

Abstract
Host defense requires the maturation and release of the pro-inflammatory cytokines interleukin (IL)-1β and IL-18 and the induction of pyroptotic cell death, which depends on the activation of inflammatory Caspases within inflammasomes by innate immune cells. Several cytosolic Pattern recognition receptors (PRRs) have been implicated in this process in response to infectious and sterile agonists. Here we summarize the current knowledge on inflammasome-organizing PRRs, emphasizing the recently described NLRP7, and their implications in human disease.
Keywords: inflammasome, pattern recognition receptor, PAMP, DAMP, Nod-like receptor, AIM2-like receptor, innate immunity, inflammation, caspase-1, hydatidiform mole, molar pregnancy
1. Introduction
To maintain tissue homeostasis, a strong defense against pathogenic microorganisms as well as the restriction of commensal bacteria is crucial. Pattern recognition receptors (PRRs) are germline-encoded “sensors” of the innate immune system that detect infections and tissue damage as a first line of defense. PRRs sense so-called conserved non-self pathogen-associated molecular patterns (PAMPs) and host-derived danger signals (damage-associated molecular patterns or DAMPs) to initiate a host defense program through activation of various signal transduction pathways, which culminates in pathogen clearance and initiation of wound healing. Most of these pathways activate a transcriptional response leading to the up-regulation of nuclear factor κB (NF-κB), mitogen-activated protein kinase (MAPK) and interferon (IFN)-dependent genes, which encode cytokines, chemokines, adhesion receptors and others. However, for the maturation of the pro-inflammatory cytokines IL-1β and IL-18 into their biologically active and secreted forms, as well as for the induction of pyroptotic cell death of infected and damaged cells, additional processing in inflammasomes is required. Although inflammasome activation is beneficial for host defense, its dysregulation and in particular, excessive and uncontrolled release of IL-1β and IL-18, is linked to an increasing number of inflammatory and metabolic diseases. Therefore, the mechanism and regulation of inflammasome activation are under active investigation and relevant for multiple disciplines. Here, we discuss PRRs that are currently known to assemble inflammasomes, including the recently characterized PRR, the nucleotide-binding domain and leucine-rich repeat containing gene family member with a pyrin domain 7 (NLRP7, also know as PYPAF3, NALP7, PAN7, NOD12, CLR19.4 and HYDM) and its role in inflammasome signaling and human disease.
2. Inflammasomes
Inflammasomes are cytosolic multi-protein complexes that link pathogen recognition by specific cytosolic PRRs, including the NOD-like receptors (NLRs) and the Absent in melanoma 2 (AIM2)-like receptors (ALRs) to the activation of pro-inflammatory Caspases, including Caspase-1, −4 and −5 in humans and Caspase-1 and −11 (the mouse Caspase-4 ortholog) in mice (Fig. 1) [1]. Caspase-1 mediates the proteolytic cleavage of pro-IL-1β and pro-IL-18, resulting in the bioactive, secreted form of these cytokines that act to initiate and perpetuate inflammatory host responses. Caspase-1 also promotes the release of other mediators, including IL-1α, IL-1Ra, HMGB1, FGF-2 and others through an unconventional secretion mechanism [2]. Infections by Gram-negative bacteria further require IFN-induced up-regulation of Caspase-11 upstream of Caspase-1 [3]. In addition, recent evidence shows that Caspase-8 can substitute for Caspase-1 under certain conditions [4,5]. Besides cytokine maturation, Caspase-1 also mediates inflammatory cell death (pyroptosis) of infected host cells [6]. Caspase-1 is recruited to activated PRRs by the central inflammasome adaptor apoptosis-associated speck-like protein containing a Caspase recruitment domain (CARD) (ASC, PyCard, TMS-1) [7,8]. Subsequently, Caspase-1 clustering causes its auto-activation through induced proximity. ASC is crucial for all inflammasomes activated by NLR family members containing a PYRIN domain (NLRPs) and ALRs (Fig. 1). However, NLRC4 (IPAF, CLAN), an NLR family member containing a CARD (NLRC), can directly recruit Caspase-1 [9–11], but ASC is nevertheless required for inflammasome activation in response to certain bacterial infections in vivo. In contrast to Caspase-1-dependent cytokine processing, which requires ASC and occurs in a singular distinct perinuclear inflammasome complex [12], Caspase-1-dependent pyroptosis proceeds independently of ASC and Caspase-1 autoproteolysis [13]. While the induced proximity mechanism of Caspase-1 activation is fairly well understood, the signaling events upstream of inflammasome-initiating PRRs of the NLRP, NLRC and ALR families are largely unknown.
Figure 1. Model for inflammasome activation.

Stimuli that activate known inflammasomes are indicated, which cause oligomerization of the respective PRR. In the case of NLRs, oligomerization is induced by NACHT domain-mediated NTP binding. Subsequently, ASC and Caspase-1 are recruited by PYD-PYD and CARD-CARD interaction, respectively, which results in assembly of the inflammasome platform and activation of Caspase-1, as depicted by an asterisk. Proteolytically active Caspase-1 then converts the pro-IL-1β and pro-IL-18 into their bioactive, mature forms.
2.1. NLR inflammasomes
22 NLRs are encoded in humans, while 34 Nlrs exist in mice. They are divided into five subfamilies based on their N-terminal effector domain: 1) NLRA (NLR containing an acidic domain), 2) NLRB (NLR containing a BIR domain), 3) NLRC, 4) NLRP and 5) NLRX (NLR with no homology to the N-terminal domain of any other NLR member). However, only a few members have so far been linked to inflammasome activation, including NLRP1, NLRP3 and NLRC4. NLRs sense and respond to a diverse array of infectious and sterile inflammatory signals with activation that promotes a conformational change, followed by receptor oligomerization, which is driven by nucleotide triphosphate binding and hydrolysis and the formation of the inflammasome platform upon recruitment of ASC-Caspase-1 and likely other proteins.
NLRP1
NLRP1 recognizes muramyl-dipeptide (MDP) and the lethal toxin (LT) from Bacillus anthraces [14,15], while the recently generated Nlrp1b deficient mice confirmed the response to LT, but failed to observe defects in the MDP response, which required Nlrp3 [16]. NLRP1 is unique among NLRs, due to the presence of an N-terminal PYRIN domain (PYD) and a C-terminal CARD, which not only allows it to recruit Caspase-1 through its PYD and ASC, but also simultaneously to bind Caspase-5 directly through its CARD [1]. Of note is that the NLRP1 gene has three paralog mouse genes, Nlrp1a, Nlrp1b and Nlrp1c, which all lack the PYD present in NLRP1, which might contribute to the conflicting observations for its requirement to the MDP-induced inflammasome response. ATPase activity within the NACHT is required for NLRP1 oligomerization and Caspase-1 activation [15].
NLRP3
NLRP3 (Cryopyrin) is clearly the best studied of the NLRs, which senses a wide variety of structurally different infectious and non-infectious agonists. Generally, NLRP3 activators affect potassium efflux, destabilization of the acidic lysosomal compartment and subsequent cathepsin B release, generation of reactive oxygen species (ROS), although the precise contribution of ROS is still controversial, and ultimately converge on the release of mitochondrial oxidized DNA and calcium mobilization from ER stores upon calcium-sensing receptor (CASR) activation [17–19]. For non-particulate activators, association of the PYD of NLRP3 with the guanylate-binding protein 5 (GBP5) is necessary for potent inflammasome activation [20]. Furthermore, depletion of intracellular levels of cAMP upon CASR activation prevents cAMP-mediated inhibition of the NACHT domain, which is also necessary for NLRP3 activation [17]. Thus, mice deficient in Nlrp3 are impaired in clearing numerous viral, microbial, fungal and protozoan pathogens due to defects in mounting an IL-1β and IL-18-based host response. The leucine-rich region (LRR) of NLRP3 is necessary for NLRP3 activation, since deletion of this domain impairs NLRP3 activation in response to monosodium urate monohydrate (MSU) crystals in vivo [21].
