

Abstract
Mitochondria are dynamic organelles that frequently undergo fusion and fission, the balance of which is critical for proper cellular functioning and viability. Most studies on mitochondrial fusion and fission mechanisms have focused on proteins thought to physically mediate the events. However, dynamic changes in membrane phospholipids also play roles in facilitating the fusion and fission events. This chapter will review the importance of lipids in mitochondrial dynamics and some of the methods that can be used to study the function of lipids in mitochondrial fusion and fission.
Keywords: Mitochondria, lipid signaling, lipid imaging, fluorescent proteins, chemiluminescence
I. Introduction
A. Mitochondrial dynamics
Mitochondrial “health” requires frequent fusion of mitochondria and subsequent division (fission). The fusion process is critical for exchanging mitochondrial contents and maintaining integrity of the mitochondrial genome, whereas fission is vital for being able to distribute mitochondria to subcellular locations where ATP is most needed or during mitosis, and for facilitating turnover of damaged mitochondria via mitophagy (Braschi and McBride, 2010; Chan, 2006; Otera and Mihara, 2011). Disturbance of the mitochondrial fusion-fission balance in humans results in multiple consequences (Kane and Youle, 2010; Westermann, 2010) including peripheral neuropathy (Zuchner et al., 2004), optic atrophy (Olichon et al., 2006), and neonatal lethality (Waterham et al., 2007).
A number of proteins critically involved in mitochondrial fusion and fission in yeast and mammals have been identified–Mitofusin 1 and 2 (Mfn1/2), Optic atrophy 1 (Opa1), human Fission 1 (hFis1) and dynamin-related protein 1 (Drp1) (Otera and Mihara, 2011). During mitochondrial fusion, the outer mitochondrial membrane proteins Mfn1/2 tether the adjacent mitochondria with their C-terminal coiled-coil domain to promote outer membrane fusion (Koshiba et al., 2004), while the inner membrane protein Opa1 faces the inter membrane space and mediates inner membrane fusion (reviewed in Chen and Chan, 2010). Conversely, hFis1 resides in the outer mitochondrial membrane and has long been thought to recruit the cytosolic protein Drp1 to sites of future fission, although recently, the proposal has been made that another outer membrane protein, mitochondrial fission factor, may be the critical component that recruits Drp1 instead of hFis1 (Gandre-Babbe and van der Bliek, 2008; Otera et al., 2010). Regardless of the recruitment mechanism, Drp1 self-assembles as an oligomer and wraps around the mitochondria to achieve membrane constriction and division (Mears et al., 2011), analogous to the role dynamin undertakes during endocytosis (Chan, 2006). Many additional proteins have been added to the list of ones that affect mitochondrial fusion and fission as a result of either screening experiments or random discoveries, complicating our understanding of the mechanisms underlying mitochondrial fusion and fission–much remains to be uncovered and explained!
B. Lipids in mitochondrial dynamics
Apart from the involvement of two membranes, many aspects of the biophysical process of mitochondrial fusion and fission are analogous to the fusion and fission of cytosolic membrane vesicles as they bud from and fuse into subcellular membrane compartments. Similarly, several lipids have been discovered to be important in potentially analogous ways as well (reviewed in Furt and Moreau, 2009; Osman et al., 2011). A role for the signaling lipid phosphatidic acid (PA) has been described in promoting Mfn-dependent mitochondrial fusion that is analogous to the role PA plays in SNARE protein-regulated exocytosis, although each fusion process uses a different lipid-modifying enzyme, MitoPLD and classic PLD, respectively, to locally produce PA (Choi et al., 2006; Huang et al., 2005; Vicogne et al., 2006; Vitale et al., 2001). More recently, another lipid, diacylglycerol (DAG), has been linked to mitochondrial fission events (Huang et al., 2011), analogous to the role of DAG in Golgi vesiculation (Fernandez-Ulibarri et al., 2007). Evidence has been presented for roles of other lipids, including cardiolipin (Khalifat et al., 2008; Montessuit et al., 2010) and cholesterol(Altmann and Westermann, 2005), in mitochondrial membrane morphodynamics (reviewed in Furt and Moreau, 2009; Osman et al., 2011). Cardiolipin, which is primarily found in the inner membrane, has been shown to play a major role cristae dynamics (Khalifat et al., 2008) and in membrane fusion through interaction with Opa1/Mgm1 (Ban et al., 2010; DeVay et al., 2009; Rujiviphat et al., 2009). On the outer membrane, where a smaller but significant amount of cardiolipin is found (Gebert et al., 2009), cardiolipin has been shown to serve as the substrate for PA production during fusion events (Choi et al., 2006). In addition, Drp1-stimulation of Bax oligomerization and cytochrome C release during apoptosis requires the presence of cardiolipin in the outer membrane, and the Drp1-cardiolipin interaction is thought to trigger membrane tethering and hemifusion (Montessuit et al., 2010) which might play also a role in fission in this context (Kozlovsky and Kozlov, 2003). For cholesterol, a mechanism of action is not clear, but many of the genes required for synthesis of cholesterol have been identified as regulators of mitochondrial morphogenesis in yeast (Altmann and Westermann, 2005).
