

Abstract
Protein misfolding and aggregation are the hallmark of a number of diseases including Alzheimer’s disease, Parkinson’s disease, Huntington’s disease, amyotrophic lateral sclerosis, and the prion diseases. In all cases, a naturally-occurring protein misfolds and forms aggregates that are thought to disrupt cell function through a wide range of mechanisms that are yet to be fully unraveled. Fourier transform infrared (FTIR) spectroscopy is a technique that is sensitive to the secondary structure of proteins and has been widely used to investigate the process of misfolding and aggregate formation. This review focuses on how FTIR spectroscopy and spectroscopic microscopy are being used to evaluate the structural changes in disease-related proteins both in vitro and directly within cells and tissues. Finally, ongoing technological advances will be presented that are enabling time-resolved FTIR imaging of protein aggregation directly within living cells, which can provide insight into the structural intermediates, time scale, and mechanisms of cell toxicity associated with aggregate formation.
Keywords: Fourier transform infrared (FTIR) spectroscopy, microspectroscopy, imaging, protein secondary structure, protein aggregation, amyloid, cell culture
1. Introduction
Many diseases involve the misfolding and aggregation of naturally occurring proteins in the body. These aggregates disrupt cellular function through a number of different mechanisms, often resulting in cell death [1]. This process is most prevalent in neurodegenerative diseases [2, 3]. For example, Alzheimer’s disease is characterized by the misfolding of the amyloid beta (Abeta) protein, which leads to amyloid plaques, whereas structural changes associated with hyperphosphorylation of the tau protein leads to the formation of neurofibrillary tangles. In Parkinson’s and Huntington’s disease, aggregates of α-synuclein and huntingtin are observed, respectively. A familial form of amyotrophic lateral sclerosis (ALS) is characterized by tiny aggregates of copper-zinc superoxide dismutase (SOD1). Prion diseases such as mad cow disease (bovine spongiform encephalopathy), Creutzfeldt-Jakob disease, scrapie, and chronic wasting disease are associated with the misfolding of the prion protein, which becomes infectious in its aberrant form. Not all protein-folding diseases occur in the central nervous system; other examples include cystic fibrosis, type II diabetes, and multiple myeloma [4]. In all cases, protein misfolding is associated with a change in secondary structure.
Fourier transform infrared spectroscopy (FTIR) has been shown to be sensitive to the secondary structure of proteins, making it a valuable technique for studying protein aggregation. A protein’s FTIR spectrum has two prominent features, the Amide I (~1650 cm−1) and Amide II (~1540 cm−1) bands, where the former arise primarily from the C=O stretching vibration and the latter is attributed to the N-H bending and C-N stretching vibrations of the peptide backbone [5]. The frequency of the Amide I band is particularly sensitive to secondary structure based on different hydrogen-bonding environments for α-helix, β-sheet, turn, and unordered conformations. For example, α-helices and β-sheets have Amide I vibrational frequencies at approximately 1655 and 1630 cm−1, respectively [6, 7]. The vibrational frequency of an aggregated protein falls around 1620 – 1625 cm−1 due to the distinct hydrophobic environment [8]. For most proteins, a mixture of secondary structures exists; thus, the Amide I band represents a combination of these components (Figure 1). As a first approximation, the Amide I band can be curve-fit to predict a protein’s secondary structure, or database approaches based on crystallographic information have also been used [9].
Figure 1.

FTIR spectrum of a typical protein illustrating the Amide I and Amide II bands at ~1650 cm−1 and ~1540 cm−1, respectively. (Inset) Expanded view of the Amide I band, which can be deconvolved into its secondary structure components.
Beyond the examination of protein secondary structure in vitro, FTIR spectroscopic microscopy can also be used to directly study protein misfolding and aggregation within cells and tissues. Protein aggregates are typically very small, where initial oligomers form at the nanoscale and the larger aggregates are only 20 – 30 μm in size. In addition, the spectral differences associated with changes in protein conformation are subtle, requiring spectra with a high signal-to-noise (S/N) ratio. These challenges have been addressed recently by taking advantage of the high brightness of a synchrotron infrared source, where protein misfolding and aggregation have been examined directly within diseased tissue in Alzheimer’s disease [8, 10–12], Parkinson’s disease [13], Huntington’s disease [14], amyotrophic lateral sclerosis [15], and scrapie [16–18].