NLRP6
NLRP6 interacts with ASC and forms an inflammasome by overexpression [22]. More recently, Nlrp6 deficient mice revealed that it is essential for restricting commensal bacteria to maintain homeostasis through promoting IL-18 release [23]. Nlrp6 deficient mice are also resistant to infection with Listeria monocytogenes, Salmonella typhimurium and Escherichia coli due to an enhanced production of MAPK- and NF-κB-dependent cytokines [24]. However, the ligands specifically sensed by NLRP6 are currently not known.
NLRC4
NLRC4 (IPAF) is essential for inflammasome activation in response to flagellated bacteria through sensing flagellin, but is also activated through a flagellin-independent mechanism (Fig. 1) [9,11]. The latter occurs through the bacterial type III and IV secretion system (T3SS and T4SS, respectively) [25]. Activation of NLRC4 requires the recognition and binding of flagellin or the T3SS rod proteins by upstream NLRB (NAIP) proteins, which causes phosphorylation of NLRC4 [26]. Although, only one NLRB exists in humans, mice encode seven Naip genes. While Naip2 interacts with T3SS rod proteins, such as PrgJ from Salmonella typhimurium and BsaK from Burkholderia, Naip5 and Naip6 bind to flagellin from Legionella pneumophila and similarly, human NAIP is necessary for sensing the T3SS needle protein CprI from Chromobacterium violaceum [27].
NLRC5
The precise function of NLRC5 is still controversial [28], but it is strongly implicated in the regulation of MHC class I gene expression [29]. Nevertheless, it has also been linked to inflammasome activation in response to bacterial infection through a mechanism involving heterodimerization with NLRP3 [30]. However, while HEK293 cell-reconstituted NLRC5 inflammasomes were active, deficiency of Nlrc5 in mouse BMDM and DC did not affect Caspase-1 activation and IL-1β release with any stimuli that are known activators of other inflammasomes, suggesting that NLRC5 likely responds to unknown pathogenic or endogenous inflammasome activators, or alternatively, functions in other, non-macrophage cells [31].
2.2. ALR inflammasomes
In humans, 4 ALRs (IFI16, IFIX, MNDA, and AIM2) have been annotated, while 13 genes are predicted to exist in mice, which are referred to as γ-IFN-inducible (Ifi) genes or the hematopoietic interferon-inducible nuclear antigens with a 200 amino acid repeat (HIN200) members 202 to 214 and the encoded p200 proteins (p202 to 214) [32]. Strikingly, these genes are highly diverse and only AIM2 (p210) is conserved between mouse, rat and humans, although Ifi204 is predicted to be the mouse IFI16 ortholog [32]. All ALRs, except for p202, contain an N-terminal PYD and contain one or two C-terminal partially conserved HIN200 DNA-binding domains [33]. However, so far only AIM2 and IFI16 have been shown to assemble an inflammasome.
AIM2
Although the ALR AIM2 is structurally different from NLRs, it functions as a cytosolic PRR involved in inflammasome activation. It contains an N-terminal PYD and one copy of the conserved HIN-200 DNA-binding domain. AIM2 directly binds to cytosolic double stranded (ds) DNA through the HIN-200 domain and forms an inflammasome in response to bacterial and viral infections, including Francisella tularensis, Listeria monocytogenes, Vaccinia virus and murine cytomegalovirus (MCMV) (Fig. 1) [34,35]. Despite its lack of a PYD, p202 has been shown to sense cytosolic DNA and to form a heterodimer with AIM2 in vitro, which is predicted to inhibit AIM2-induced clustering of the adaptor protein ASC and Caspase-1 activation [36].
IFI16
The ALR IFI16 is a predominantly nuclear protein also involved in DNA binding and recognition, but contains two copies of the HIN-200 domain (Fig. 1). IFI16 is not encoded in mice, but has a putative orthologous gene, ifi204. In contrast to infection by MCMV, infection with another herpes virus, the Kaposi sarcoma-associated herpes virus (KSHV), which replicates and establishes a latent infection in the nucleus, does not trigger a cytosolic DNA response but initially activates a nuclear IFI16 inflammasome [37].
2.3. Other inflammasomes
Pyrin
Pyrin interacts with various inflammasome components and is able to negatively regulate inflammasome-responses [38,39]. However, Pyrin has also been implicated in inflammasome activation upon binding to PSTPIP1 and in particular to PAPA syndrome-linked PSTPIP1 mutants [40,41]. An inflammasome activating function of Pyrin is further supported by the recently generated knock-in mice expressing the constitutive active, FMF-linked mutation of Pyrin (MefvM680I, MefvM694V and MefvV726A), which show constitutive and ASC-dependent Caspase-1 activation and elevated IL-1β [42].
3. NLRP7 and its link to inflammasomes
There is emerging evidence that NLRP7 is able to regulate inflammasomes. However, conflicting reports describe NLRP7 as either an inflammasome activator or as an inhibitor of Caspase-1-dependent production of mature IL-1β and IL-18 (Fig. 2) [43–45]. The NLRP7 gene, also referred to as PYPAF3, NALP7, PAN7, NOD12, CLR19.4 and HYDM is located on the long arm of human chromosome 19 at position q13.42, but no true mouse orthologous gene exists. Nlrp2 is the closest relative and NLRP2 is located adjacent to NLRP7 on human chromosome 19. Similar to NLRP3, NLRP7 transcription is induced in response to pro-inflammatory stimuli, including LPS and IL-1β in peripheral blood mononuclear cells (PBMC) and is abundantly expressed in thymus, spleen and bone marrow, suggesting a role in inflammation and host defense. However, its expression is not restricted to the immune system and is also high in the nervous system and testis [44]. The domain architecture of NLRP7 is reminiscent of other NLR family members and contains an N-terminal PYD followed by a NACHT domain and 11 LRRs at the C-terminus. As observed for other NLR family members, NLRP7 is subject to differential splicing (Fig. 3), which primarily affects the number of LRRs, but one alternative transcript also lacks the NACHT domain, likely yielding a dominant negative protein due to defective oligomerization.
Figure 2. Mechanisms of NLRP7 function in inflammation and host defense.

The top panel depicts the proposed mechanisms by which NLRP7 inhibits IL-1β release. 1) NLRP7 inhibits NF-κB activation by an unknown mechanism, which may involve FAF1, which prevents transcription of pro-IL-1β. 2) NLRP7 directly interacts with pro-Caspase-1 and pro-IL-1β, which might prevent their activation or recruitment into inflammasomes. 3) NLRP7 localizes to the Golgi and was predicted to affect trafficking of mature IL-1β containing vesicles necessary for release. The bottom panel depicts NLRP7 inflammasome formation in response to sensing bacterial acylated lipoproteins (acLP).
Figure 3. Differential splicing of NLRP7.
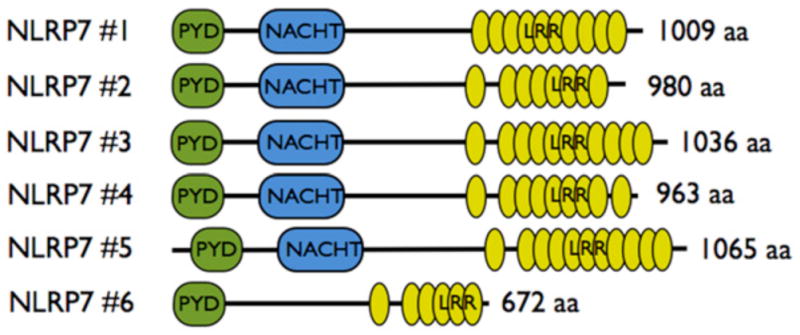
Several alternative transcripts exist for NLRP7, which encode a varying number of LRRs or delete the NACHT, further supporting the importance of these domains for the function of NLRP7.
