because the target species of this field design was the Siberian chipmunk (Tamias sibiricus) which is diurnal. Abundance of wood mice and bank voles were similar at sites 1 to 4 in April 2008. Sacrificed rodents were frozen at −20°C before analysis. In the laboratory, the spleen was removed under sterile conditions, and stored at −80°C until they were to be used for Bartonella culture and/or DNA PCR detection.
Table 1. Distribution of species and clusters of Bartonella in the rodent community in the Forest of Sénart.
| Species | n | taylorii | doshiae | grahamii | rochalimae-like | ||||
| Clusters | A | B | C | D | E | ||||
| Site 1–4 (April 2008) | |||||||||
| A. sylvaticus (wood mice) | 70 | 3 | 0 | 0 | 0 | 6 | 0 | 0 | 0 |
| M. glareolus (bank voles) | 69 | 12 | 4 | 8 | 8 | 0 | 2 | 1 | 1 |
| Site 5 (2007–2008) | |||||||||
| M. glareolus (bank voles) | 402 | 38 | 14 | 23 | 34 | 0 | 4 | 28 | 0 |
| Mi. Agrestis (field voles) | 9 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 |
Bacterial isolates and culture conditions
Bacterial isolation was performed by grinding a piece of spleen in 500 µl of F-12 nutrient mixture medium (Invitrogen), and then plating 250 µl of the mixture onto Colombia agar containing 5% defibrinated sheep's blood (CBA), which was then incubated at 35°C in a humidified atmosphere with 5% CO2 up to 30 days (plates being checked every day from day 5).
PCR amplification and sequencing of protein-coding genes
DNA was extracted from rodent spleen samples as previously described [17], and bacterial isolates was heated in 200 µl suspension at 95°C for 10 min, followed by a 5 min centrifugation step of 6,000 g at 4°C. The supernatant was then collected and used for PCR. The presence of Bartonella DNA was determined by using a portion of the gltA gene as previously described [17]. For all positive samples, five other protein-coding gene sequences were amplified for MLSA analysis: cell division protein gene (ftsZ), NADH dehydrogenase gamma-subunit gene (nuoG), 60 kDa heat-shock protein gene (groEL), riboflavin synthase gene (ribC), and RNA polymerase beta-subunit gene (rpoB) using primers previously described [28], [31]–[34].
For virB5 analysis, the 447–522 bp region was amplified and sequenced from 63 DNA samples (or isolate when avalaible) representing the diversity of Bartonella strains recovered in this study. To amplify the complete virB5 gene, primers were designed as follows: the forward primer virB5F-B4: 5′-GCA-GAA-CTY-AAY-TTA-CGK-GG-3′ was complementary to the 3′ end of the virB4 gene (accession numbers, NC_012846, NC_010161, NC_014932, NC_005956), and the reverse primer virB5R3-B6: 5′-GCA-TTY-GTT-GCC-ATT-GTT-GTC-AC-3′ complemented the 5′ end of the virB6 gene (accession numbers, NC_012846, NC_010161, NC_014932, NC_005956), resulting in complete coverage of the virB5 gene.
PCR amplification was performed in a final volume of 25 µl containing 0.5 µM of each primer, 3% (vol/vol) of dimethyl sulfoxide, 200 µM of each deoxynucleotide triphosphate, 1x PhusionTM GC buffer, 0.4 U of PhusionTM DNA polymerase (Finnzymes) and 20–50 ng of genomic DNA. The PCR amplification conditions were as follows: an initial cycle of 95°C for 5 min; 37 amplification cycles, each consisting of 95°C for 30 s, 54°C (gltA), 55°C (ftsZ), 54°C (groEL), 53°C (rpoB), 48°C (ribC), 53°C (nuoG) and 48°C (virB5) for 30 s, followed by an elongation step of 72°C for 1 min; and a final incubation at 72°C for 10 min. Amplification products were analyzed by electrophoresis on 2.5% agarose gels with 0.1 mg/ml of ethidium bromide and verified under UV light. PCR products were then purified and sequenced on both strands by Eurofins MWG Operon (Germany).
To validate all amplified sequences, we repeated DNA extraction, amplification and sequencing for each spleen sample and isolate.
Phylogenetic analysis and recombination tests
Sequences were edited and aligned using GeneiousTM v5.6.4 software. Nucleotide diversity indices and the polymorphic level were calculated using DNAsp v5.10 [35]. Neighbor-joining tree analysis for each individual gene was performed using MEGA v5 [36]. For the MLSA approach, individual gene sequences were concatenated. The best model of nucleotide substitution was determined with jModelTest [37]. A phylogeny was constructed based on the concatenates by using the maximum-likelihood method implemented in PhyML software [38]. Missing genes were considered as deletions. Bartonella reference strain sequences are listed in Table S1 in File S1.
To build the phylogeny of virB5 sequences, the best model of nucleotide substitution was calculated with jModelTest, and the phylogeny constructed using the maximum-likelihood method with MEGA v5.
A phylogenetic tree was also inferred using ClonalFrame v1.1 [39] with the number of MCMC iterations set to 200,000, following a burn-in period of 100,000 iterations. ClonalFrame is a Bayesian inference method which jointly reconstructs the clonal relationships between the isolates in a sample, as well as the location of recombination events that have disrupted the clonal signal.
Allelic profiles were identified for each individual Bartonella strain, and an allele number was assigned to every distinct sequence variant for each of the six loci, using the BioNumerics v6.5 software (Applied-Maths, Sint Maartens-Latem, Belgium). To test for linkage disequilibrium between alleles of the six analyzed loci, the Index of association (Ia) between alleles [40] was calculated using the START program (http://pubmlst.org).
Recombination test calculations were performed using RDP3 [41]. The Neighbor-Net implemented in the software SplitsTree 4.12 [42] with 1,000 bootstrap replicates was used to create the phylogenic network for the concatenated sequences. Furthermore, we used the pairwise homoplasy index (PHI) [43] in SplitsTree 4.0 in order to test the role of past recombination events in generating allelic variation. To estimate the relative contribution of recombination and mutation events among Bartonella genotypes, we performed two independent runs of the analysis tool ClonalFrame [39], each consisting of 200,000 MCMC iterations, discarding the first half as burn-in. We used the linkage model in Structure v2.3 [44] to identify populations with distinct allele frequencies present in our data and estimate genetic exchange among these populations. We performed five runs, each using a different value of between two and eight for the number of populations (K), each with 100,000 MCMC iterations, following a burn-in period of 50,000 iterations. These tests indicated that the model probability was highest at K = 4.