In this review, we will describe how FTIR spectroscopy and microscopy have been used to examine protein misfolding and aggregation both in vitro and in situ. The challenges associated with data collection and spectral interpretation will be discussed along with recent technological developments and opportunities for future studies.
2. FTIR Spectroscopy of Protein Structure in vitro
Since the Amide I band is sensitive to protein secondary structure, FTIR spectroscopy is frequently used to study the process of protein misfolding and aggregation in vitro. Due to unique hydrogen bonding environments for the different secondary structure elements, shifts are observed in the frequency of the Amide I band [6, 19]. And by studying protein structure in vitro, the effects of temperature, pH, solvent, and protein conformation can be systematically examined.
The major difficulty with measuring FTIR spectra of proteins in water arises from the strong water bending mode that overlaps the Amide I vibrational mode. Thus, transmission measurements of protein solutions are typically performed in D2O, where the O-D bending mode is shifted to lower frequency (Figure 2). Alternatively, spectra can be collected in water with very thin pathlengths (< 10 μm), but this requires a higher protein concentration. For FTIR spectroscopy of protein solutions in D2O, protein concentrations of 0.5 – 10 mM are common. Protein solutions are placed in a sandwich cell between two IR-transparent windows (typically CaF2 or BaF2), separated by a thin spacer (typically 25 – 50 μm thick). The reference (background) spectrum is collected through the buffer solution.
Figure 2.
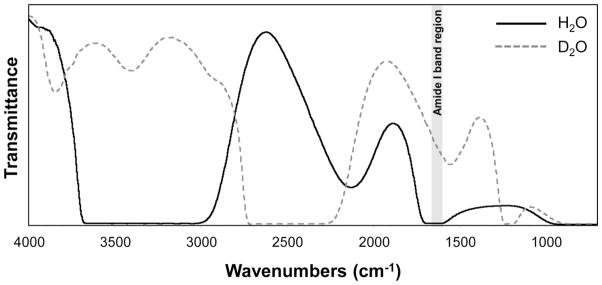
FTIR transmission spectra of H2O and D2O. As can be seen, an O-H bending mode overlaps with the protein Amide I band at ~1650 cm−1. With deuteration, the O-D bending mode falls at a lower frequency (~1225 cm−1), increasing the IR transmission in the region of the Amide I band and enabling better background (i.e. solvent) subtraction.
Secondary structure analysis of the Amide I band has been performed in a number of ways, including second derivatives, Fourier self-deconvolution, curve-fitting, and neural networks [6, 7, 9, 19–23]. Most frequently, second-derivative analysis is used to identify the frequencies of the underlying spectral components. Then spectral deconvolution (i.e. curve-fitting) is performed on the Amide I band to define the intensities of the components (see Figure 1). While absolute secondary structure quantification can be done, the best results are achieved when structural changes are studied as a function of an external perturbation, as has been demonstrated for the amyloid-beta protein [24].
2.1 Alzheimer’s disease
Protein misfolding and aggregation has been studied extensively in vitro using FTIR spectroscopy. For example, Alzheimer’s disease (AD) is a neurodegenerative disorder characterized by the accumulation of senile plaques and neurofibrillary tangles in gray matter areas of the brain. Plaque formation is brought about by the transformation of a small peptide known as amyloid-beta (Abeta) from a soluble form through an oligomeric intermediate to an aggregated, fibrillary structure [25].
The mechanism behind the structural changes and toxicity of Abeta during aggregate formation has been the subject of numerous in vitro studies. The seminal studies utilized circular dichroism (CD) and NMR to show the structural conversion of Abeta from a soluble α-helical protein to a fibrillar β-sheet protein [26–29]. More recently, FTIR spectroscopy has been used to study the specific alignment of β-sheet strands within Abeta fibrils. Using isotopically-labeled Abeta(16–22), Petty and coworkers showed conclusively that the β-sheets are antiparallel and in alignment across all strands. This realignment of the peptide strands is important for therapeutics because they form more stable amyloid fibrils [30].