II. Imaging mitochondrial tubules in overexpression or knockdown samples
Since directly manipulating lipids on mitochondrial membranes via introduction of liposomes or other approaches is almost impossible in living cells, controlling expression levels and/or localization of lipid-modifying enzymes is the methodology used to test the importance of individual lipids in mitochondrial morphology. The approaches used should include overexpression and inactivation (via RNAi, small molecule inhibitors, or genetic ablation) of the lipid-generating enzyme, re-targeting of enzymes that can generate the lipid but that are normally found elsewhere in the cell (Komatsu et al., 2010), and overexpression or re-targeting of enzymes that consume the lipid (Choi et al., 2006; Huang et al., 2011). Since each of these approaches has inherent caveats, use of more than one approach when possible is preferable. As well, some thought needs to be given to potential compensatory issues; there are multiple pathways for generation and consumption of most lipids, and long-term removal of a specific pathway (e.g. via genetic ablation) may have a diminished or different effect than acute removal (e.g. via a small molecule inhibitor (e.g. Su et al., 2009) or chemically-induced rapid re-targeting of a lipid modifying enzyme (e.g. Nishioka et al., 2010)).
A. Choice of cell types
To assess mitochondrial morphology in mammalian cells, in theory, any cell line that is easy to culture and transfect can be used. We routinely use HeLa, NIH-3T3, or mouse embryo fibroblast (MEF) cells. However, for investigators who are interested in mitochondrial dynamics in specific cell types such as neurons or muscle cells, specialized cell types can be used as well. It is advisable to check multiple cell lines at the beginning of the intended studies, since different cell lines can have unique characteristics that result in different mitochondrial tubule lengths under basal conditions. When the intent is to study a lipid involved in mitochondrial fission, it is better for some purposes to start off with a cell line that has longer tubules such as HeLa cells in order to easily detect overexpression phenotypes, and conversely, cell lines with shorter tubules are preferred for lipids that promote fusion. However, since gain and loss-of-function experiments will yield opposite effects, the general ideal is to start with cells with intermediate length tubules.
Some cells are relatively challenging to study because of specific characteristics. For example, the OP9 and NIH3T3-L1 pre-adipocyte cell lines are relatively flat and well-spread on coverslips and easily imaged. However, upon differentiation, the cells become spherical, filled with lipid droplets, and have relatively scant cytoplasm, making it very challenging to discriminate changes in mitochondrial morphology using confocal microscopy. In some cases, however, use of stronger-adhering coverslip coatings (for example, poly-L-lysine), may elicit sufficient flattening and spreading of the cells that the imaging can be performed.
B. Mitochondrial labeling methods
There are multiple ways to label the mitochondrial tubules–mitochondrial-targeted fluorescent proteins (MitoFPs), MitoTracker dyes, and immunostaining using mitochondria-specific protein antibodies. The MitoFPs, constructed by fusing any fluorescent protein with a mitochondrial localization sequence (MLS), can be co-transfected with the lipid enzyme or co-expressed in an IRES vector (a number of such constructs are available from Clontech). However, when a specific cell line has to be used and achieving high transfection efficiency is not possible, the MitoTracker dyes and antibody staining are preferred since the mitochondria in all the cells will be labeled equally well.
There are a range of MitoTracker dyes with different wavelengths available from Invitrogen. Most dyes, such as MitoTracker Orange and MitoTracker Red, are sensitive to mitochondrial membrane potential, which is useful as an indicator of mitochondrial function, but potentially problematic if manipulation of the lipid under study might result in compromised membrane potential. In contrast, MitoTracker Green accumulates in mitochondria regardless of membrane potential. However, it should be noted that cells labeled with MitoTracker Green should be fixed with cold acetone instead of aldehyde-based fixatives.
Finally, immunostaining with mitochondria-specific antibodies requires more procedure steps and time, but is versatile, offers a broad choice of secondary antibodies, and avoids the risk of altering the morphology of mitochondria through introducing a foreign protein or dye into them. Common and abundant mitochondria-specific proteins used for imaging with commercially-available antibodies include cytochrome c, cytochrome c oxidase IV, Tom 20, Tom 22, Tom 70 and porin. Worth noting, when multiple proteins of interest need to be detected at the same time, it can be preferable to use MitoFPs or MitoTracker dyes to visualize the mitochondrial population, since there are only a limited number of species that secondary antibodies are available for, limiting the number of primary antibodies that can be simultaneously detected.