Nucleotide sequences
Sequences of the six protein-coding genes and the virB5 gene were deposited in GenBank/EMBL/DDBJ databases under the accession numbers JX846090 to JX846493 (Table S2 in File S1).
Results
Among the 550 analyzed rodents, 195 DNA extracts were amplified using Bartonella-gltA specific primers (i.e., 35.5% of the rodent community). Considering each rodent species, prevalence was 11% for field voles (1/9); 39,3% for bank voles (185/471) and 12,8% for wood mice (9/70). Bacterial isolation was possible from 43 out of the 195 gltA-positive spleen samples.
Phylogenetic relationships of Bartonella genotypes, delineation of genetic clusters
To characterize the diversity of Bartonella circulating in the rodent community, we first performed a phylogenetic analysis based on the alignment of the concatenated sequences of the six housekeeping genes, using the 195 Bartonella gltA-positive DNA extracts and the corresponding Bartonella strains when available. The phylogenetic tree (Figure 1) was generated using maximum likelihood (PhyML) with a K81 substitution model (estimated using jModelTest). Only one representative of each genotype was reported on the phylogenetic tree. This analysis revealed the phylogenetic relationships of the 63 unique genotypes with three known Bartonella species: B. taylorii, B. doshiae, and B. grahamii; and a genotype (A296, cluster H) closely related to B. rochalimae and B. clarridgeiae (8.2 and 9.3% nucleotide divergence respectively). B. grahamii (cluster G) did not exhibit an obvious internal phylogenetic structure, except for one genotype (A621) recovered from a single bank vole, which was genetically diverged from all the other B. grahamii genotypes by 2.4 to 2.9%. In contrast, within the B. taylorii species, the phylogenetic tree showed a clear demarcation of five clades (A, B, C, D and E) with high bootstrap support (81–100%). Each clade formed distinct compact clusters separated by genetic distances ranging from 1.3 to 3.5%. As a MLSA-based genetic distance below 5% is sufficient to join a genotype to a known species, all these clusters clearly belong to B. taylorii species [45]. Two genotypes were associated with the species B. doshiae (cluster F), of which one (A340) was more closely related to the R18 reference strain with a genetic distance of 0.7%, and another (A538) more distant which diverged by a distance of 4.5%. Distribution of Bartonella species and clusters in the rodent community is summarized in Table 1. The 63 different genotypes, the rodent species from where they were detected or isolated (as well the number of animal in which they were recoververd) and the corresponding Bartonella species and clusters are descripted in Table 2.
Figure 1. Phylogenetic analysis of representative genotypes circulating in the rodent community.

Maximum-likelihood analysis of 63 Bartonella unique genotypes detected in this study and 19 Bartonella species using the alignment of concatenated sequences of six loci (ftsZ, gltA, groEL, nuoG, ribC and rpoB). The numbers at the nodes correspond to bootstrap values higher than 80%.
Table 2. Description of the 63 genotypes of Bartonella identified in rodents.
| Related genotypes | Strain isolation | Rodent species | Bartonella sp. (cluster) | Number of infected animals |
| A132 | Yes | M. glareolus | B. taylorii (D) | 2 |
| A140 | M. glareolus/A. sylvaticus | B. taylorii (A) | 2 | |
| A144 | M. glareolus | B. taylorii (A) | 1 | |
| A145 | M. glareolus/M. agrestis | B. taylorii (D) | 7 | |
| A148 | Yes | M. glareolus | B. grahamii (G) | 1 |
| A149 | M. glareolus | B. grahamii (G) | 3 | |
| A150 | M. glareolus | B. taylorii (A) | 1 | |
| A181 | Yes | M. glareolus | B. grahamii (G) | 2 |
| A190 | M. glareolus | B. taylorii (D) | 1 | |
| A193 | Yes | M. glareolus | B. taylorii (D) | 1 |
| A195 | M. glareolus | B. taylorii (B) | 1 | |
| A197 | Yes | M. glareolus | B. grahamii (G) | 1 |
| A201 | M. glareolus | B. taylorii(D) | 1 | |
| A202 | Yes | M. glareolus | B. grahamii (G) | 2 |
| A205 | M. glareolus | B. taylorii (A) | 1 | |
| A213 | Yes | M. glareolus | B. grahamii (G) | 4 |
| A216 | Yes | M. glareolus | B. grahamii (G) | 2 |
| A235 | M. glareolus | B. taylorii (B) | 1 | |
| A238 | M. glareolus | B. taylorii (B) | 1 | |
| A241 | M. glareolus | B. taylorii (D) | 1 | |
| A265 | M. glareolus | B. taylorii (A) | 1 | |
| A266 | M. glareolus | B. taylorii (D) | 7 | |
| A286 | Yes | A. sylvaticus | B. taylorii (E) | 6 |
| A296 | Yes | M. glareolus | B. rochalimae-like (H) | 1 |
| A340 | Yes | M. glareolus | B. doshiae (F) | 2 |
| A357 | Yes | M. glareolus | B. taylorii (B) | 13 |
| A358 | M. glareolus | B. grahamii (G) | 5 | |
| A368 | Yes | M. glareolus | B. grahamii (G) | 1 |
| A393 | Yes | M. glareolus | B. taylorii (A) | 30 |
| A419 | Yes | M. glareolus | B. taylorii (D) | 1 |
| A440 | M. glareolus | B. taylorii (A) | 1 | |
| A444 | M. glareolus | B. taylorii (D) | 1 | |
| A445 | Yes | M. glareolus | B. grahamii (G) | 2 |
| A446 | Yes | M. glareolus | B. taylorii (C) | 8 |
| A447 | Yes | M. glareolus/A. sylvaticus | B. taylorii (A) | 13 |
| A45 | M. glareolus | B. taylorii (A) | 1 | |
| A452 | M. glareolus | B. taylorii (A) | 1 | |
| A454 | M. glareolus | B. taylorii (A) | 1 | |
| A46 | M. glareolus | B. taylorii (A) | 1 | |
| A468 | M. glareolus | B. taylorii (D) | 3 | |
| A471 | M. glareolus | B. taylorii (A) | 1 | |
| A475 | M. glareolus | B. taylorii (D) | 1 | |
| A476 | M. glareolus | B. taylorii (A) | 1 | |
| A52 | Yes | M. glareolus | B. taylorii (A) | 1 |
| A527 | M. glareolus | B. taylorii (A) | 1 | |
| A538 | Yes | M. glareolus | B. doshiae (F) | 4 |
| A550 | Yes | M. glareolus | B. taylorii (D) | 6 |
| A554 | Yes | A. sylvaticus | B. taylorii (E) | 3 |
| A578 | M. glareolus | B. taylorii (C) | 1 | |
| A592 | M. glareolus | B. taylorii (C) | 11 | |
| A597 | M. glareolus | B. taylorii (B) | 1 | |
| A610 | Yes | M. glareolus | B. grahamii (G) | 1 |
| A612 | M. glareolus | B. taylorii (B) | 1 | |
| A614 | M. glareolus | B. taylorii (D) | 8 | |
| A615 | M. glareolus | B. taylorii (C) | 3 | |
| A619 | M. glareolus | B. grahamii (G) | 2 | |
| A620 | M. glareolus | B. taylorii (A) | 1 | |
| A621 | M. glareolus | B. grahamii (G) | 2 | |
| A622 | M. glareolus | B. taylorii (C) | 8 | |
| A641 | M. glareolus | B. taylorii (B) | 1 | |
| A66 | M. glareolus | B. taylorii (A) | 1 | |
| A70 | M. glareolus | B. taylorii (D) | 1 | |
| A77 | Yes | M. glareolus | B. grahamii (G) | 1 |
The description includes the rodent species and the number of animals from where gentoypes were detected or isolated, as well as the Bartonella species and clusters of the corresponding genotype.