Since Abeta is derived from the membrane-bound amyloid precursor protein (APP), it has been suggested that oligomeric Abeta forms pore-like structures in lipid membranes that disrupt ion homeostasis and cause cell death. Using FTIR spectroscopy, Abeta oligomers have been shown to exhibit an antiparallel β-sheet structure, where the spectrum was very similar to that of bacterial outer membrane porins [31]. With polarized attenuated total internal reflection FTIR (ATR-FTIR) spectroscopy, it was also shown that membranes promote β-sheet formation [32] and that membranes containing oxidatively damaged phospholipids accumulated Abeta significantly faster than membranes containing only unoxidized or saturated phospholipids [33].
In addition to the Abeta protein, FTIR spectroscopy has also been instrumental in understanding the structural conformation of the tau protein – a neuronal microtubule protein that becomes hyperphosphorylated and forms paired helical filaments (PHFs) in the AD brain. Conflicting views on the structure of the tau “tangles” have been proposed, ranging from mainly α-helical structure to mainly β-sheet, or a mixture of mostly random coil and β-sheet. In vitro FTIR analysis showed that the soluble tau protein is a natively unfolded protein dominated by random coil structure, whereas Alzheimer PHFs show an increase in the level of β-structure [34, 35]. The results support a model in which the repeat domain of tau (which lies within the core of PHFs) adopts an increasing level of β-structure during aggregation, whereas the N- and C-terminal domains projecting away from the PHF core are mostly random coil.
2.2 The prion diseases
The transmissible spongiform encephalopathies (TSEs), also termed prion diseases, are a group of fatal, neurodegenerative diseases characterized by the misfolding and accumulation of the prion protein (PrPSc). This protein differs in secondary structure from its normal, cellular isoform (PrPC), which is physiologically expressed mostly by neurons. Importantly, prions are considered infectious because PrPSc is capable of causing the normal PrPC protein to misfold into the disease-related form, resulting in a catalytic process that produces a large amount of aberrant protein. Thus, only a small amount of PrPSc is needed to initiate disease, which is always fatal.
Scrapie is a prion disease first described in the 18th century in sheep and goats and a rodent model has been established to study the pathogenesis and pathology of the disease. FTIR spectroscopy of the isolated prion protein showed that PrPC was predominantly α-helical in structure whereas PrPSc was primarily β-sheet [16]. Similar structural differences were observed in scrapie-infected ganglia, although not all neurons were affected [18].
Early diagnosis of the prion diseases is paramount to successful treatment. FTIR spectroscopy and artificial neural networks were used to screen blood serum from terminal scrapie-infected hamsters and consistently yielded test sensitivities and specificities of 97% and 100%, respectively [36]. A similar classification algorithm was also developed for terminal bovine spongiform encephalopathy (BSE) with a sensitivity and specificity of 96% and 92%, respectively [37]. And more recently, preclinical scrapie (100 days post infection) was reliably detected from blood serum [38].
Another substantial challenge in prion research is the discrimination of different TSE strains that cause scrapie, BSE, or Creutzfeldt-Jakob disease. In a study by Thomzig and coworkers, four different isolates of hamster-adapted scrapie were infected into Syrian hamsters and probed by FTIR spectroscopy [39]. Results showed different secondary structures between the TSE agents, even in cases where immunobiochemical typing failed to detect structural differences. Thus, FTIR spectroscopy may provide a promising analytical tool for molecular strain typing without antibodies and without restrictions to specific TSEs or mammalian species.
2.3 Parkinson’s disease
Parkinson’s disease is a neurodegenerative disorder characterized by the formation of abnormal protein aggregates called Lewy bodies in the neurons of the brain. The principle component of Lewy bodies is the α-synuclein protein, which is a protein of unknown function that is normally found as a soluble, unordered structure in the cytosol of neurons.