C. Data analysis
Mitochondria display diverse morphologies even within the same cell line. Therefore, it is critical to take high quality images of mitochondrial morphology for many (often one to several hundred) cells for each experimental condition (generally using confocal microscopy) and then quantify the results. Nonetheless, mitochondrial morphology is a subjective phenotype, and it can be difficult to cleanly categorize them. Therefore, before analyzing the data, it is important to become familiar with the possible mitochondrial phenotypes that can exist under different conditions, some of which are shown in Figure 1. Mitochondria normally appear tubular, intermediate, or in a minority of cases, fragmented; however, in extreme conditions when a mitochondrial fusion protein or fission protein is overexpressed, the mitochondria can aggregate in the peri-nuclear region or fragment into small spherical or oval mitochondria, respectively. Software analysis programs such as ImageJ (freely available from NIH) can be used to compare mitochondrial tubule lengths using the “analyze particles” function. However, the disadvantage of automated programs is that they can only distinguish very obvious phenotypes such as mitochondrial aggregation or mitochondrial fragmentation. When it comes to subtle changes in tubule length, none of the programs are as sophisticated as the trained human eye.
Fig. 1.
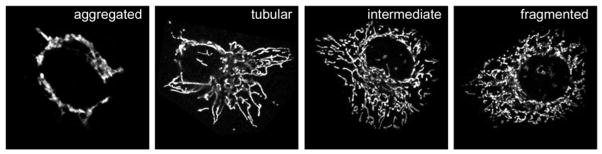
Examples of mitochondrial morphology. HeLa cells display different mitochondrial morphologies, as shown for cells overexpressing MitoPLD that have aggregated mitochondria (left), or others with tubular, intermediate, or fragmented mitochondria as seen under basal conditions. Mitochondria are stained with anti-cytochrome c antibody.
With sufficient numbers of images of mitochondrial morphology generated from three or more independent experiments for each treatment, the data can be analyzed (ideally by a blinded investigator) by categorizing each cell as tubular, intermediate or fragmented, and then comparing the distribution of the categories for each sample by calculating the percentage of cells exhibiting each phenotype. Statistical analysis (t-test for two treatments or one-way ANOVA for more than two treatments) is then used to determine whether there is a significant difference between the experimental conditions. When the lipid has a profound effect on the mitochondrial morphology, the categories can be assigned differently to suit the extreme conditions. For example, when PA is generated by MitoPLD overexpression, it induces mitochondrial aggregation at high levels of expression and mitochondrial elongation at low levels(Choi et al., 2006; Huang et al., 2011). These morphological phenotypes can be tabulated as different categories. Similarly, other manipulations can cause tubulation or fragmentation (Choi et al., 2006; Huang et al., 2011).
III. A quantitative assay for mitochondrial fusion
Through the above experiments, the investigators should have an idea whether the lipids they are interested in help to elongate or shorten mitochondrial tubule lengths. However, the mitochondrial morphology is determined by the balance of the opposing fusion and fission events. Therefore, assessing morphological changes alone only indicates shifts in the balance of fusion and fission. Longer tubules could be result either from more fusion or from fewer fission events. To determine which, mitochondrial fusion assays can be employed to assess whether the cells have abnormal rates of fusion. If the lipid is suspected to be involved in mitochondrial fission, there are other methods that have been described recently (Molina and Shirihai, 2009).
Multiple versions of mitochondrial fusion assays are available. The classic mitochondrial fusion assay introduces two different fluorescent proteins into two populations of the cells, mixes the cells and then induces cell fusion with polyethylene glycerol (PEG) or a hemagglutinating virus. The subsequent mitochondrial fusion after cell fusion is assayed several hours later by scoring the extent of coincidence of the two fluorescent proteins (Nunnari et al., 1997). A second mitochondrial fusion assay was developed to take advantage of photoactivatable GFP (PA-GFP). By co-labeling the mitochondria with matrix-targeted PA-GFP and DsRed2, the fusion and fission of mitochondria can be quantitatively recorded after irradiation at regions of interest in a chosen cell(Twig et al., 2008; Twig et al., 2006). In brief, the DsRed2 protein visualizes all of the mitochondria using red fluorescence; when the PA-GFP is activated in a restricted region of the cell, mitochondria in that area become yellow (as a combination of the DsRed and the newly fluorescent PA-GFP). As fusion subsequently occurs, mixing of matrix contents transfers the activated PA-GFP into previously red-only mitochondria, resulting in spread and dilution of the yellow color, both of which can be quantitated to describe mitochondrial dynamics (motility and fusion). Both of the above invaluable mitochondrial fusion assays are described in reviews elsewhere (Ingerman et al., 2007; Molina and Shirihai, 2009). Here, we will discuss the quantitative assay for mitochondrial fusion that was newly developed by our lab (Huang et al., 2010).