Although the function of NLRP7 is poorly understood, several proteins have been identified that interact with NLRP7, including pro-Caspase-1, pro-IL-1β, ASC and Fas associated factor 1 (FAF1), which likely influence the functional aspects attributed to NLRP7 as discussed below.
3.1. Negative regulation of inflammasome responses by NLRP7
Several NLR family members trigger inflammasome formation and regulate NF-κB activation, but the initial in vitro characterization of the NLR family member NLRP7 revealed an inhibitory function of NLRP7 on IL-1β secretion. Utilizing an inflammasome reconstitution assay in HEK293 cells, Kinoshita and colleagues demonstrate that NLRP7 (PYPAF3) inhibits NLRP3 and Caspase-1 mediated IL-1β release, as well as pro-Caspase-1 and pro-IL1β processing without affecting ASC-mediated NF-κB activation [44]. The proposed mechanism for inflammasome inhibition of NLRP7 is through its direct interaction with pro-Caspase-1 and pro-IL-1β, which was shown by co-immunoprecipitation, but not through inhibition of IL-1β transcription, since NF-κB was not impaired. In addition, stable expression of an N-terminal fragment of NLRP7 prevents LPS-induced IL-1β release in THP-1 cells. However, it should be noted that this NLRP7 fragment lacked some of the functional domains of NLRP7 and could therefore represent a dominant negative form of this protein functioning as an inflammasome inhibitor.
Another study of NLRP7 by Messaed and colleagues focuses on the functional consequences of NLRP7 mutations found in patients with recurrent hydatidiform moles (HM) [43]. Interestingly, IL-1β and TNFα secretion is diminished in response to LPS in PBMCs containing HM-associated NLRP7 mutations compared to PBMCs from healthy controls. However, processing of intracellular pro-IL-1β into mature IL-1β is not affected and all but one HM patients have normal to higher protein levels of intracellular pro- and mature IL-1β, which implies an inflammasome independent function of NLRP7, but suggests a function of NLRP7 in cytokine secretion. In PBMCs, NLRP7 co-localizes to the Golgi apparatus and the microtubule-organizing center, where mutated NLRP7 may affect cytokine trafficking and secretion by interfering with the classical and non-classical secretory pathways. While expression levels of NLRP7 and mutated NLRP7 in HM patients are similar, overexpression of NLRP7 or HM-associated missense mutants of NLRP7 in HEK293 cells causes reduced protein levels of overexpressed intracellular pro- and mature IL-1β. However, HM associated nonsense mutants of NLRP7, which harbor a stop codon shortly after the PYD, are unable to decrease intracellular pro- and mature IL-1β. Similarly, overexpression of several NLRP7 domain or domain-deletion constructs all cause reduced intracellular pro- and mature IL-1β, except for the PYD [43]. Overall, NLRP7 is expected to down-regulate intracellular pro- and mature IL-1β, while HM-associated mutations in NLRP7 prevent this function and lead to increased intracellular pro- and mature IL-1β. The precise mechanism for regulating intracellular IL-1βas well as the mechanism describing NLRP7 involvement in IL-1β and TNFαsecretion remain unclear.
Several NLRs have an anti-inflammatory function including NLRP6, NLRP10 (PYNOD) and NLRP12 (Monarch-1), which are able to prevent NF-κB activation and subsequent cytokine production, although an anti-inflammatory role for Nlrp10 is not observed in vivo. NLRP7 could also be linked to the inhibition of inflammation by interacting with NF-κB regulatory proteins, such as FAF1, which interacts with several NLRs, including NLRP2, NLRP3, NLRP7, NLRP10 and NLRP12 and promotes apoptosis and inhibits NF-κB activation [46]. However, a study investigating the effects of several NLRs, including NLRP7, on NF-κB activation failed to detect an inhibitory role of NLRP7 on NF-κB activation in a COS-7L cell system and silencing of NLRP7 in human macrophages also does not modulate cytokine and LPS-induced NF-κB activation [22,45].
Collectively, in vitro evidence supports a role of NLRP7 in inhibiting inflammation by several possible mechanisms, including direct binding and inhibition of inflammasome components, impairing transcription of pro-IL-1β and modulating the trafficking and release of IL-1β (Fig. 2). However, as most of these studies were performed in a reconstitution system in the absence of the essential inflammasome adaptor ASC, their findings will require further confirmation.
3.2. Positive regulation of inflammasome responses by NLRP7
Overexpression of NLRP7 inhibits inflammation through several mechanisms, as discussed above. However, there is also evidence for a pro-inflammatory role of NLRP7 in human macrophages. Using an siRNA-based screen, NLRP7 was specifically identified as an intracellular sensor for bacterial acylated lipoproteins, which is required for IL-1β release (Fig. 2) [45]. While Toll-like receptor (TLR) 2 heterodimers are responsible for mediating NF-κB activation and subsequent transcription of pro-IL-1β in response to bacterial acylated lipoproteins, NLRP7 is essential for bacterial acylated lipoprotein-mediated Caspase-1 activation and maturation of IL-1β and IL-18. Similar to other inflammasome-activating NLRs, also NLRP7 shifts into a large, high molecular weight complex, where it interacts with ASC and Caspase-1 and promotes Caspase-1 activation [45]. Even though the PYD of NLRP7 lacks a positively charged electrostatic interaction patch, which promotes interaction with ASC, endogenous NLRP7 and ASC are able to interact [45,47]. More importantly, NLRP7 is also crucial for restricting growth of intracellular bacteria, including Staphylococcus aureus and Listeria monocytogenes in macrophages [45]. L. monocytogenes is also sensed by NLRC4, NLRP3 and AIM2, suggesting that several distinct NLRs form inflammasomes to control bacterial infections. Although NLRP3 does not sense bacterial acylated lipoproteins, a similar mechanism involving lysosomal damage upstream of NLRP7 is required for inflammasome activation. Thus, there is compelling evidence that NLRP7 acts as a direct or indirect cytosolic sensor of microbial acylated lipopeptides during infection.
While these findings appear to be contradictive, it is conceivable that NLRP7 prevents inflammasome formation and IL-1β release in quiescent cells, while in the response to a proper stimulus, such as bacterial infection, NLRP7 is activated, recruits ASC and assembles an inflammasome to promote Caspase-1 activation, IL-1β and IL-18 release for bacterial clearance. This mechanism could ensure that cytokine release occurs only when required. NLRP7 may also exhibit these opposing functions in a cell-type-specific manner. However, the molecular details and the specificity of NLRP7 sensing bacterial acylated proteins are still elusive. While NLRP3 and its role in inflammasome activation is well established, we are just now beginning to unravel the mechanism of its activation. Similar to NLRP3 disease-causing mutations, NLRP7 mutations are also linked to human disease. Therefore elucidating the function of NLRP7 is important for a better understanding of innate immune responses as well as NLRP7-linked disease.
4. Inflammasomes and disease
4.1. Mutations in inflammasome proteins
Due to their critical role in host defense, mutations in NLRs and the resulting dysregulated inflammasome activation is also linked to several human autoinflammatory diseases, which are characterized by unprovoked episodes of inflammation in the absence of high-titer autoantibodies and antigen-specific T cells, and are commonly referred to as inflammasomopathies [48]. After positional cloning identified Pyrin (marenostrin, MEFV) as the underlying cause for one of the most common autoinflammatory disease, Familial Mediterranean fever (FMF), more than 80 mutations of Pyrin have been identified in FMF patients. Recently generated mouse models implicate excessive inflammasome activation in the pathogenesis of FMF [42]. Similarly, positional cloning for several related autoinflammatory diseases, including familial cold autoinflammatory syndrome (FCAS), Muckle-Wells syndrome (MWS) and neonatal-onset multisystem inflammatory disease (NOMID or CINCA), identified NLRP3 (Cryopyrin, CIAS1), and subsequently mutations in NLRP3, as the underlying cause for these diseases, which are now referred to as Cryopyrinopathies (CAPS) [49]. Similar to Pyrin, knock-in of mutant NLRP3 in mice caused excessive inflammasome activation and release of IL-1β, and confirmed that CAPS originate from NLRP3 mutations causing dysregulated inflammasome activation [50,51]. However, these mice also displayed a skewed Th17 response and further support a role of the NLRP3 inflammasome in regulating adaptive immunity.