There is considerable debate about the mechanism behind the fibrillation of α-synuclein during Lewy body formation and subsequent disease toxicity. FTIR and CD spectroscopy have shown that α-synuclein fibrils are antiparallel β-sheet in structure, similar to fibrillar Abeta [40]. Fibrillation likely takes place via long-range intramolecular interactions between the N- and C-termini of α-synuclein [41]. Two homologous proteins, β- and γ-synuclein, are also natively unfolded but do not form fibrils unless exposed to metal ions such as Cu2+, Zn2+, and Pb2+ [42]. It has also been shown by FTIR spectroscopy that mixtures of the synucleins can prevent the fibrillation of α-synuclein [43].
2.4 Huntington’s disease
Huntington’s disease is a neurodegenerative disorder that is caused by a genetic mutation of the Huntingtin gene (HTT), which codes for the huntingtin protein. More specifically, Huntington’s disease is a trinucleotide repeat disorder, where the HTT gene codes for a long chain of glutamine residues – called the polyQ region – in the huntingtin protein. Disease symptoms occur when the polyQ region is longer than ~36 residues.
The disease is characterized by neuronal inclusion bodies that are primarily composed of aggregates formed from fragments of polyQ region of the mutant huntingtin protein. Using FTIR spectroscopy of the purified huntingtin fragments, results showed that globular and protofibrillar intermediates participated in the genesis of mature fibrils [44]. The intermediates were high in β-sheet structure. Moreover, Congo Red – a dye commonly used to assay for amyloid fiber formation – prevented the assembly of mature huntingtin fibrils, possibly by binding to the β-structure of the protofibril.
3. FTIR Microspectroscopy of Protein Structure in Fixed Cells and Tissues
FTIR microscopes have been used to examine the composition of biological cells and tissues for over 20 years. However, the long wavelengths of infrared light result in diffraction effects that limit the spatial resolution that can be achieved to 2 – 10 μm in the mid-infrared region. In addition, the brightness of conventional thermal (globar) IR sources are inherently limited near the diffraction limit.
Examination of protein misfolding and aggregation presents a unique challenge for FTIR spectroscopic microscopy (FTIRM) because the aggregates are typically very small, i.e. initial oligomers form at the nanoscale and the larger aggregates are only 20 – 30 μm in size. In addition, the spectral differences associated with changes in protein conformation are subtle, requiring spectra with a high S/N ratio. Over recent years, dramatic improvements have been realized through the utilization of a synchrotron infrared source, which is 100 – 1000 times brighter than a conventional thermal source [45]. This brightness advantage is not because the synchrotron produces more power, but because the effective source size is small and the light is emitted into a narrow range of angles. The high brightness (i.e. flux density) of the synchrotron source allows smaller regions to be probed with an acceptable S/N ratio [46, 47].
One of the earliest uses of a synchrotron-based IR microscope was to examine the secondary structure of Abeta directly within the brain tissue of an Alzheimer’s case [10]. Interestingly, these findings revealed spectral differences between the in vitro, solution-state protein and the in situ, amyloid structure of AD plaques, indicating that the misfolding of Abeta depends on factors present in brain tissue environment. More recently, synchrotron-based FTIRM showed that the density of dense-core plaques was approximately 1.5 times higher than the surrounding brain tissue [12] and the Amide I maximum was observed at 1623 cm−1 [8], significantly lower than Abeta aggregates in solution, and characteristic of aggregated protein in a highly hydrophobic environment (Figure 3). In contrast, diffuse plaques were not associated with IR detectable changes in protein secondary structure [11].
Figure 3.
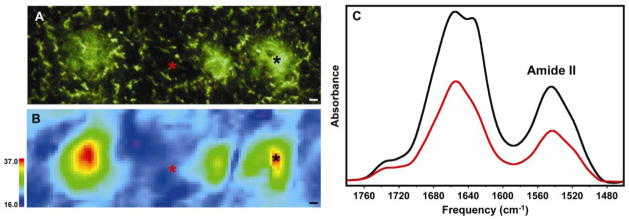
FTIRM of amyloid plaques in a PSAPP mouse model of Alzheimer’s disease. (A) Thioflavin S-stained brain tissue showing three plaques. (B) Infrared image of the same tissue showing the distribution of protein measured by the Amide II band. (C) Infrared spectra collected from the areas marked with asterisks in (A) and (B), showing the relative amount of protein in the center of a plaque (black) and the surrounding tissue (red). The increase in β-sheet content within the plaques is evident from the shoulder at ~1625 cm−1 in the plaque (black) spectrum. All scale bars are 5 μm. With permission from [12].