Host specificity of Bartonella genotypes
When considering the specific association between Bartonella species and their rodent hosts, all B. grahamii, B. doshiae and the B. rochalimae related species genotypes were only recovered from bank voles. At the species level, B. taylorii strains were recovered from field voles, bank voles and wood mice with an apparent host preference for bank voles (93.3 % of B. taylorii strains were isolated from bank voles). However, data seem to indicate host specificity at the genotype level with B. taylorii clusters B and C only recovered from bank voles, while cluster E has been only isolated from wood mice. Clusters A and D have a larger host range and are detected in bank voles, wood mice (cluster A) or field voles (cluster D) (Figure 1, Table 2). Interestingly, genotypes infecting wood mice (A140 and A447) from cluster A and genotypes infecting field voles (A145) from cluster D can also infect bank voles (Figure 1, Table 2).
Genetic polymorphism and recombination within rodent adapted Bartonella species
We examined sequence polymorphism of the six housekeeping genes in all genotypes corresponding to B. taylorii and B. grahamii. B. doshiae and B. rochalimae related species were excluded from the study because of the low number of genotypes. Following alignment of the 3448 nucleotides of the six gene fragments, a total of 793 (23%) polymorphic sites were found, discriminating 41 individual alleles (Table 3). The number of polymorphic sites on a given locus varied from 56 (17.2%) for nuoG to 217 (26.72%) for groEL. The number of individual alleles for each of the six protein-coding genes ranged from 8 for ftsZ to 28 for groEL. Most polymorphisms resulted in synonymous substitutions, and the ratio of non-synonymous to synonymous substitutions (dN/dS) varied from 0.016 (for nuoG) to 0.24 (for ribC) (Table 3). These low ratios indicate a combination of purifying selection on amino-acid variation and a lack, or a very limited contribution, of positive environmental selection to the sequence variation in the six analyzed loci. These genes are therefore assumed to be appropriate for a population genetic study.
Table 3. Polymorphism and nucleotide diversity of six housekeeping protein-coding genes among Bartonella genotypes.
| Genes | Size | N. | Polymorphic (non synonymous) sites | dN | dS | dN/dS | π (%) | |
| (bp) | alleles | N | % | |||||
| All genotypes (n = 64) | ||||||||
| ftsZ | 788 | 8 | 147 | 18.65 | 0.0064 | 0.1623 | 0.0396 | 4.405 |
| gltA | 326 | 11 | 73 | 22.39 | 0.0128 | 0.2064 | 0.0619 | 5.419 |
| groEL | 812 | 28 | 217 | 26.72 | 0.0118 | 0.1520 | 0.0775 | 4.625 |
| nuoG | 325 | 16 | 56 | 17.23 | 0.0037 | 0.2262 | 0.0164 | 5.801 |
| ribC | 408 | 18 | 138 | 33.82 | 0.0428 | 0.1816 | 0.2358 | 7.562 |
| rpoB | 788 | 9 | 162 | 20.56 | 0.0080 | 0.1785 | 0.0449 | 4.952 |
| Concatenate | 3448 | 41 | 793 | 23.00 | 0.0127 | 0.1758 | 0.0724 | 5.182 |
| B. taylorii (n = 48) | ||||||||
| ftsZ | 788 | 4 | 26 | 3.30 | 0.0001 | 0.0376 | 0.0037 | 0.920 |
| gltA | 326 | 7 | 32 | 9.82 | 0.0065 | 0.0790 | 0.0820 | 2.202 |
| groEL | 812 | 22 | 112 | 13.79 | 0.0101 | 0.0175 | 0.5784 | 1.199 |
| nuoG | 325 | 14 | 51 | 15.69 | 0.0047 | 0.1583 | 0.0296 | 4.208 |
| ribC | 408 | 13 | 98 | 24.02 | 0.0158 | 0.0597 | 0.2652 | 2.618 |
| rpoB | 788 | 6 | 90 | 11.42 | 0.0020 | 0.0570 | 0.0351 | 1.542 |
| Concatenate | 3448 | 33 | 409 | 11.86 | 0.0058 | 0.0548 | 0.1062 | 1.760 |
| B. grahamii (n = 16) | ||||||||
| ftsZ | 788 | 2 | 1 | 0.13 | 0.0002 | 0.0000 | NA | 0.016 |
| gltA | 326 | 2 | 2 | 0.61 | 0.0005 | 0.0018 | 0.2722 | 0.077 |
| groEL | 812 | 4 | 9 | 1.11 | 0.0002 | 0.0114 | 0.0175 | 0.296 |
| nuoG | 325 | 1 | 0 | 0.00 | 0.0000 | 0.0000 | NA | 0.000 |
| ribC | 408 | 3 | 17 | 4.17 | 0.0050 | 0.0070 | 0.7133 | 0.547 |
| rpoB | 788 | 2 | 1 | 0.13 | 0.0000 | 0.0024 | 0.0000 | 0.058 |
| Concatenate | 3448 | 7 | 30 | 0.87 | 0.0007 | 0.0043 | 0.1733 | 0.159 |
π, average number of nucleotide differences per site; dS, number of synonymous changes per synonymous site; dN, number of non-synonymous changes per non-synonymous site; NA, not applicable.