The quantitative assay for mitochondrial fusion works by labeling two populations of cells with split-Renilla luciferase (RLuc) fragments that reconstitute and perform chemiluminescence when the mitochondria fuse together following chemical induced cell fusion (Huang et al., 2010). Therefore, the first steps for the assay are to choose a desired cell line and to introduce each split-RLuc construct into the cells to select for stable expressing cells, following which the assay can be performed to compare the luciferase activity between different samples.
A. Generation of stable cells
1. Choice of cell lines
In situations where small molecule inhibitors can be used, the most important consideration is the relevance of the cell type to investigator’s interest in the functions that ultimately need to be assessed. In other situations, e.g. where manipulation of a lipid by overexpressing or knocking-down a lipid enzyme is needed, one has to balance between the relevance of the cell type and the ease of transfection. For the assay to be valid using transient transfection approaches, at least 80% transfection efficiency needs to be achieved. If this is not feasible using standard plasmid transfection approaches, alternative ways to introduce the DNA can include methods such as viral infection. Another solution is to generate stable expression cell lines first for the lipid modifying enzyme of interest, and then introduce the split-RLuc constructs into both control and the stable-expressing cell lines.
2. Generation of stable cell lines
Since the split-RLuc constructs were generated in a retroviral backbone (Huang et al., 2010), generation of stable cell lines can be achieved by producing the retrovirus in a virus packaging cell line, infecting the target cells with the virus, and then selecting stable cell populations with appropriate antibiotics, in this case puromycin. The detailed protocol varies with the virus packaging cell line, each of which has its own instructions and manuals with the protocols needed to generate stable cell lines.
B. The quantitative assay for mitochondrial fusion
Once the stable cell lines are ready, two more steps that need to be carried out before performing the mitochondrial fusion assay are to determine how many cells are needed for the assay and what concentration of cycloheximide (CHX) to use during the assay. CHX is required to prevent de novo expression and mitochondrial targeting of reporter proteins subsequent to cell fusion, which would generate a false positive readout for the mitochondrial fusion assay (Huang et al., 2010).
To determine the cell number, a range in numbers of cells from 105 to 106 should be plated in a 12-well plate and examined the following day to identify the well that has reached 80–100% confluency. If transfection of plasmid constructs or RNAi oligos is required, it will be necessary to plate a smaller number of cells, perform the transfection the following day, and wait for 1–3 days to achieve expression / knockdown of a specific mRNA and protein. The assay can be scaled up or down in size according to need. To determine the optimal CHX concentration, the cell mixture containing each split-RLuc construct are co-plated at the cell density determined above. The next day, the cells are treated with different concentrations of CHX, fused with PEG 1500, incubated for three hours and then assayed for luciferase activity (refer to detailed protocols below). The relative light unit (RLU) of luciferase activity will decrease with increased CHX concentration until a baseline is reached, signifying the level at which de novo protein production has ceased. The lowest CHX concentration that achieves baseline Luciferase activity should be chosen as that used for the assay. In practice, this concentration will vary for different types of cell lines. Due to the extreme sensitivity of the luciferase activity measurement and thus the fluctuations in the readings, each sample should be measured at least three times to determine the optimal CHX concentration. Concentrations of 0, 50 μg/ml, 100 μg/ml, 200μg/ml, and 400 μg/ml represent a range that will cover most cell types.
General protocol for mitochondrial fusion assay
Mix and plate cells containing each split-RLuc construct in 12-well plates with the predetermined cell number. Number of wells to plate (N=a×b×c) depends on the treatment numbers (a), time points numbers (b) and repeat numbers (c). Experiments should be conducted at the minimum in triplicate.
(Optional) The next day, transfect or infect the cells with constructs or oligos as desired.
When the cells reach 80–100% confluence, pre-treat with small molecule inhibitors as desired and then add CHX at the predetermined concentration 30 minutes before cell fusion.
At the end of the CHX incubation period, remove the media from the wells and add 300 μl per well pre-warmed 50% PEG 1500 for exactly 60 seconds. Practice first to see how many samples can be handled at the same time. Divide samples into several groups if necessary.
After 60 seconds, leaving the PEG in the well, immediately add 1 ml of complete media containing cycloheximide to each well. Swirl well to get rid of the PEG and wash two more times with complete media containing cycloheximide.