4.2. Inflammasomes and crystalline arthropathies
A number of crystalline and particulate substances have been identified as agonists for the NLRP3 inflammasome. Hence, the presence of these substances causes inflammasome activation, excessive IL-1β release and the development of crystalline arthropathies. In gout and pseudogout the NLRP3 inflammasome is activated in response to monosodium urate (MSU) and calcium pyrophosphate dihydrate (CPPD) crystals and responds responds to asbestos and silica fibers in asbestosis and silicosis, respectively [52–55]. Similarly, amyloid-β fibrils and islet amyloid polypeptide (IAPP) activate the NLRP3 inflammasome, which may contribute to Alzheimer’s disease and the progression of type 2 diabetes, respectively [56,57]. Interestingly, cholesterol crystals are also sensed by NLRP3, which may contribute to the chronic vascular inflammation caused by cholesterol crystal deposition at the artery wall and the development of atherosclerotic lesions. Hence, inflammasome deficient mice develop reduced atherosclerotic lesions compared to wild type mice [58]. In addition the vaccine adjuvant aluminum hydroxide (alum) and hemozoin crystals, produced by malaria-causing parasites, are able to induce an NLRP3 dependent inflammasome response [59–62].
4.3. Inflammasomes and metabolic disease
Recent evidence revealed that inflammasomes are also central in maintaining metabolic homeostasis, and that defects in this inflammasome-mediated surveillance of the gastrointestinal tract contribute to obesity, insulin resistance, type 2 diabetes, atherosclerosis, non alcoholic fatty liver disease (NAFLD) and eventually development of non-alcoholic steatohepatitis (NASH). This is not surprising, given that chronic inflammation is a key factor driving metabolic disease. In particular, NLRP6 was discovered to play a central role within the gastrointestinal tract to restrict colitogenic microbiota species, specifically members of the Prevotellaceae and TM7 environmental bacteria families, which have also been linked to increased body mass index in humans [23]. Restricting these colitogenic species is dependent on IL-18, rather than IL-1β and dysbiosis causes increased PAMP-perpetuated inflammation, eventually leading to systemic metabolic dysfunction [23,63]. Similar results were also obtained for mice deficient in Nlrp3 and Nlrc4, indicating that several PRRs collaborate also in this context [63–65].
Increasing evidence also supports a contribution of NLRP3 to obesity-initiated insulin resistance and type 2 diabetes, since increased lipid storage in hypertrophic adipocytes supplies NLRP3-activating DAMPs, including the saturated fatty acid palmitate and saturated fatty acid metabolism-derived ceramide [66–68]. Subsequently, excessive production of IL-1β impairs glucose tolerance and insulin sensitivity in peripheral insulin target tissues, and also directly promotes cell death of pancreatic β-cells. Further, the NLRP3 activator IAPP is deposited in the pancreas of type 2 diabetes patients which, in combination with high glucose levels, promotes inflammasome priming [57].
4.4. Inflammasomes and adaptive immunity
Inflammasomes are most significant for host defense in innate immune cells, such as macrophages and DC as a first line of defense. IL-1β, the IL-1RI, and IL-18 are also required for the induction of Th17 cells and IL-17 production by unconventional γ∂ T cells in the presence of commensal bacteria and in several auto-immune disease models, including experimental autoimmune encephalomyelitis (EAE) [69,70]. Inflammasome-derived IL-1β also promotes GM-CSF production from CD4+ and γ∂ T cells, which has an essential role in EAE [71]. IL-22, which has protective, but pro-inflammatory properties, is produced by innate lymphoid cells, Th17 cells and Th22 cells and is regulated through its soluble receptor, IL-22BP. In turn, IL-22BP is inhibited through IL-18 produced at mucosal surfaces by NLRP3 and NLRP6 inflammasomes [72]. Synergistically with IL-12, IL-18 stimulates Th1-mediated and IFN-γ-dependent immune responses, but also promotes auto-immunity [73]. Thus, increasing evidence supports the notion that inflammasome activation in antigen presenting cells provides the necessary cues for regulating adaptive immunity in models of contact skin hypersensitivity, EAE and aluminium hydroxide adjuvant-mediated T-cell priming, extending the role of inflammasomes from innate to adaptive immunity. The contribution of IL-1β and IL-18 in the context of alum-induced inflammasome-mediated regulation of adaptive immunity remains controversial [60].
5. NLRP7 and disease
Until now, the only clinical evidence for the involvement of NLRP7 in human disease originated from genetic studies mapping the locus responsible for hydatidiform mole (HM), an abnormal human pregnancy referred to as molar pregnancy to human chromosome 19q13, and subsequently linked to several mutations within the NLRP7 (NALP7) gene [74]. In addition, the lack of NLRP7 mutations in a large control cohort and co-segregation of mutated alleles with disease phenotypes, supports a contribution of NLRP7 in the development of HM [74]. While recurrent molar pregnancies occur in 1 in 150 to 1 in 1000 pregnancies, depending on the ethnic group, familial recurrent HM (FRHM) is exceptionally rare. However, women who had a previous mole have a higher risk of recurrence (1.2–1.4%), which increases to 20% after the second mole. HM is a form of trophoblastic neoplasia characterized by cystic degeneration of the chorionic villi and abnormal or lack of embryonic growth. It is a heterogeneous condition, which is classified into two major groups according to karyotypic and histopathological features: complete HM and partial HM. Both HM subtypes result in reproductive wastage and/or spontaneous abortion. However, while 5–25% of complete HMs eventually develop into gestational choriocarcinoma, only 2–3% of partial HMs develop this highly aggressive cancer, which may spread to the lungs, lower genital tract, brain, liver, kidney and gastrointestinal tract via the bloodstream or lymphatic vessels [75]. So far 214 unique sequence variants of NLRP7 have been associated with this group of diseases [76]. While HM patients harbor homozygous, compound heterozygous and heterozygous NLRP7 genotypes, FRHM can also occur in the absence of NLRP7 mutations, which underlines the multifactorial nature of the disease [77]. Although mutations are found throughout most exonic and intronic regions of NLRP7, the region encoding the LRR represents a particularly common location of missense mutations and many identified nonsense mutations yield NLRP7 proteins lacking the LRR domain, emphasizing the importance of this domain for the function of NLRP7 (Fig. 4) [78]. Recently, the importance of the NLRP7 LRR was also recognized for sensing bacterial acylated lipoproteins in vitro, since NLRP7ΔLRR failed to respond to bacterial acylated lipoproteins, compared to wild type NLRP7 [45]. Similarly, deletion of the Nlrp3 LRR rendered the protein unresponsive to MSU crystals in vivo, resulting in ameliorated airpouch synovitis [21]. Overall the LRR seems to play an important functional role for NLRs.
Figure 4. NLRP7 Mutations and linkage to reproductive disease.