The average nucleotide diversity (π) within all Bartonella genotypes was 5.2%±0.471, ranging from 4.4% to 7.5% per gene. Within the species B. grahamii, the average nucleotide distance was restricted (π = 0.159%±0.054, Table 3), indicating that the core genome of B. grahamii is relatively homogeneous, whereas B. taylorii (π = 1.76%±0.211) was diverse. We also noted striking intergene variation in levels of diversity within B. taylorii and B. grahamii genotypes: nuoG (π = 4.2%±0.458) sequences were found to be more variable than sequences of gltA (2.2%±0.255 on average) within B. taylorii species; within B. grahamii, groEL sequences (0.3±0.077) were more heterogeneous than those of gltA (0.077%±0.065), whereas nuoG sequences (1 allele, no polymorphic site) were homogeneous.
A quantitative analysis of the association between alleles from the six loci, by calculating the Index of Association (Ia) [40], estimated the contribution of recombination to the genetic structure among individuals of a population. Including all the Bartonella strains in the analysis, significant linkage disequilibrium was detected (Ia = 1.999±0.846), as expected for distinct species. Within B. taylorii genotypes, Ia was calculated as 1.847±0.988, confirming the significant linkage disequilibrium between alleles and hence a deviation from a random-mixing population within this species. In contrast, among B. grahamii strains, with a value of Ia at 0.128±0.761, no significant linkage disequilibrium was detected indicating frequent homologous recombination events.
Relationships between host specificity of rodent adapted Bartonella species and homologous recombination among clusters
Throughout the entire Bartonella genotypes, the inferred recombination to mutation value (r/m ratio) calculated with ClonalFrame was estimated at 4.06 (CI95 = [2.28; 6.38]), suggesting that nucleotide changes in housekeeping genes occur more frequently by recombination than by de novo mutation. Among the rodent community's two most abundant Bartonella species, recombination rather than mutation was found to have played a more important role within B. taylorii as compared to B. grahamii (r/m = 6.81 and 3.77, respectively). To examine any possible influences of recombination on the tree topology (Figure 1), we inferred a phylogenetic tree of the complete dataset using ClonalFrame, which takes recombination into account during tree building. Analysis resulted in a tree with the same major clades as identified by MLSA (Figure 2). We then evaluated whether recombination was detected between clusters by using several different approaches. Firstly, we individually compared the phylogeny of each gene and demonstrated that Bartonella genotype position varied depending upon the gene (Figure S1). Indeed, individual ftsZ, gltA, ribC and rpoB phylogenies showed strong congruence among clusters, but with some remarkable discrepancies. For example, as opposed to the concatenate or to five other genes, gltA did not strongly associate all of the genotypes belonging to cluster A. In contrast for this phylogeny, genotypes of cluster A were separated into two clades, of which one merged in a short branch with cluster B and C genotypes (Figure S1). This observation could be attributed to the horizontal transfer of the gltA gene from a donor related to cluster A, into an ancestral strain of clusters B and C. The rpoB sequence of the genotype A538 (B. doshiae) was identical to cluster A of B. taylorii sequences. Likewise, in ribC phylogeny, we identified a different position for 10 genotypes among clusters A, C, D (B. taylorii), G (B. grahamii) and H (B. rochalimae-like). For example, the ribC sequences of genotypes A296 (cluster H), A132 and A193 (cluster D) were almost identical to B. grahamii sequences, whereas the ribC sequence of A621 (B. grahamii) was identical to that of B. taylorii cluster A genotypes (Figure S1). GroEL and nuoG phylogenies were more discordant in their phylogenetic placement of genotypes. For the nuoG phylogeny, cluster C genotypes were distributed amongst all clusters, and genotype A296 was associated with cluster D. The groEL phylogeny showed an association of B. taylorii and B. doshiae genotypes to the B. birtlesii reference strain IBS 325, strongly supported by high bootstrap (Figure S1). These results, which were confirmed by re-sequencing of the six genes from new DNA extracts, demonstrated the occurrence of horizontal gene transfer between distantly related Bartonella species, as has been previously described [28].
Figure 2. 50% consensus tree from ClonalFrame.

Secondly, in order to detect those more subtle recombination events which result in detectable footprints of foreign DNA incorporation, nucleotide polymorphisms within the six genes were analyzed with the Structure software [44]. Structure recognized four clusters (A, D, E and G), and most strains within the identified clusters were homogeneous in terms of their ancestral polymorphism source (Figure 3). However, as expected, the transfer of gene sequences from other clusters were visualized by Structure, as is evident from the mixed color of the corresponding genotype column (Figure 3). Moreover, Structure revealed a number of genotypes that appeared to have a partially mixed origin, such that a mosaic pattern was seen where recombination of a large contiguous part of a gene had occurred. For example, a 323 bp segment of ribC from genotype A132 (B. taylorii, cluster D, recovered from a bank vole) was nearly identical to B. grahamii sequences (cluster G specific to bank voles), while a 293 bp segment of groEL was identical to sequences of B. taylorii cluster E genotypes (specific to wood mice). Furthermore, genotype A550 (B. taylorii, cluster D recovered from bank voles) contained a 313 bp ribC segment nearly identical to B. taylorii cluster A sequences (recovered from both bank voles and wood mice). Likewise, genotype A621 (B. grahamii, cluster G) exhibited a mixed origin, with ribC sequences matching those in B. taylorii clusters A and E (344 and 556 bp respectively). Interestingly, a number of B. taylorii and B. grahamii sequences originally obtained from bank voles were likely to have a mixed origin, as sequences were found to be identical to those from B. taylorii cluster E, which were exclusively found in wood mice. This sequence mosaicism was supported by at least three different algorithms in RDP3 and SplitsTree (p<0.05). Thus, Structure analysis disclosed evidence of recombination events among B. taylorii related genotypes, as well as between B. taylorii and B. grahamii genotypes, independent of the rodent species from which they were recovered.
Figure 3. Recombination events between genotypes.