The misfolding and aggregation of the infectious prion protein has also been studied in the dorsal root ganglia of a hamster model of scrapie. Synchrotron-based FTIRM was used to show that, at pre-clinical time points, the scrapie-infected animals exhibited a significant increase in protein expression, but the β-sheet protein content was significantly lower than controls, suggesting that the pre-clinical stages of scrapie were characterized by an overexpression of proteins low in β-sheet content [17, 18]. As the disease progressed, the β-sheet content increased significantly but immunostaining confirmed that this increase was partly - but not solely - due to the formation of PrPSc in the tissue and indicated that other proteins high in β-sheet were produced, either by overexpression or misfolding (Figure 4). These dramatic changes in protein content and structure, especially at pre-clinical time points, emphasize the possibility for identifying other proteins involved in early pathogenesis, which are important for a further understanding and early diagnosis of the disease.
Figure 4.

FTIRM of the misfolded prion protein in a hamster model of scrapie showing results from a preclinical stage 100 days post infection (left column, 100 dpi) and at the first clinical signs of the disease (right column, fcs). Photomicrographs of the areas investigated with FTIRM are shown in the 1st row (unstained) and 2nd row (3F4-antibody stained for the prion protein). The β-sheet and α-helix distributions are represented in the 3rd and 4th rows, respectively. At the earliest stages, some areas show high β-sheet as well as high α-helix (*), which was not observed at the terminal stage. Areas with misfolded prion protein deposition as seen in the 3F4 stained sections showed elevated β-sheet (arrows), while some areas showed high β-sheet but no aggregate deposition (arrowheads). All scale bars are 20 μm. With permission from [17].
Other FTIRM studies have examined subcellular protein compositional changes in tissue but not specifically focusing on aggregate formation. For example, the protein composition in the substantia nigra in Parkinson’s disease revealed significant protein structural changes in the nerve cell bodies prior to the formation of Lewy body aggregates, suggesting that disturbances of normal functioning neurons appear well before their morphological atrophy [13]. In a rat model of Huntington’s disease, misfolding of the huntingtin protein was evidenced by the appearance of β-sheet-rich structures in striatal neurons [14]. At the same time, other spectral regions demonstrated a reduction in unsaturated lipid content and an increase protein phosphorylation, which are consistent with myelination deficiency and early pro-apoptotic events, respectively, and demonstrate that a range of compositional changes can be assessed and correlated with protein misfolding in situ.
4. FTIR Spectroscopic Imaging of Protein Structure in Living Cells
As can be seen, FTIR spectroscopy and microspectroscopy have been widely used to examine the mechanisms behind the pathology of a wide range of protein-folding diseases. These studies have been performed in vitro, in cell culture, and in situ on histological tissue sections. However to date, the vast majority of these experiments have been performed on static systems.
In the field of neurodegenerative protein-folding diseases, a number of recent studies suggest that the neuron toxicity arises from the initial oligomeric species, rather than the larger protein plaques. The mechanism of oligomer toxicity is still under debate, but includes the formation of transmembrane pores that disrupt ion transport through cells. Pore-forming oligomeric structures have been described with Abeta in Alzheimer’s disease [48] and α-synuclein in Parkinson’s disease [49]. Over the years, a number of static studies have demonstrated that FTIR spectroscopy can be used to study the formation of amyloid in vitro. However, given the evidence that the oligomeric state is likely the toxic species, future in vitro experiments are likely to include time-resolved measurements in order to study the initial misfolding, oligomerization, and fibrillization of protein aggregates. One such example used a sub-millisecond pH jump to monitor the kinetics of the structural transition of Abeta(1–28) and showed an initial rapid transition from a random coil to an oligomeric β-sheet conformation followed by a second slower transition, which yielded larger aggregates [50].