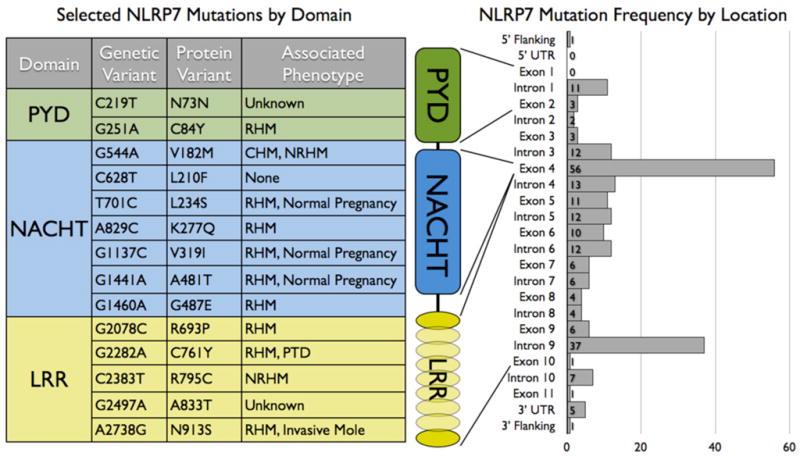
(left) Selected characterized genetic variations in exonic regions of the NLRP7 locus and the resulting amino acid changes in NLRP7 are listed by domain. Associated phenotypes for each individual mutation and mutations with higher frequency are indicated that occur in more than five unrelated families. (right) Histogram of mutations in the NLRP7 locus, which are organized by frequency within both coding- and non-coding regions and location to the individual NLRP7 domains. RHM: recurrent hydatidiform mole; CHM: complete hydatidiform mole; NRHM: non-recurrent hydatidiform mole; PTD: persistent trophoblastic disease.
The functional consequence of NLRP7 mutation and its effect on reproductive fidelity is not understood. The distinctive and arguably opposing models for NLRP7 function in inflammatory signaling allows for different interpretation of its possible role in molar pregnancy. Whether IL-1β, IL-18 or any inflammation, infection or commensal bacteria contribute to HM at all is currently not known. However, histopathological analysis of placentas from HM patients revealed an increased incidence of chorioamnionitis, necrosis and chorangiosis, potentially implicating excessive tissue inflammation [79]. Analysis of PBMC from several HM patients revealed a spectrum of IL-1β levels ranging from lower to higher than controls, with a similar broad range that has also been observed in earlier studies in the general population [43]. While wild-type NLRP7 inhibits pro-IL-1β transcription/translation in vitro, all tested missense mutations of NLRP7 do not affect this function, suggesting that this function of NLRP7 is not affected by HM mutations [43]. A potential loss-of-function could prevent NLRP7 to regulate physiological inflammation and may allow for increased insult from commensal and other pathogens in the lower reproductive tract, or potentially renders HM patients to tolerate growth of moles due to impaired immune surveillance [43]. Conversely, a pro-inflammatory role of NLRP7 in response to S. aureus infection could also imply a gain-of-function, in which NLRP7 mutants are hyperactive and contribute to excessive inflammation. Indeed, gain-of-function mutations have been classified in other NLR family members, such as NLRP3, where mutations cause Cryopyrinopathies [49]. In fact, several common HM-linked NLRP7 mutations show hyperactivity in vitro, which, however, has not yet been confirmed in a more physiological context [45]. Thus, a contribution of inflammatory signaling in the development of molar pregnancy has still to be firmly established. In addition, inflammation-independent functions for NLRP7 may potentially contribute to the development of HM. Interestingly, expression levels of NLRP7 are highly elevated in embryonal carcinomas and testicular seminomas, when compared to normal testis, implying a potential role in germ-cell proliferation. Further, RNAi-mediated silencing of NLRP7 causes growth suppression of testicular germ-cell tumors, suggesting a role in cell proliferation, which could contribute to HM [80].
6. Concluding remarks
Despite an emerging role of NLRP7 in inflammation, the precise role by which it impacts inflammasome activity is still poorly understood and thus, additional studies are necessary to unravel the mechanism of action of NLRP7. In particular, the mechanism of ligand recognition by NLRP7 and its potential conversion from an inhibitor to an activator of inflammasomes and the consequences on inflammatory signaling requires further studies. Moreover, since a significant percentage of HM patients lack mutations in NLRP7, additional studies with wild type and HM-linked NLRP7 mutants are necessary to elucidate the detailed mechanism, by which potential gain- or loss-of-function defects in NLRP7 contribute to the multifactorial HM disease.
Acknowledgments
This work was supported by grants from the NIH to C.S (R01GM071723, R21AI092490, R21HL097183, R01AR064349, R01AI083506 and P30AR057216) and A.D (R03AR057532). L.d.A. is supported by a postdoctoral fellowship from the American Heart Association (11POST585000).
Footnotes
The authors have no conflicting financial interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Martinon F, Burns K, Tschopp J. The Inflammasome: A molecular platform triggering activation of inflammatory caspases and processing of proIL-1beta. Mol Cell. 2002;10:417–426. doi: 10.1016/s1097-2765(02)00599-3. [DOI] [PubMed] [Google Scholar]
- 2.Keller M, Ruegg A, Werner S, Beer HD. Active caspase-1 is a regulator of unconventional protein secretion. Cell. 2008;132:818–831. doi: 10.1016/j.cell.2007.12.040. [DOI] [PubMed] [Google Scholar]
- 3.Rathinam VAK, Vanaja SK, Waggoner L, Sokolovska A, Becker C, Stuart LM, Leong JM, Fitzgerald KA. TRIF licenses caspase-11-dependent NLRP3 inflammasome activation by gram-negative bacteria. Cell. 2012;150:606–619. doi: 10.1016/j.cell.2012.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gringhuis SI, Kaptein TM, Wevers BA, Theelen B, van der Vlist M, Boekhout T, Geijtenbeek TBH. Dectin-1 is an extracellular pathogen sensor for the induction and processing of IL-1β via a noncanonical caspase-8 inflammasome. Nat Immunol. 2012;13:246–254. doi: 10.1038/ni.2222. [DOI] [PubMed] [Google Scholar]
- 5.Pierini R, Juruj C, Perret M, Jones CL, Mangeot P, Weiss DS, Henry T. AIM2/ASC triggers caspase-8-dependent apoptosis in Francisella-infected caspase-1-deficient macrophages. Cell Death Differ. 2012;19:1709–1721. doi: 10.1038/cdd.2012.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fink SL, Cookson BT. Caspase-1-dependent pore formation during pyroptosis leads to osmotic lysis of infected host macrophages. Cell Microbiol. 2006;8:1812–1825. doi: 10.1111/j.1462-5822.2006.00751.x. [DOI] [PubMed] [Google Scholar]
- 7.Srinivasula SM, Poyet JL, Razmara M, Datta P, Zhang Z, Alnemri ES. The PYRIN-CARD protein ASC is an activating adaptor for Caspase-1. J Biol Chem. 2002;277:21119–21122. doi: 10.1074/jbc.C200179200. [DOI] [PubMed] [Google Scholar]
- 8.Stehlik C, Lee SH, Dorfleutner A, Stassinopoulos A, Sagara J, Reed JC. Apoptosis-associated speck-like protein containing a caspase recruitment domain is a regulator of procaspase-1 activation. J Immunol. 2003;171:6154–6163. doi: 10.4049/jimmunol.171.11.6154. [DOI] [PubMed] [Google Scholar]
- 9.Franchi L, Amer A, Body-Malapel M, Kanneganti TD, Ozoren N, Jagirdar R, Inohara N, Vandenabeele P, Bertin J, Coyle A, Grant EP, Nunez G. Cytosolic flagellin requires Ipaf for activation of caspase-1 and interleukin-1beta in Salmonella-infected macrophages. Nat Immunol. 2006;7:576–582. doi: 10.1038/ni1346. [DOI] [PubMed] [Google Scholar]
- 10.Mariathasan S, Newton K, Monack DM, Vucic D, French DM, Lee WP, Roose-Girma M, Erickson S, Dixit VM. Differential activation of the inflammasome by caspase-1 adaptors ASC and Ipaf. Nature. 2004;430:213–218. doi: 10.1038/nature02664. [DOI] [PubMed] [Google Scholar]