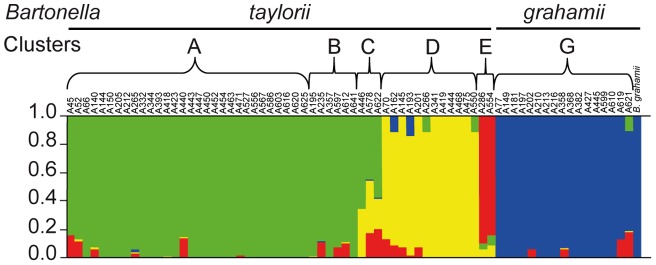
Results of Structure analysis showing the inferred proportion of nucleotides from four ancestral populations for each strain. The plot shows one vertical line for each strain, and the length of the colored segments indicates the proportions of nucleotides from each of the four ancestral populations.
These independent recombination events were further confirmed by performing a phylogenetic network analysis using the Neighbor-Net method. Evidence of extensive homologous recombination within the different genotypes, including cluster E, was supported by visual inspection of the bushy network structure with complex parallelogram formation (Figure 4).
Figure 4. Neighbor-Net graph based on concatenated sequences of the six housekeeping genes of Bartonella genotypes.

Note the bushy network structure among Bartonella strains indicative of pervasive homologous recombination.
Diversity and allele sharing of virB5 coding sequences among Bartonella clusters
Due to the important role of the VirB/D4 type IV SS in host adaptability of Bartonella species [5], we investigated the diversity and homologous recombination events among the pilus component-coding virB5 sequences.
virB5 sequences analyse was performed using genotypes retrieved from bank voles and wood mice only. Overall, the coding sequences of virulence factor virB5 showed 12 alleles undergoing a distinctive pattern of evolution. In relation to the entire virB5 gene, calculation of the dN/dS ratio (1.145) disclosed a weak purifying selection and/or diversifying selection. Differences between virB5 alleles were due to mutation, recombination events and the loss/gain of indels [46]. In all, the inferred VirB5 product ranged from 149 to 174 amino acids (aa) in length.
A phylogenetic tree (Figure 5) was generated using maximum likelihood with a GTR substitution model. virB5 sequences were clustered into seven clades (from clusters 1 to 7, in Figure 5). In contrast to housekeeping genes, phylogenetic analysis of virB5 sequences merged those sequences related to B. taylorii cluster C (specific to bank voles) with that of B. taylorii cluster D (infecting mainly bank voles and to a lesser extent, field mice), with 1.1% nucleotide divergence (see cluster 3 in Figure 5). This phylogenetic analysis also uncovered a conspicuous pattern of homologous recombination of short intragenic regions and entire virB5 sequences among the clusters, resulting in mosaic-like structures. The virB5 sequence of genotype A550 (B. taylorii, cluster D with MLSA, recovered from bank voles) was identical to cluster A sequences (infecting mainly bank voles, and wood mice to a lesser extent). Likewise, the virB5 sequence of genotype A554 (cluster E with MLSA, infecting wood mice) was merged with those from cluster A. Moreover, the virB5 sequences of genotype A612 and A357 (cluster B infecting bank voles) were nearly identical to those of B. grahamii (exclusively infecting bank voles).
Figure 5. Phylogeny of virB5 gene sequences using Maximum likelihood, with a GTR substitution model.
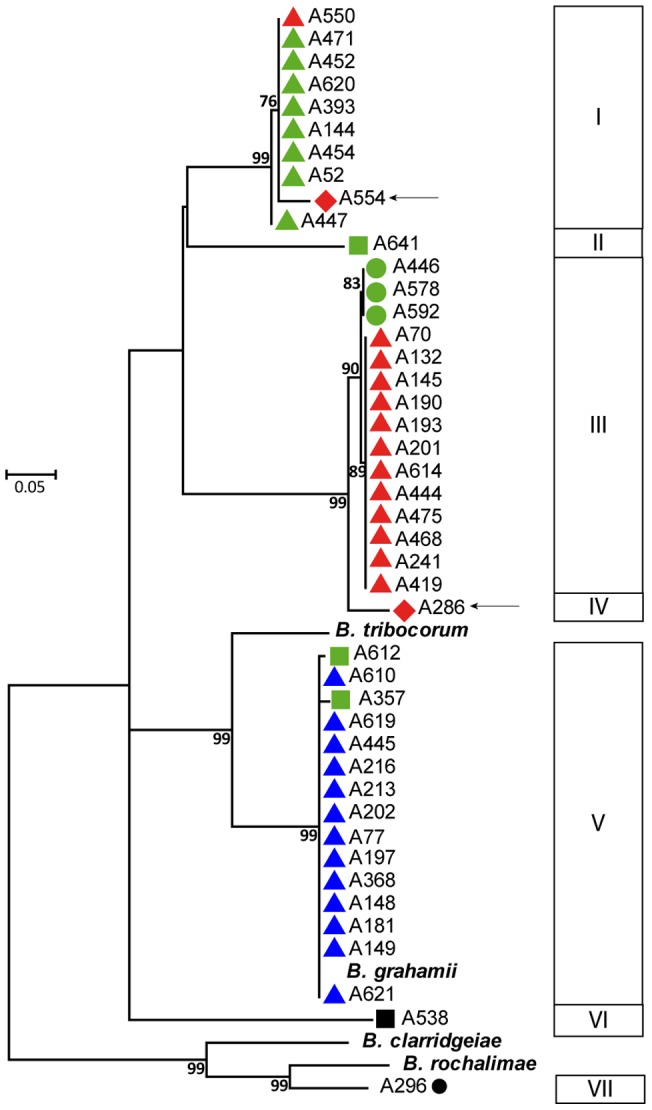
Clusters in which virB5 alleles were found are indicated by colors and shapes identical to Figure 1 (see legend). Bootstrap values higher than 80% are given at the nodes. Arrows indicate virB5 sequences amplified and sequenced from strains obtained from wood mice.
These results indicated that similar virb5 sequences are distributed across the clades identified with MLSA analysis, clearly demonstrating that virB5 alleles are not structured according to rodent host species.
Discussion
By studying the diversity of Bartonella strains recovered from a rodent community comprising two main rodent species, we provide evidence of strong host-specific associations between Bartonella genotypes and their hosts. Interestingly, we also highlight the existence of many recombination events between different Bartonella species and genotypes, demonstrating that even though these genotypes do not share the same rodent host species, they should co-exist during their life cycle, probably within their arthropod vectors, providing opportunities for recombination.