Similar studies are being imagined in cell culture, where protein misfolding and aggregation are probed in real time. However, experimental challenges such as overcoming the water absorption and maximizing the subcellular spatial resolution must be addressed. One promising method for achieving these goals is synchrotron-based infrared imaging with a full-field focal plane array (FPA) detector. By utilizing a high magnification IR microscope objective, the pixel resolution of the detector can be increased dramatically, enabling pixel oversampling and image deconvolution. However, with a conventional thermal IR source, the flux incident on each pixel may be more than 100× smaller, limiting the spectral quality [51]. The ~1000-fold brightness of the synchrotron source improves the S/N ratio and has recently been used to demonstrate sub-micron spatial resolution [52]. Thus, by taking advantage of the synchrotron-based full-field imaging approach, spectra can be collected rapidly at high resolution, providing time-resolved images of small structural changes over time.
In addition to improvements in spatial resolution and spectral quality, widespread adoption of time-lapsed FTIR imaging (FTIRI) requires the development of environmental chambers that are compatible with the stringent requirements of the sample and IR microscope optics. In particular, high-magnification Schwarzschild objectives have short working distances that are typically less than a few millimeters, and high numerical apertures that necessitate wide opening angles. For biological cells, viability must be maintained by carefully controlling the atmosphere, pH, and temperature within the chamber. Moreover, the path length of the cell must be limited to <10 μm to allow for the transmission of the IR beam through the aqueous media.
Ongoing developments of versatile chambers for in vivo FTIRI are underway. For example, one chamber design uses thin diamond film as windows, which are IR and visible light transparent, to aid in minimizing the chamber thickness [53]. Others have used lithography to pattern spacers of precise thickness onto calcium fluoride windows [54] and microfluidics to flow media to the cells [55]. Additionally, modifications have been made to commercially available flow cells to accommodate the needs of live cells with thinner spacers and micro-patterned channels for flow [56]. The use of attenuated total internal reflectance (ATR), a technique that can improve the spatial resolution by taking advantage of ATR crystals with a high index of refraction, has also been implemented to study cancer cell growth [57, 58]. In a more recent study, the ATR crystal was attached to the IR objective allowing data to be taken directly from a Petri dish [59]. These chambers vary significantly in design, but they all maintain a low profile to accommodate small working distances and minimize water absorption, and they flow nutrients to the cells.
To demonstrate the coupling of an FTIR-compatible environmental chamber with a full-field FPA IR detector, we have recently performed time-lapsed infrared imaging of aggregate formation in a cell culture model of amyotrophic lateral sclerosis (ALS). Specifically, ALS is a neurodegenerative disease affecting the motor neurons of the spinal cord. While most ALS cases are sporadic with no known cause, some are linked to mutations in the antioxidant protein copper-zinc superoxide dismutase (SOD1), which is associated with the formation of small SOD1 aggregates in motor neurons. Interestingly, there are over 166 individual mutations that have been shown to cause the formation of SOD1 aggregates in motor neurons [60, 61].
It is widely hypothesized that the SOD1 aggregates are comprised of immature SOD1 protein, i.e. prior to metallation or post-translational modification. This hypothesis is primarily based on the high stability of many of the holo-SOD1 mutations, making them unlikely to aggregate once they are metallated [62]. Additional experiments have shown that detergent-insoluble SOD1 aggregates extracted from mice overexpressing human SOD1 mutations lack the post-translational modifications [63] and do not contain the copper and zinc ions [64] expected of a mature protein. In vitro studies using FTIR spectroscopy have shown that both metallated and apo-SOD1 showed similar changes to the natural β-sheet structure of the protein, regardless of the aggregation method [65]. It has also been shown that disulfide-reduced SOD1 readily induces oligomerization under physiologically relevant conditions [66]. In addition, there is a wide range of aggregation propensities among the numerous disease-conferring SOD1 mutations, and to date, no common factor has been found to predict the rate of aggregation of a given mutation based on its known biochemical and biophysical properties [67]. Despite many extensive studies on SOD1 aggregation, the mechanism(s) of aggregation remain unclear.