- 11.Miao EA, Alpuche-Aranda CM, Dors M, Clark AE, Bader MW, Miller SI, Aderem A. Cytoplasmic flagellin activates caspase-1 and secretion of interleukin-1beta via Ipaf. Nat Immunol. 2006;7:569–575. doi: 10.1038/ni1344. [DOI] [PubMed] [Google Scholar]
- 12.Bryan NB, Dorfleutner A, Rojanasakul Y, Stehlik C. Activation of inflammasomes requires intracellular redistribution of the apoptotic speck-like protein containing a caspase recruitment domain. J Immunol. 2009;182:3173–3182. doi: 10.4049/jimmunol.0802367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Broz P, von Moltke J, Jones JW, Vance RE, Monack DM. Differential requirement for Caspase-1 autoproteolysis in pathogen-induced cell death and cytokine processing. Cell Host Microbe. 2010;8:471–483. doi: 10.1016/j.chom.2010.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Boyden ED, Dietrich WF. Nalp1b controls mouse macrophage susceptibility to anthrax lethal toxin. Nat Genet. 2006;38:240–244. doi: 10.1038/ng1724. [DOI] [PubMed] [Google Scholar]
- 15.Faustin B, Lartigue L, Bruey JM, Luciano F, Sergienko E, Bailly-Maitre B, Volkmann N, Hanein D, Rouiller I, Reed JC. Reconstituted NALP1 inflammasome reveals two-step mechanism of caspase-1 activation. Mol Cell. 2007;25:713–724. doi: 10.1016/j.molcel.2007.01.032. [DOI] [PubMed] [Google Scholar]
- 16.Kovarova M, Hesker PR, Jania L, Nguyen M, Snouwaert JN, Xiang Z, Lommatzsch SE, Huang MT, Ting JPY, Koller BH. NLRP1-dependent pyroptosis leads to acute lung injury and morbidity in mice. J Immunol. 2012;189:2006–2016. doi: 10.4049/jimmunol.1201065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lee GS, Subramanian N, Kim AI, Aksentijevich I, Goldbach-Mansky R, Sacks DB, Germain RN, Kastner DL, Chae JJ. The calcium-sensing receptor regulates the NLRP3 inflammasome through Ca2+ and cAMP. Nature. 2012;492:123–127. doi: 10.1038/nature11588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Murakami T, Ockinger J, Yu J, Byles V, McColl A, Hofer AM, Horng T. Critical role for calcium mobilization in activation of the NLRP3 inflammasome. Proc Natl Acad Sci U S A. 2012;109:11282–11287. doi: 10.1073/pnas.1117765109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shimada K, Crother TR, Karlin J, Dagvadorj J, Chiba N, Chen S, Ramanujan VK, Wolf AJ, Vergnes L, Ojcius DM, Rentsendorj A, Vargas M, Guerrero C, Wang Y, Fitzgerald KA, Underhill DM, Town T, Arditi M. Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity. 2012;36:401–414. doi: 10.1016/j.immuni.2012.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shenoy AR, Wellington DA, Kumar P, Kassa H, Booth CJ, Cresswell P, MacMicking JD. GBP5 promotes NLRP3 inflammasome assembly and immunity in mammals. Science. 2012;336:481–485. doi: 10.1126/science.1217141. [DOI] [PubMed] [Google Scholar]
- 21.Hoffman HM, Scott P, Mueller JL, Misaghi A, Stevens S, Yancopoulos GD, Murphy A, Valenzuela DM, Liu-Bryan R. Role of the leucine-rich repeat domain of cryopyrin/NALP3 in monosodium urate crystal-induced inflammation in mice. Arthritis Rheum. 2010;62:2170–2179. doi: 10.1002/art.27456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Grenier JM, Wang L, Manji GA, Huang WJ, Al-Garawi A, Kelly R, Carlson A, Merriam S, Lora JM, Briskin M, DiStefano PS, Bertin J. Functional screening of five PYPAF family members identifies PYPAF5 as a novel regulator of NF-kappaB and caspase-1. FEBS Lett. 2002;530:73–78. doi: 10.1016/s0014-5793(02)03416-6. [DOI] [PubMed] [Google Scholar]
- 23.Elinav E, Strowig T, Kau AL, Henao-Mejia J, Thaiss CA, Booth CJ, Peaper DR, Bertin J, Eisenbarth SC, Gordon JI, Flavell RA. NLRP6 inflammasome regulates colonic microbial ecology and risk for colitis. Cell. 2011;145:745–757. doi: 10.1016/j.cell.2011.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Anand PK, Malireddi RKS, Lukens JR, Vogel P, Bertin J, Lamkanfi M, Kanneganti TD. NLRP6 negatively regulates innate immunity and host defence against bacterial pathogens. Nature. 2012;488:389–393. doi: 10.1038/nature11250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Miao EA, Mao DP, Yudkovsky N, Bonneau R, Lorang CG, Warren SE, Leaf IA, Aderem A. Innate immune detection of the type III secretion apparatus through the NLRC4 inflammasome. Proc Natl Acad Sci U S A. 2010;107:3076–3080. doi: 10.1073/pnas.0913087107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Qu Y, Misaghi S, Izrael-Tomasevic A, Newton K, Gilmour LL, Lamkanfi M, Louie S, Kayagaki N, Liu J, Kömüves L, Cupp JE, Arnott D, Monack D, Dixit VM. Phosphorylation of NLRC4 is critical for inflammasome activation. Nature. 2012;490:539–542. doi: 10.1038/nature11429. [DOI] [PubMed] [Google Scholar]
- 27.Kofoed EM, Vance RE. NAIPs: building an innate immune barrier against bacterial pathogens. NAIPs function as sensors that initiate innate immunity by detection of bacterial proteins in the host cell cytosol. Bioessays. 2012;34:589–598. doi: 10.1002/bies.201200013. [DOI] [PubMed] [Google Scholar]
- 28.Meissner TB, Li A, Kobayashi KS. NLRC5: a newly discovered MHC class I transactivator (CITA) Microbes Infect. 2011;14:477–484. doi: 10.1016/j.micinf.2011.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Meissner TB, Li A, Biswas A, Lee KH, Liu YJ, Bayir E, Iliopoulos D, van den Elsen PJ, Kobayashi KS. NLR family member NLRC5 is a transcriptional regulator of MHC class I genes. Proc Natl Acad Sci U S A. 2010;107:13794–13799. doi: 10.1073/pnas.1008684107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Davis BK, Roberts RA, Huang MT, Willingham SB, Conti BJ, Brickey WJ, Barker BR, Kwan M, Taxman DJ, Accavitti-Loper MA, Duncan JA, Ting JP. Cutting edge: NLRC5-dependent activation of the inflammasome. J Immunol. 2011;186:1333–1337. doi: 10.4049/jimmunol.1003111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kumar H, Pandey S, Zou J, Kumagai Y, Takahashi K, Akira S, Kawai T. NLRC5 deficiency does not influence cytokine induction by virus and bacteria infections. J Immunol. 2010;186:994–1000. doi: 10.4049/jimmunol.1002094. [DOI] [PubMed] [Google Scholar]
- 32.Cridland JA, Curley EZ, Wykes MN, Schroder K, Sweet MJ, Roberts TL, Ragan MA, Kassahn KS, Stacey KJ. The mammalian PYHIN gene family: Phylogeny, evolution and expression. BMC Evol Biol. 2012;12:140. doi: 10.1186/1471-2148-12-140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Albrecht M, Choubey D, Lengauer T. The HIN domain of IFI-200 proteins consists of two OB folds. Biochem Biophys Res Commun. 2005;327:679–687. doi: 10.1016/j.bbrc.2004.12.056. [DOI] [PubMed] [Google Scholar]