Until now, the majority of studies describing Bartonella populations within rodent communities used a gltA gene sequence to identify and characterize Bartonella genotypes. As shown in this study, many recombination events occur within this gene. Thus, only using this gene might lead to biases resulting in the false identification of genotypes [28]. To obtain unbiased and more discriminatory Bartonella genotype identification, we used six housekeeping genes, as well as the host adaptability VirB5-encoding gene, enabling a more precise overview of Bartonella strain diversity from distinct rodent species. The present work unambiguously confirmed the high diversity of Bartonella genotypes recovered from woodland rodents as demonstrated in previous studies [16], [18], [47], with 63 different Bartonella genotypes retrieved from 195 infected animals among 550 captured. These genotypes were separated into three known Bartonella species: B. taylorii, B. grahamii and B. doshiae, commonly found in Europe, and one genotype related to B. rochalimae-like isolates obtained from wood mice in Sweden and Spain [15], [48]. The percentage of nucleotide divergence between this genotype and B. rochalimae is 8.2%, based on MLSA. As a nucleotide divergence greater than 5% within housekeeping genes is now considered as equivalent to the 70% DNA-DNA re-association criteria used for species demarcation, B.rochalimae-like strains isolated here and elsewhere in Europe thus correspond to a new species. Unfortunately, we failed to obtain a pure microbiological culture, therefore it will be named candidatus Bartonella senartensis, “Sénart” being the name of the forest from where the rodents were collected.
To highlight specific associations between Bartonella and their mammalian hosts, our strategy was to study the diversity of Bartonella strains isolated from different sympatric rodent species. Globally, the B. taylorii population, identified in 160 individuals (bank voles, field voles and wood mice), was much larger than the B. grahamii population, which was only recovered from 29 bank voles. B. doshiae and B. rochalimae-like sequences were the least abundant genotypes, recovered from six and one bank vole(s) respectively. Interestingly, we identified much higher diversity within the five B. taylorii clades (1.76% using MLSA), compared to B. grahamii (0.2%) which were grouped into a single cluster. These large differences in diversity were also corroborated by the virB5 phylogeny, which distinguished nine different alleles for B. taylorii, while only one virB5 sequence was identified for B. grahamii. Similar homogeneity within B. grahamii strains has been observed in both the UK and Sweden (0.1%) [26] while high polymorphism of this species has been observed in Asia [25]. Different hypothesis could explain this low diversity of B. grahamii in Europe. The first one is that B. grahamii originated in Asia and then spread recently in Europe by the introduction of its hosts. A second hypothesis might be that B. grahamii experienced in Europe a severe bottleneck (i.e., a reduction in population size) relatively recently, with too little time having elapsed for polymorphisms to re-accumulate. This bottleneck could be the consequence of a host shift from a sympatric rodent species not sampled in this study and bank voles. The low polymorphism rate within B. grahamii is also consistent with the simultaneous diversification of B. taylorii in multiple lineages, due to a possible rapid population expansion in order to occupy empty ecological niches following the proposed B. grahamii bottleneck.
Despite the small number of wood mice and field voles compared to bank voles, our study tends to demonstrate that the Bartonella population hosted by bank voles was more diverse than those retrieved from wood mice or field voles. Our results also strongly suggest that B. taylorii (clusters B-C), B. grahamii, B. doshiae and candidatus Bartonella senartensis might present host specificity for bank voles, and cluster E of B. taylorii for wood mice. A broader host range has been identified for some genotypes belonging to clusters A and D (of B. taylorii), that can infect both bank voles or wood mice, or bank voles and field voles respectively. Seasonal host density is known to drive which Bartonella species infects sympatric rodents [47]. However, in our study, vole and mouse concomitantly trapped on the same grids in April 2008 presented equivalent population abundances (Dr B. Pisanu, personal communication). Our results thus suggest that host density plays a minor role in explaining the differences in Bartonella infection between voles and mice. Rather, host susceptibility might have a major impact on the different patterns of Bartonella diversity according to rodent species. This hypothesis should be confirmed by a longitudinal analysis of Bartonella diversity in wood mice co-inhabiting with bank voles in Sénart.
Interestingly, the ratio of recombination to mutation (r/m) across the entire Bartonella population was estimated to be 4.06, which was much higher compared to that of B. henselae (0.1) [27]. In the particular case of B. grahamii, we obtained the high r/m ratio of 3.77, compared to 1.7 for B. grahamii strains published in the study by Berglund et al. [26]. This difference may be due to the population of B. grahamii isolates sampled (i.e., B. grahamii genotypes collected worldwide versus B. grahamii genotypes sampled from a small area in our study). Interestingly, even though B. grahamii strains had a low polymorphism rate, we did identify recombination events, suggesting that perhaps the resulting recombinants did not persist within bank voles, due to a possible inability to adapt to their host. Another finding was that the contribution of recombination in generating diversity among B. taylorii (r/m = 6.81) was almost twice as high as that seen within B. grahamii (r/m = 3.77). These results reflect the greater diversity of B. taylorii strains compared to B. grahamii, and are also consistent with the wider host range of B. taylorii.
Our analysis also demonstrated DNA exchanges between Bartonella genotypes infecting the same rodent species such as B. taylorii clusters A and D; B. taylorii cluster D and B. grahamii cluster G. Interestingly, recombination between wood mice-specific genotypes (cluster E) and bank vole-specific genotypes from B. taylorii and B. grahamii has also been highlighted, suggesting their co-occurrence during their life cycle, possible in a common arthropod vector. Interestingly, different rodent species in the Sénart forest had equivalent diversity of rodent flea communities, supporting the hypothesis of genetic exchange within the vector [49].
As for the mechanisms responsible for host specificity, our results clearly indicate that the virB5 phylogeny is not sufficient to completely account for the pattern of rodent host association, and therefore must depend on additional more complex systems. In order to evaluate the potentially increased risk for humans to be infected with rodent-adapted Bartonella species due to possible host-shifting, understanding the molecular mechanisms of host specificity in areas where humans encounter rodents is extremely important. As suggested by recent genomic surveys, many putative host adaptability genes are essential for blood stream infection [5], [8] including other T4SS, adhesins, autotransporters. In this context, global analysis of all genomes is thus required in order to elucidate the subtle differences between closely related genotypes with different host ranges. Our study provides closely related genotypes harboring different host specificity patterns that should allow to further elucidate Bartonella host range preference at the molecular level by global genome comparison.