Add complete media containing cycloheximide and then replace the cells in the incubator. Collect cells at 0, 30 minutes, 1 hour, 2 hours, 3 hours, 5 hours, and 7 hours, or more simply, 0, 1 hours and 3 hours. To collect individual samples, take out the plate from incubator, add 500 μl 5 mM EDTA in PBS containing cycloheximide, wait 1 minute for the cells to come off the plate and then collect the cells into an eppendorf tube. Put the plate back in the incubator. Spin down the cells at 14,000 rpm for 1 minute and aspirate off the supernatant. Immediately store the cell pellets in −20 °C.
When all the samples are collected, lyse the cell pellets with 50 μl 1× Renilla luciferase assay lysis buffer on ice for 15 min. Vortex for 2 seconds.
Freshly prepare an adequate volume of luciferase substrate to perform the desired number of Renilla luciferase assays (20 μl reagent per assay sample). Add 1 volume of 100× Renilla luciferase substrate to 100 volumes of Renilla luciferase assay buffer.
Set up the luminometer with integration time of five seconds.
Add 20 μl of Renilla luciferase assay solution into a transparent eppendorf tube and place the tube in the luminometer.
Add 3 μl of the lysate sample into the solution. Pipette up and down to mix well and then initiate measurement.
Proceed with the next sample from step 10.
C. Data analysis
For analysis, plot time (x) versus RLU (y) and compare the resulting curves for each experimental condition. Potential outcomes of the plots are described below (Figure 2). Each curve should also be compared with the control experimental condition, which indicates the baseline for fusion rates for that cell type. The Luciferase signal increases over time in parallel to the progression of fusion, but then eventually begins to fall, as the reporters being to undergo degradation.
Fig. 2.

Data analysis. Hypothetical examples of data representing reduced mitochondrial fusion (A), lack of mitochondrial fusion (B), and increased mitochondrial fusion (C).
Example A: Each early time point in the experimental sample reveals a lower luciferase activity than is observed for the corresponding control sample, but the curves eventually merge, indicating that cells in the experimental condition are undergoing mitochondrial fusion at a slower rate than the cells in the control conditions.
Example B: Luciferase activity is dramatically lower in comparison to the control sample, with a peak reading at the 1 hour time point instead of at 3 hours, indicating that there is little ongoing fusion. The residual luciferase activity is thought to come from reconstitution of a small fraction of the reporter proteins in the cytoplasm, or release of the reporter proteins during apoptosis.
Example C: Each time point in the experimental sample exhibits a higher luciferase activity than that of the corresponding control sample with the peak luciferase activity occurring earlier, suggesting that the cells are undergoing mitochondrial fusion at an increased rate.
IV Visualizing lipids in cells
Morphological analysis of mitochondrial length and the fusion assay described above provide function information concerning the consequences of manipulating the levels of individual lipids on mitochondrial fusion or fission. Complementing these approaches, lipid sensors exist for a number of kinds of lipids that permit direct visualization of the lipids on the mitochondrial surface (readers should also be directed to another chapter in this volume by Sarantis and Grinstein). Sensors for PA and DAG have been successfully employed to detect these lipids on the mitochondrial surface (Choi et al., 2006; Gallegos et al., 2006; Huang et al., 2011; Sato et al., 2006).
A. Lipid sensors
Standard lipid sensors consist of the lipid-recognizing domains of known lipid-binding proteins fused with fluorescent proteins of various colors, which enables live cell imaging of lipid dynamics to study the spatio-temporal aspects of lipid signaling. This approach has been utilized to generate sensors for a wide variety of lipids including PA (Nakanishi et al., 2004; Rizzo et al., 2000), DAG (Dries et al., 2007; Giorgione et al., 2006) and different species of phosphoinositides (Varnai and Balla, 2006), using a variety of protein domains. These sensors are very useful for visualizing the relative distribution of individual lipids within the cell. However, it should be noted that it is quite challenging to use such sensors for quantitative measurements of the lipids, in contrast to more technically challenging approaches using fluorescence resonance energy transfer (FRET)-based lipid sensors that have been developed specifically to this end. Different approaches have been developed utilizing the FRET principle to generate signals based on the lipid levels. So far, the most sophisticated method is to attach both fluorophores to the same lipid-binding domain, taking advantage of the conformational change that occurs upon lipid binding that alters the distance between the fluorophores, resulting in changes in energy transfer. Examples of such lipid sensors include FRET-based DAG (Gallegos et al., 2006; Sato et al., 2006), PA (Nishioka et al., 2010), and PIP3 sensors (Sato et al., 2006; Tanimura et al., 2004).