- 34.Fernandes-Alnemri T, Yu JW, Juliana C, Solorzano L, Kang S, Wu J, Datta P, McCormick M, Huang L, McDermott E, Eisenlohr L, Landel CP, Alnemri ES. The AIM2 inflammasome is critical for innate immunity to Francisella tularensis. Nat Immunol. 2010;11:385–393. doi: 10.1038/ni.1859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rathinam VA, Jiang Z, Waggoner SN, Sharma S, Cole LE, Waggoner L, Vanaja SK, Monks BG, Ganesan S, Latz E, Hornung V, Vogel SN, Szomolanyi-Tsuda E, Fitzgerald KA. The AIM2 inflammasome is essential for host defense against cytosolic bacteria and DNA viruses. Nat Immunol. 2010;11:395–402. doi: 10.1038/ni.1864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Roberts TL, Idris A, Dunn JA, Kelly GM, Burnton CM, Hodgson S, Hardy LL, Garceau V, Sweet MJ, Ross IL, Hume DA, Stacey KJ. HIN-200 proteins regulate caspase activation in response to foreign cytoplasmic DNA. Science. 2009;323:1057–1060. doi: 10.1126/science.1169841. [DOI] [PubMed] [Google Scholar]
- 37.Kerur N, Veettil MV, Sharma-Walia N, Bottero V, Sadagopan S, Otageri P, Chandran B. IFI16 acts as a nuclear pathogen sensor to induce the inflammasome in response to Kaposi Sarcoma-associated Herpesvirus infection. Cell Host Microbe. 2011;9:363–375. doi: 10.1016/j.chom.2011.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chae JJ, Wood G, Masters SL, Richard K, Park G, Smith BJ, Kastner DL. The B30.2 domain of pyrin, the familial Mediterranean fever protein, interacts directly with caspase-1 to modulate IL-1beta production. Proc Natl Acad Sci U S A. 2006;103:9982–9987. doi: 10.1073/pnas.0602081103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Papin S, Cuenin S, Agostini L, Martinon F, Werner S, Beer HD, Grutter C, Grutter M, Tschopp J. The SPRY domain of Pyrin, mutated in familial Mediterranean fever patients, interacts with inflammasome components and inhibits proIL-1beta processing. Cell Death Differ. 2007;14:1457–1466. doi: 10.1038/sj.cdd.4402142. [DOI] [PubMed] [Google Scholar]
- 40.Shoham NG, Centola M, Mansfield E, Hull KM, Wood G, Wise CA, Kastner DL. Pyrin binds the PSTPIP1/CD2BP1 protein, defining familial Mediterranean fever and PAPA syndrome as disorders in the same pathway. Proc Natl Acad Sci USA. 2003;100:13501–13506. doi: 10.1073/pnas.2135380100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yu JW, Fernandes-Alnemri T, Datta P, Wu J, Juliana C, Solorzano L, McCormick M, Zhang Z, Alnemri ES. Pyrin activates the ASC pyroptosome in response to engagement by autoinflammatory PSTPIP1 mutants. Mol Cell. 2007;28:214–227. doi: 10.1016/j.molcel.2007.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chae JJ, Cho YH, Lee GS, Cheng J, Liu PP, Feigenbaum L, Katz SI, Kastner DL. Gain-of-function Pyrin mutations induce NLRP3 protein-independent interleukin-1beta activation and severe autoinflammation in mice. Immunity. 2011;34:755–768. doi: 10.1016/j.immuni.2011.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Messaed C, Akoury E, Djuric U, Zeng J, Saleh M, Gilbert L, Seoud M, Qureshi S, Slim R. NLRP7, a nucleotide oligomerization domain-like receptor protein, is required for normal cytokine secretion and co-localizes with Golgi and the microtubule-organizing center. J Biol Chem. 2011;286:43313–43323. doi: 10.1074/jbc.M111.306191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kinoshita T, Wang Y, Hasegawa M, Imamura R, Suda T. PYPAF3, a PYRIN-containing APAF-1-like protein, is a feedback regulator of caspase-1-dependent interleukin-1beta secretion. J Biol Chem. 2005;280:21720–21725. doi: 10.1074/jbc.M410057200. [DOI] [PubMed] [Google Scholar]
- 45.Khare S, Dorfleutner A, Bryan NB, Yun C, Radian AD, de Almeida L, Rojanasakul Y, Stehlik C. An NLRP7-containing inflammasome mediates recognition of microbial lipopeptides in human macrophages. Immunity. 2012;36:464–476. doi: 10.1016/j.immuni.2012.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kinoshita T, Kondoh C, Hasegawa M, Imamura R, Suda T. Fas-associated factor 1 is a negative regulator of PYRIN-containing Apaf-1-like protein 1. Int Immunol. 2006;18:1701–1706. doi: 10.1093/intimm/dxl104. [DOI] [PubMed] [Google Scholar]
- 47.Pinheiro AS, Proell M, Eibl C, Page R, Schwarzenbacher R, Peti W. Three-dimensional structure of the NLRP7 pyrin domain: insight into pyrin-pyrin-mediated effector domain signaling in innate immunity. J Biol Chem. 2010;285:27402–27410. doi: 10.1074/jbc.M110.113191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Masters SL, Simon A, Aksentijevich I, Kastner DL. Horror autoinflammaticus: the molecular pathophysiology of autoinflammatory disease. Annu Rev Immunol. 2009;27:621–668. doi: 10.1146/annurev.immunol.25.022106.141627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Aksentijevich I, Remmers DPCEF, Mueller JL, Le J, Kolodner RD, Moak Z, Chuang M, Austin F, Goldbach-Mansky R, Hoffman HM, Kastner DL. The clinical continuum of cryopyrinopathies: novel CIAS1 mutations in North American patients and a new cryopyrin model. Arthritis Rheum. 2007;56:1273–1285. doi: 10.1002/art.22491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Meng G, Zhang F, Fuss I, Kitani A, Strober W. A mutation in the Nlrp3 gene causing inflammasome hyperactivation potentiates Th17 cell-dominant immune responses. Immunity. 2009;30:860–874. doi: 10.1016/j.immuni.2009.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Brydges SD, Mueller JL, McGeough MD, Pena CA, Misaghi A, Gandhi C, Putnam CD, Boyle DL, Firestein GS, Horner AA, Soroosh P, Watford WT, O’Shea JJ, Kastner DL, Hoffman HM. Inflammasome-mediated disease animal models reveal roles for innate but not adaptive immunity. Immunity. 2009;30:875–887. doi: 10.1016/j.immuni.2009.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Martinon F, Petrilli V, Mayor A, Tardivel A, Tschopp J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature. 2006;440:237–241. doi: 10.1038/nature04516. [DOI] [PubMed] [Google Scholar]
- 53.Dostert C, Petrilli V, Van Bruggen R, Steele C, Mossman BT, Tschopp J. Innate immune activation through Nalp3 inflammasome sensing of asbestos and silica. Science. 2008;320:674–677. doi: 10.1126/science.1156995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hornung V, Bauernfeind F, Halle A, Samstad EO, Kono H, Rock KL, Fitzgerald KA, Latz E. Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat Immunol. 2008;9:847–856. doi: 10.1038/ni.1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cassel SL, Eisenbarth SC, Iyer SS, Sadler JJ, Colegio OR, Tephly LA, Carter AB, Rothman PB, Flavell RA, Sutterwala FS. The Nalp3 inflammasome is essential for the development of silicosis. Proc Natl Acad Sci U S A. 2008;105:9035–9040. doi: 10.1073/pnas.0803933105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Halle A, Hornung V, Petzold GC, Stewart CR, Monks BG, Reinheckel T, Fitzgerald KA, Latz E, Moore KJ, Golenbock DT. The NALP3 inflammasome is involved in the innate immune response to amyloid-beta. Nat Immunol. 2008;9:857–865. doi: 10.1038/ni.1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Masters SL, Dunne A, Subramanian SL, Hull RL, Tannahill GM, Sharp FA, Becker C, Franchi L, Yoshihara E, Chen Z, Mullooly N, Mielke LA, Harris J, Coll RC, Mills KH, Mok KH, Newsholme P, Nunez G, Yodoi J, Kahn SE, Lavelle EC, O’Neill LA. Activation of the NLRP3 inflammasome by islet amyloid polypeptide provides a mechanism for enhanced IL-1beta in type 2 diabetes. Nat Immunol. 