Supporting Information
Individual phylogenies of Bartonella genotypes constructed using internal sequences of six protein-coding genes (ftsZ, gltA, groEL, nuoG, ribC and rpoB).
(PDF)
This Includes Table S1 and Table S2. Table S1. GenBank accession numbers for ftsZ, gltA, groEL, ribC, rpoB and nuoG sequences of Bartonella reference strains. Table S2. Accession numbers of sequences of the six protein-coding genes and the virB5 gene from the 63 representative genotypes in GenBank/EMBL/DDBJ.
(DOC)
Acknowledgments
We thank Dr Jean-François Cosson for critical reading of the manuscript, and the group Tiques et Maladies à Tiques group (TMT) of Réseau Ecologie des Interactions Durables for stimulating discussion and support.
Funding Statement
This study was partially funded by the Regional Council of Ile-de-France, the Animal Health department of INRA and the EU grant FP7-261504 EDENext. It is catalogued by the EDENext Steering Committee as EDENext 078G (http://www.edenext.eu). The contents of this publication are the sole responsibility of the authors and do not necessarily reflect the views of the European Commission. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Finlay BB, Cossart P (1997) Exploitation of mammalian host cell functions by bacterial pathogens. Science 276: 718–725. [DOI] [PubMed] [Google Scholar]
- 2. Vayssier-Taussat M, Le Rhun D, Bonnet S, Cotte V (2009) Insights in Bartonella host specificity. Ann N Y Acad Sci 1166: 127–132. [DOI] [PubMed] [Google Scholar]
- 3. Deng H, Le Rhun D, Buffet JP, Cotte V, Read A, et al. (2012) Strategies of exploitation of mammalian reservoirs by Bartonella species. Vet Res 43: 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Dehio C (2004) Molecular and cellular basis of bartonella pathogenesis. Annu Rev Microbiol 58: 365–390. [DOI] [PubMed] [Google Scholar]
- 5. Engel P, Salzburger W, Liesch M, Chang CC, Maruyama S, et al. (2011) Parallel evolution of a type IV secretion system in radiating lineages of the host-restricted bacterial pathogen Bartonella. PLoS Genet 7: e1001296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Engel P, Dehio C (2009) Genomics of host-restricted pathogens of the genus bartonella. Genome Dyn 6: 158–169. [DOI] [PubMed] [Google Scholar]
- 7. Vayssier-Taussat M, Le Rhun D, Deng HK, Biville F, Cescau S, et al. (2010) The Trw type IV secretion system of Bartonella mediates host-specific adhesion to erythrocytes. PLoS Pathog 6: e1000946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Saenz HL, Engel P, Stoeckli MC, Lanz C, Raddatz G, et al. (2007) Genomic analysis of Bartonella identifies type IV secretion systems as host adaptability factors. Nat Genet 39: 1469–1476. [DOI] [PubMed] [Google Scholar]
- 9. Kosoy M, Murray M, Gilmore RD Jr, Bai Y, Gage KL (2003) Bartonella strains from ground squirrels are identical to Bartonella washoensis isolated from a human patient. J Clin Microbiol 41: 645–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Jardine C, Appleyard G, Kosoy MY, McColl D, Chirino-Trejo M, et al. (2005) Rodent-associated Bartonella in Saskatchewan, Canada. Vector Borne Zoonotic Dis 5: 402–409. [DOI] [PubMed] [Google Scholar]
- 11. Inoue K, Kabeya H, Hagiya K, Kosoy MY, Une Y, et al. (2011) Multi-locus sequence analysis reveals host specific association between Bartonella washoensis and squirrels. Vet Microbiol 148: 60–65. [DOI] [PubMed] [Google Scholar]
- 12. Bai Y, Calisher CH, Kosoy MY, Root JJ, Doty JB (2011) Persistent infection or successive reinfection of deer mice with Bartonella vinsonii subsp. arupensis. Appl Environ Microbiol 77: 1728–1731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ying B, Kosoy MY, Maupin GO, Tsuchiya KR, Gage KL (2002) Genetic and ecologic characteristics of Bartonella communities in rodents in southern China. Am J Trop Med Hyg 66: 622–627. [DOI] [PubMed] [Google Scholar]
- 14. Birtles RJ, Hazel SM, Bennett M, Bown K, Raoult D, et al. (2001) Longitudinal monitoring of the dynamics of infections due to Bartonella species in UK woodland rodents. Epidemiol Infect 126: 323–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Holmberg M, Mills JN, McGill S, Benjamin G, Ellis BA (2003) Bartonella infection in sylvatic small mammals of central Sweden. Epidemiol Infect 130: 149–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Berglund EC, Ehrenborg C, Vinnere Pettersson O, Granberg F, Naslund K, et al. (2010) Genome dynamics of Bartonella grahamii in micro-populations of woodland rodents. BMC Genomics 11: 152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Buffet JP, Marsot M, Vaumourin E, Gasqui P, Masseglia S, et al.. (2012) Co-infection of Borrelia afzelii and Bartonella spp. in bank voles from a suburban forest. Comp Immunol Microbiol Infect Dis. [DOI] [PubMed]
- 18. Paziewska A, Harris PD, Zwolinska L, Bajer A, Sinski E (2012) Differences in the ecology of Bartonella infections of Apodemus flavicollis and Myodes glareolus in a boreal forest. Parasitology 139: 881–893. [DOI] [PubMed] [Google Scholar]
- 19. Berglund EC, Frank AC, Calteau A, Vinnere Pettersson O, Granberg F, et al. (2009) Run-off replication of host-adaptability genes is associated with gene transfer agents in the genome of mouse-infecting Bartonella grahamii. PLoS Genet 5: e1000546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Arvand M, Feil EJ, Giladi M, Boulouis HJ, Viezens J (2007) Multi-locus sequence typing of Bartonella henselae isolates from three continents reveals hypervirulent and feline-associated clones. PLoS One 2: e1346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Chaloner GL, Harrison TG, Coyne KP, Aanensen DM, Birtles RJ (2011) Multilocus sequence typing of Bartonella henselae in the United Kingdom indicates that only a few, uncommon sequence types are associated with zoonotic disease. J Clin Microbiol 49: 2132–2137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bouchouicha R, Durand B, Monteil M, Chomel BB, Berrich M, et al. (2009) Molecular epidemiology of feline and human Bartonella henselae isolates. Emerg Infect Dis 15: 813–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Foucault C, La Scola B, Lindroos H, Andersson SG, Raoult D (2005) Multispacer typing technique for sequence-based typing of Bartonella quintana. J Clin Microbiol 43: 41–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Arvand M, Raoult D, Feil EJ (2010) Multi-locus sequence typing of a geographically and temporally diverse sample of the highly clonal human pathogen Bartonella quintana. PLoS One 5: e9765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Inoue K, Kabeya H, Kosoy MY, Bai Y, Smirnov G, et al. (2009) Evolutional and geographical relationships of Bartonella grahamii isolates from wild rodents by multi-locus sequencing analysis. Microb Ecol 57: 534–541. [DOI] [PubMed] [Google Scholar]