B. Assay design
Most of the lipid sensors that have been described are openly available from the investigators that generated them. It’s worth noting that some of the sensors appear to work better on some membrane surfaces than others (e.g., plasma membrane versus mitochondrial surface), either because the presentation of the lipid is different in the different environment due to the presence of other lipids or differences in the fatty acid composition of the lipid, or because the sensors also interact with proteins on the surface, rather than purely reacting to the target lipid. Therefore, failure to detect altered distribution of a specific lipid sensor after manipulating the lipid level on a specific membrane surface in the cell may not necessarily mean there is no change there in the lipid level. Controls that can be used for this approach include mutant lipid sensors that are unable to bind to the lipid, and inactive lipid-modifying enzymes that do not change the lipid levels. Reviews on technical and theoretical aspects of FRET measurements have been reviewed elsewhere (Sekar and Periasamy, 2003; Thaler et al., 2005; van Rheenen et al., 2004).
Finally, it should be noted that such sensors can only be used to probe the mitochondrial surface, since the inner leaflet of the outer membrane and both sides of the inner membrane are essentially incapable of being accessed by these protein-based sensors. Thus, some caution should be used when attempting to compare the information obtained using sensors to biochemical analyses of lipid content that assay additional leaflets and membranes.
C. Biochemical approaches
Although the list of lipid sensors has been expanding continuously, sensors are not available for all types of lipids, and some of the available sensors are not very sensitive(readers should also be directed to another chapter in this volume by Sarantis and Grinstein). As an alternate approach, biochemical approaches such as Liquid Chromatography/Mass Spectrometry-Mass Spectrometry (LC/MS-MS) can be utilized to measure changes in concentrations of individual mitochondrial lipids(Bird et al., 2011; Choi et al., 2007). However, in addition to the issue cited above regarding the additional leaflets and membranes, it should also be noted that the sensors report on lipid that is accessible to binding; lipid already bound by other proteins can not be imaged by the sensor–whereas the MS approach will identify all of the lipid present. On the other hand, fairly large amounts of material are required for MS approaches (Bird et al., 2011), and in some cases the lipids may undergo modification or degradation during the purification and recovery steps needed to prepare them for analysis.
V. Summary
With the discovery of the involvement of lipids such as PA, DAG, cholesterol, and cardiolipin in mitochondrial fusion and fission dynamics, there is increased awareness of the need to study other lipids for their roles in the process of mitochondrial morphodynamics.
Acknowledgments
Supported by NIH GM071520and GM084251.
References
- Altmann K, Westermann B. Role of essential genes in mitochondrial morphogenesis in Saccharomyces cerevisiae. Mol Biol Cell. 2005;16:5410–7. doi: 10.1091/mbc.E05-07-0678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ban T, et al. OPA1 disease alleles causing dominant optic atrophy have defects in cardiolipin-stimulated GTP hydrolysis and membrane tubulation. Hum Mol Genet. 2010;19:2113–22. doi: 10.1093/hmg/ddq088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bird SS, et al. Lipidomics profiling by high-resolution LC-MS and high-energy collisional dissociation fragmentation: focus on characterization of mitochondrial cardiolipins and monolysocardiolipins. Anal Chem. 2011;83:940–9. doi: 10.1021/ac102598u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braschi E, McBride HM. Mitochondria and the culture of the Borg: understanding the integration of mitochondrial function within the reticulum, the cell, and the organism. Bioessays. 2010;32:958–66. doi: 10.1002/bies.201000073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan DC. Mitochondrial fusion and fission in mammals. Annual Review of Cell and Developmental Biology. 2006;22:79–99. doi: 10.1146/annurev.cellbio.22.010305.104638. [DOI] [PubMed] [Google Scholar]
- Chen H, Chan DC. Physiological functions of mitochondrial fusion. Ann N Y Acad Sci. 2010;1201:21–5. doi: 10.1111/j.1749-6632.2010.05615.x. [DOI] [PubMed] [Google Scholar]
- Choi SY, et al. Cardiolipin deficiency releases cytochrome c from the inner mitochondrial membrane and accelerates stimuli-elicited apoptosis. Cell Death Differ. 2007;14:597–606. doi: 10.1038/sj.cdd.4402020. [DOI] [PubMed] [Google Scholar]
- Choi SY, et al. A common lipid links Mfn-mediated mitochondrial fusion and SNARE-regulated exocytosis. Nature Cell Biology. 2006;8:1255–U29. doi: 10.1038/ncb1487. [DOI] [PubMed] [Google Scholar]