2010;11:897–904. doi: 10.1038/ni.1935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Duewell P, Kono H, Rayner KJ, Sirois CM, Vladimer G, Bauernfeind FG, Abela GS, Franchi L, Nunez G, Schnurr M, Espevik T, Lien E, Fitzgerald KA, Rock KL, Moore KJ, Wright SD, Hornung V, Latz E. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature. 2010;464:1357–1361. doi: 10.1038/nature08938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Eisenbarth SC, Colegio OR, O’Connor W, Sutterwala FS, Flavell RA. Crucial role for the Nalp3 inflammasome in the immunostimulatory properties of aluminium adjuvants. Nature. 2008;453:1122–1126. doi: 10.1038/nature06939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Franchi L, Nunez G. The Nlrp3 inflammasome is critical for aluminium hydroxide-mediated IL-1beta secretion but dispensable for adjuvant activity. Eur J Immunol. 2008;38:2085–2089. doi: 10.1002/eji.200838549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Li H, Willingham SB, Ting JP, Re F. Cutting edge: inflammasome activation by alum and alum’s adjuvant effect are mediated by NLRP3. J Immunol. 2008;181:17–21. doi: 10.4049/jimmunol.181.1.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kool M, Petrilli V, De Smedt T, Rolaz A, Hammad H, van Nimwegen M, Bergen IM, Castillo R, Lambrecht BN, Tschopp J. Cutting Edge: alum adjuvant stimulates inflammatory dendritic cells through activation of the NALP3 inflammasome. J Immunol. 2008;181:3755–3759. doi: 10.4049/jimmunol.181.6.3755. [DOI] [PubMed] [Google Scholar]
- 63.Henao-Mejia J, Elinav E, Jin C, Hao L, Mehal WZ, Strowig T, Thaiss CA, Kau AL, Eisenbarth SC, Jurczak MJ, Camporez JP, Shulman GI, Gordon JI, Hoffman HM, Flavell RA. Inflammasome-mediated dysbiosis regulates progression of NAFLD and obesity. Nature. 2012;482:179–185. doi: 10.1038/nature10809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ayres JS, Trinidad NJ, Vance RE. Lethal inflammasome activation by a multidrug-resistant pathobiont upon antibiotic disruption of the microbiota. Nat Med. 2012;18:799–806. doi: 10.1038/nm.2729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Franchi L, Kamada N, Nakamura Y, Burberry A, Kuffa P, Suzuki S, Shaw MH, Kim Y-G, Núñez G. NLRC4-driven production of IL-1β discriminates between pathogenic and commensal bacteria and promotes host intestinal defense. Nat Immunol. 2012;13:449–456. doi: 10.1038/ni.2263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Stienstra R, Joosten LA, Koenen T, van Tits B, van Diepen JA, van den Berg SA, Rensen PC, Voshol PJ, Fantuzzi G, Hijmans A, Kersten S, Muller M, van den Berg WB, van Rooijen N, Wabitsch M, Kullberg BJ, van der Meer JW, Kanneganti T, Tack CJ, Netea MG. The inflammasome-mediated caspase-1 activation controls adipocyte differentiation and insulin sensitivity. Cell Metab. 2010;12:593–605. doi: 10.1016/j.cmet.2010.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wen H, Gris D, Lei Y, Jha S, Zhang L, Huang MT, Brickey WJ, Ting JP. Fatty acid-induced NLRP3-ASC inflammasome activation interferes with insulin signaling. Nat Immunol. 2011;12:408–415. doi: 10.1038/ni.2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Vandanmagsar B, Youm YH, Ravussin A, Galgani JE, Stadler K, Mynatt RL, Ravussin E, Stephens JM, Dixit VD. The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nat Med. 2011;17:179–188. doi: 10.1038/nm.2279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Shaw MH, Kamada N, Kim YG, Núñez G. Microbiota-induced IL-1β, but not IL-6, is critical for the development of steady-state TH17 cells in the intestine. J Exp Med. 2012;209:251–258. doi: 10.1084/jem.20111703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sutton CE, Lalor SJ, Sweeney CM, Brereton CF, Lavelle EC, Mills KHG. Interleukin-1 and IL-23 induce innate IL-17 production from γδ T cells, amplifying Th17 responses and autoimmunity. Immunity. 2009;31:331–341. doi: 10.1016/j.immuni.2009.08.001. [DOI] [PubMed] [Google Scholar]
- 71.Lukens JR, Barr MJ, Chaplin DD, Chi H, Kanneganti TD. Inflammasome-derived IL-1β regulates the production of GM-CSF by CD4+ T cells and γ δ T cells. J Immunol. 2012;88:3107–3115. doi: 10.4049/jimmunol.1103308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Huber S, Gagliani N, Zenewicz LA, Huber FJ, Bosurgi L, Hu B, Hedl M, Zhang W, O’Connor W, Murphy AJ, Valenzuela DM, Yancopoulos GD, Booth CJ, Cho JH, Ouyang W, Abraham C, Flavell RA. IL-22BP is regulated by the inflammasome and modulates tumorigenesis in the intestine. Nature. 2012;491:259–263. doi: 10.1038/nature11535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Dinarello CA. Interleukin 1 and interleukin 18 as mediators of inflammation and the aging process. Am J Clin Nutr. 2006;83:447S–455S. doi: 10.1093/ajcn/83.2.447S. [DOI] [PubMed] [Google Scholar]
- 74.Murdoch S, Djuric U, Mazhar B, Seoud M, Khan R, Kuick R, Bagga R, Kircheisen R, Ao A, Ratti B, Hanash S, Rouleau GA, Slim R. Mutations in NALP7 cause recurrent hydatidiform moles and reproductive wastage in humans. Nat Genet. 2006;38:300–302. doi: 10.1038/ng1740. [DOI] [PubMed] [Google Scholar]
- 75.Slim R, Mehio A. The genetics of hydatidiform moles: new lights on an ancient disease. Clin Genetics. 2007;71:25–34. doi: 10.1111/j.1399-0004.2006.00697.x. [DOI] [PubMed] [Google Scholar]
- 76.Milhavet F, Cuisset L, Hoffman HM, Slim R, El-Shanti H, Aksentijevich I, Lesage S, Waterham H, Wise C, Sarrauste de Menthiere C, Touitou I. The infevers autoinflammatory mutation online registry: update with new genes and functions. Hum Mutat. 2008;29:803–808. doi: 10.1002/humu.20720. [DOI] [PubMed] [Google Scholar]
- 77.Dixon PH, Trongwongsa P, Abu-Hayyah S, Ng SH, Akbar SA, Khawaja NP, Seckl MJ, Savage PM, Fisher RA. Mutations in NLRP7 are associated with diploid biparental hydatidiform moles, but not androgenetic complete moles. J Med Genet. 2012;49:206–211. doi: 10.1136/jmedgenet-2011-100602. [DOI] [PubMed] [Google Scholar]
- 78.Wang CM, Dixon PH, Decordova S, Hodges MD, Sebire NJ, Ozalp S, Fallahian M, Sensi A, Ashrafi F, Repiska V, Zhao J, Xiang Y, Savage PM, Seckl MJ, Fisher RA. Identification of 13 novel NLRP7 mutations in 20 families with recurrent hydatidiform mole; missense mutations cluster in the leucine-rich region. J Med Genet. 2009;46:569–575. doi: 10.1136/jmg.2008.064196. [DOI] [PubMed] [Google Scholar]
- 79.Williams D, Hodgetts V, Gupta J. Recurrent hydatidiform moles. Eur J Obstet Gynecol Reprod Biol. 2010;150:3–7. doi: 10.1016/j.ejogrb.2010.01.003. [DOI] [PubMed] [Google Scholar]
- 80.Okada K, Hirota E, Mizutani Y, Fujioka T, Shuin T, Miki T, Nakamura Y, Katagiri T. Oncogenic role of NALP7 in testicular seminomas. Cancer Sci. 2004;95:949–954. doi: 10.1111/j.1349-7006.2004.tb03182.x. [DOI] [PMC free article] [PubMed] [Google Scholar]