- 26. Berglund EC, Ellegaard K, Granberg F, Xie Z, Maruyama S, et al. (2010) Rapid diversification by recombination in Bartonella grahamii from wild rodents in Asia contrasts with low levels of genomic divergence in Northern Europe and America. Mol Ecol 19: 2241–2255. [DOI] [PubMed] [Google Scholar]
- 27. Vos M, Didelot X (2009) A comparison of homologous recombination rates in bacteria and archaea. ISME J 3: 199–208. [DOI] [PubMed] [Google Scholar]
- 28. Paziewska A, Harris PD, Zwolinska L, Bajer A, Sinski E (2011) Recombination within and between species of the alpha proteobacterium Bartonella infecting rodents. Microb Ecol 61: 134–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Pisanu B, Marsot M, Marmet J, Chapuis JL, Reale D, et al. (2010) Introduced Siberian chipmunks are more heavily infested by ixodid ticks than are native bank voles in a suburban forest in France. Int J Parasitol 40: 1277–1283. [DOI] [PubMed] [Google Scholar]
- 30. Marmet J, Pisanu B, Chapuis J (2011) Natal dispersal of introduced Siberian chipmunks, Tamias sibiricus, in a suburban forest. Journal of Ethology 29: 23–29. [Google Scholar]
- 31. Zeaiter Z, Liang Z, Raoult D (2002) Genetic classification and differentiation of Bartonella species based on comparison of partial ftsZ gene sequences. J Clin Microbiol 40: 3641–3647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zeaiter Z, Fournier PE, Ogata H, Raoult D (2002) Phylogenetic classification of Bartonella species by comparing groEL sequences. Int J Syst Evol Microbiol 52: 165–171. [DOI] [PubMed] [Google Scholar]
- 33. Renesto P, Gouvernet J, Drancourt M, Roux V, Raoult D (2001) Use of rpoB gene analysis for detection and identification of Bartonella species. J Clin Microbiol 39: 430–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Colborn JM, Kosoy MY, Motin VL, Telepnev MV, Valbuena G, et al. (2010) Improved detection of Bartonella DNA in mammalian hosts and arthropod vectors by real-time PCR using the NADH dehydrogenase gamma subunit (nuoG). J Clin Microbiol 48: 4630–4633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Librado P, Rozas J (2009) DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25: 1451–1452. [DOI] [PubMed] [Google Scholar]
- 36.Tamura K, Peterson D, Peterson N, Stecher G, Nei M, et al.. (2011) MEGA5: Molecular Evolutionary Genetics Analysis using Maximum Likelihood, Evolutionary Distance, and Maximum Parsimony Methods. Mol Biol Evol. [DOI] [PMC free article] [PubMed]
- 37. Posada D (2008) jModelTest: phylogenetic model averaging. Mol Biol Evol 25: 1253–1256. [DOI] [PubMed] [Google Scholar]
- 38. Guindon S, Gascuel O (2003) A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol 52: 696–704. [DOI] [PubMed] [Google Scholar]
- 39. Didelot X, Falush D (2007) Inference of bacterial microevolution using multilocus sequence data. Genetics 175: 1251–1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Smith JM, Smith NH, O'Rourke M, Spratt BG (1993) How clonal are bacteria? Proc Natl Acad Sci U S A 90: 4384–4388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Martin DP, Lemey P, Lott M, Moulton V, Posada D, et al. (2010) RDP3: a flexible and fast computer program for analyzing recombination. Bioinformatics 26: 2462–2463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Huson DH, Bryant D (2006) Application of phylogenetic networks in evolutionary studies. Mol Biol Evol 23: 254–267. [DOI] [PubMed] [Google Scholar]
- 43. Bruen TC, Philippe H, Bryant D (2006) A simple and robust statistical test for detecting the presence of recombination. Genetics 172: 2665–2681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Hubisz MJ, Falush D, Stephens M, Pritchard JK (2009) Inferring weak population structure with the assistance of sample group information. Mol Ecol Resour 9: 1322–1332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Palys T, Nakamura LK, Cohan FM (1997) Discovery and classification of ecological diversity in the bacterial world: the role of DNA sequence data. Int J Syst Bacteriol 47: 1145–1156. [DOI] [PubMed] [Google Scholar]
- 46. Paziewska A, Sinski E, Harris PD (2012) Recombination, diversity and allele sharing of infectivity proteins between bartonella species from rodents. Microb Ecol 64: 525–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Telfer S, Begon M, Bennett M, Bown KJ, Burthe S, et al. (2007) Contrasting dynamics of Bartonella spp. in cyclic field vole populations: the impact of vector and host dynamics. Parasitology 134: 413–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Gil H, Garcia-Esteban C, Barandika JF, Peig J, Toledo A, et al. (2010) Variability of Bartonella genotypes among small mammals in Spain. Appl Environ Microbiol 76: 8062–8070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Pisanu B, Marmet J, Beaucournu JC, Chapuis JL (2008) [Fleas community in introduced Siberian chipmunks (Tamias sibiricus Laxmann) in Forest of Senart, France]. Parasite 15: 35–43. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Individual phylogenies of Bartonella genotypes constructed using internal sequences of six protein-coding genes (ftsZ, gltA, groEL, nuoG, ribC and rpoB).
(PDF)
This Includes Table S1 and Table S2. Table S1. GenBank accession numbers for ftsZ, gltA, groEL, ribC, rpoB and nuoG sequences of Bartonella reference strains. Table S2. Accession numbers of sequences of the six protein-coding genes and the virB5 gene from the 63 representative genotypes in GenBank/EMBL/DDBJ.
(DOC)