- DeVay RM, et al. Coassembly of Mgm1 isoforms requires cardiolipin and mediates mitochondrial inner membrane fusion. J Cell Biol. 2009;186:793–803. doi: 10.1083/jcb.200906098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dries DR, et al. A single residue in the C1 domain sensitizes novel protein kinase C isoforms to cellular diacylglycerol production. J Biol Chem. 2007;282:826–30. doi: 10.1074/jbc.C600268200. [DOI] [PubMed] [Google Scholar]
- Fernandez-Ulibarri I, et al. Diacylglycerol is required for the formation of COPI vesicles in the Golgi-to-ER transport pathway. Molecular Biology of the Cell. 2007;18:3250–3263. doi: 10.1091/mbc.E07-04-0334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furt F, Moreau P. Importance of lipid metabolism for intracellular and mitochondrial membrane fusion/fission processes. Int J Biochem Cell Biol. 2009;41:1828–36. doi: 10.1016/j.biocel.2009.02.005. [DOI] [PubMed] [Google Scholar]
- Gallegos LL, et al. Targeting protein kinase C activity reporter to discrete intracellular regions reveals spatiotemporal differences in agonist-dependent signaling. J Biol Chem. 2006;281:30947–56. doi: 10.1074/jbc.M603741200. [DOI] [PubMed] [Google Scholar]
- Gandre-Babbe S, van der Bliek AM. The novel tail-anchored membrane protein Mff controls mitochondrial and peroxisomal fission in mammalian cells. Mol Biol Cell. 2008;19:2402–12. doi: 10.1091/mbc.E07-12-1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gebert N, et al. Mitochondrial cardiolipin involved in outer-membrane protein biogenesis: implications for Barth syndrome. Curr Biol. 2009;19:2133–9. doi: 10.1016/j.cub.2009.10.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giorgione JR, et al. Increased membrane affinity of the C1 domain of protein kinase C delta compensates for the lack of involvement of its C2 domain in membrane recruitment. Journal of Biological Chemistry. 2006;281:1660–1669. doi: 10.1074/jbc.M510251200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H, et al. A quantitative assay for mitochondrial fusion using Renilla luciferase complementation. Mitochondrion. 2010;10:559–66. doi: 10.1016/j.mito.2010.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H, et al. piRNA-associated germline nuage formation and spermatogenesis require MitoPLD pro-fusogenic mitochondrial-surface lipid signaling. Developmental Cell. 2011 doi: 10.1016/j.devcel.2011.01.004. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang P, et al. Insulin-stimulated plasma membrane fusion of Glut4 glucose transporter-containing vesicles is regulated by phospholipase D1. Mol Biol Cell. 2005;16:2614–23. doi: 10.1091/mbc.E04-12-1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingerman E, et al. In vitro assays for mitochondrial fusion and division. Methods Cell Biol. 2007;80:707–20. doi: 10.1016/S0091-679X(06)80032-4. [DOI] [PubMed] [Google Scholar]
- Kane LA, Youle RJ. Mitochondrial fission and fusion and their roles in the heart. J Mol Med. 2010;88:971–9. doi: 10.1007/s00109-010-0674-6. [DOI] [PubMed] [Google Scholar]
- Khalifat N, et al. Membrane deformation under local pH gradient: mimicking mitochondrial cristae dynamics. Biophys J. 2008;95:4924–33. doi: 10.1529/biophysj.108.136077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komatsu T, et al. Organelle-specific, rapid induction of molecular activities and membrane tethering. Nat Methods. 2010;7:206–8. doi: 10.1038/nmeth.1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koshiba T, et al. Structural basis of mitochondrial tethering by mitofusin complexes. Science. 2004;305:858–62. doi: 10.1126/science.1099793. [DOI] [PubMed] [Google Scholar]
- Kozlovsky Y, Kozlov MM. Membrane fission: model for intermediate structures. Biophys J. 2003;85:85–96. doi: 10.1016/S0006-3495(03)74457-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mears JA, et al. Conformational changes in Dnm1 support a contractile mechanism for mitochondrial fission. Nat Struct Mol Biol. 2011;18:20–6. doi: 10.1038/nsmb.1949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molina AJ, Shirihai OS. Monitoring mitochondrial dynamics with photoactivatable [corrected] green fluorescent protein. Methods Enzymol. 2009;457:289–304. doi: 10.1016/S0076-6879(09)05016-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montessuit S, et al. Membrane remodeling induced by the dynamin-related protein Drp1 stimulates Bax oligomerization. Cell. 2010;142:889–901. doi: 10.1016/j.cell.2010.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakanishi H, et al. Positive and negative regulation of a SNARE protein by control of intracellular localization. Mol Biol Cell. 2004;15:1802–15. doi: 10.1091/mbc.E03-11-0798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishioka T, et al. Heterogeneity of phosphatidic acid levels and distribution at the plasma membrane in living cells as visualized by a Foster resonance energy transfer (FRET) biosensor. J Biol Chem. 2010;285:35979–87. doi: 10.1074/jbc.M110.153007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nunnari J, et al. Mitochondrial transmission during mating in Saccharomyces cerevisiae is determined by mitochondrial fusion and fission and the intramitochondrial segregation of mitochondrial DNA. Molecular Biology of the Cell. 1997;8:1233–1242. doi: 10.1091/mbc.8.7.1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olichon A, et al. Mitochondrial dynamics and disease, OPA1. Biochimica Et Biophysica Acta-Molecular Cell Research. 2006;1763:500–509. doi: 10.1016/j.bbamcr.2006.04.003. [DOI] [PubMed] [Google Scholar]
- Osman C, et al. Making heads or tails of phospholipids in mitochondria. J Cell Biol. 2011;192:7–16. doi: 10.1083/jcb.201006159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otera H, Mihara K. Molecular mechanisms and physiologic functions of mitochondrial dynamics. J Biochem. 2011;149:241–51. doi: 10.1093/jb/mvr002. [DOI] [PubMed] [Google Scholar]
- Otera H, et al. Mff is an essential factor for mitochondrial recruitment of Drp1 during mitochondrial fission in mammalian cells. J Cell Biol. 2010;191:1141–58. doi: 10.1083/jcb.201007152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizzo MA, et al. The recruitment of Raf-1 to membranes is mediated by direct interaction with phosphatidic acid and is independent of association with Ras. J Biol Chem. 2000;275:23911–8. doi: 10.1074/jbc.M001553200. [DOI] [PubMed] [Google Scholar]
- Rujiviphat J, et al. Phospholipid association is essential for dynamin-related protein Mgm1 to function in mitochondrial membrane fusion. J Biol Chem. 2009;284:28682–6. doi: 10.1074/jbc.M109.044933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato M, et al. Imaging diacylglycerol dynamics at organelle membranes. Nat Methods. 2006;3:797–9. doi: 10.1038/nmeth930. [DOI] [PubMed] [Google Scholar]
- Sekar RB, Periasamy A. Fluorescence resonance energy transfer (FRET) microscopy imaging of live cell protein localizations. J Cell Biol. 2003;160:629–33. doi: 10.1083/jcb.200210140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su W, et al. FIPI, a Phospholipase D pharmacological inhibitor that alters cell spreading and inhibits chemotaxis. Mol Pharmacol. 2009;75:437–46. doi: 10.1124/mol.108.053298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanimura A, et al. Fluorescent biosensor for quantitative real-time measurements of inositol 1,4,5-trisphosphate in single living cells. J Biol Chem. 2004;279:38095–8. doi: 10.1074/jbc.C400312200. [DOI] [PubMed] [Google Scholar]
- Thaler C, et al. Quantitative multiphoton spectral imaging and its use for measuring resonance energy transfer. Biophys J. 2005;89:2736–49. doi: 10.1529/biophysj.105.061853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Twig G, et al. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. Embo Journal. 2008;27:433–446. doi: 10.1038/sj.emboj.7601963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Twig G, et al. Tagging and tracking individual networks within a complex mitochondrial web with photoactivatable GFP. American Journal of Physiology-Cell Physiology. 2006;291:C176–C184. doi: 10.1152/ajpcell.00348.2005. [DOI] [PubMed] [Google Scholar]
- van Rheenen J, et al. Correcting confocal acquisition to optimize imaging of fluorescence resonance energy transfer by sensitized emission. Biophys J. 2004;86:2517–29. doi: 10.1016/S0006-3495(04)74307-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varnai P, Balla T. Live cell imaging of phosphoinositide dynamics with fluorescent protein domains. Biochim Biophys Acta. 2006;1761:957–67. doi: 10.1016/j.bbalip.2006.03.019. [DOI] [PubMed] [Google Scholar]
- Vicogne J, et al. Asymmetric phospholipid distribution drives in vitro reconstituted SNARE-dependent membrane fusion. Proc Natl Acad Sci U S A. 2006;103:14761–6. doi: 10.1073/pnas.0606881103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitale N, et al. Phospholipase D1: a key factor for the exocytotic machinery in neuroendocrine cells. EMBO J. 2001;20:2424–34. doi: 10.1093/emboj/20.10.2424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterham HR, et al. A lethal defect of mitochondrial and peroxisomal fission. New England Journal of Medicine. 2007;356:1736–1741. doi: 10.1056/NEJMoa064436. [DOI] [PubMed] [Google Scholar]
- Westermann B. Mitochondrial fusion and fission in cell life and death. Nat Rev Mol Cell Biol. 2010;11:872–84. doi: 10.1038/nrm3013. [DOI] [PubMed] [Google Scholar]
- Zuchner S, et al. Mutations in the mitochondrial GTPase mitofusin 2 cause Charcot-Marie-Tooth neuropathy type 2A. Nature Genetics. 2004;36:449–451. doi: 10.1038/ng1341. [DOI] [PubMed] [Google Scholar]