In addition to understanding protein aggregation, the process by which aggregation is associated with neuron death remains illusive and necessitates more in vivo studies. In particular, it is possible that toxicity arises due to pore-forming oligomers analogous to those described for Abeta in Alzheimer’s disease [48] and α-synuclein in Parkinson’s disease [49]. The transmembrane pore structures are rich in antiparallel β-sheet structure. Alternatively or in conjunction, fibril formation may (also) be the culprit, which can be identified by the presence of parallel β-sheet structures. Since FTIR spectroscopy can readily distinguish parallel vs. antiparallel β-sheet structure [31], time-lapsed FTIRI of SOD1 aggregation can provide insight into the structural intermediates, time scale, and mechanism(s) of cell toxicity associated with aggregate formation.
In cell culture, transient transfection has been used to overexpress mutant SOD1 and form protein aggregates [67]. In order to visualize the aggregates, SOD1 is co-expressed with yellow fluorescent protein (YFP) [68]. Results have shown different aggregation patterns and time scales that depend upon the specific mutation, suggesting that multiple misfolding pathways may exist [67].
Chinese hamster ovary (CHO-K1) cells were transfected to express the mutant G37R SOD1-YFP with Lipo-D 293 transfection reagent. The G37R mutation is located in the β-barrel and forms a moderate amount of punctuate aggregates within 24 hours of transfection [67]. Transfected cells were imaged at Beamline U10B at the National Synchrotron Light Source (NSLS) at Brookhaven National Laboratory using a Bruker Hyperion 3000 FTIR microscope equipped with a 128×128 pixel focal plane array detector. The cells were grown on calcium fluoride windows and transfected in the Petri dish in a conventional incubator. Ten hours after the transfection was initiated, the cells were transferred to custom FTIR-compatible incubator and ultra-buffered (20 mM HEPES) F12K medium was circulated through the flow cell. A field of approximately twelve cells was selected based on the presence of YFP fluorescence and FTIR images were taken every hour from 11 – 16 hours post-transfection.
Figure 5 shows the visible and epifluorescence images of SOD1-YFP after ten hours, showing two transfected cells as indicated by the green fluorescence. FTIR spectra of the transfected (green) vs. non-transfected (colorless) cells after 16 hours shows a shift in the Amide I band to higher frequency, suggesting an increase in antiparallel β-sheet structure. FTIR images of the transfected cells confirm the development of antiparallel β-sheet structure through a decrease in the 1630/1695 cm−1 ratio over time, which can be observed as a shift from red/yellow to blue pixels in the false-color image (Figure 5). In contrast, the protein structure in the surrounding non-transfected cells remains relatively constant. Thus, the initial stages of aggregate formation in the G37A mutant reveal a spectral signature that is consistent with the formation of pore-forming oligomeric structures. Moreover, it is also likely that the antiparallel β-sheet structure is an intermediate conformation for fibril formation. Further studies at longer time scales and with other SOD1 mutants will be needed to provide a complete picture of the folding intermediates and pathway to cell toxicity in SOD1-ALS.
Figure 5.

Time-lapsed FTIR imaging of CHO-K1 cells transiently transfected to overexpress the mutant, G37R-SOD1, over the course of 16 hours. (Top row) Photomicrographs of a field of ~12 cells showing a positive transfection in 2 cells as identified by the yellow-green fluorescence from the fusion tag, YFP, and outlined in red. (Middle row) FTIRI images from the living cells in a FTIR-compatible incubator at 12, 14, and 16 hours post transfection. False color images show the ratio of 1630 /1695 cm−1, which represents the parallel/antiparallel β-sheet structure. As can be seen, this ratio decreases with time, indicating an increase in the antiparallel β-sheet structure over time, which suggests the formation of pore-forming oligomeric structures. (Bottom) FTIR spectra from the transfected (red) and non-transfected (black) cells, also illustrating an increase in the antiparallel β-sheet conformation in the transfected cells. All scale bars are 10 μm.
5. Conclusion and Outlook
In summary, FTIR spectroscopy and microspectroscopy have been an integral part of understanding the mechanisms behind protein misfolding diseases including Alzheimer’s disease, Parkinson’s disease, Huntington’s disease, the prion diseases, and amyotrophic lateral sclerosis. The field is expected to continue its growth by incorporating new in vivo and imaging technologies for time-resolved FTIR studies of protein oligomerization, fibril formation, and aggregation. In the future, we expect that continued advances in FTIR spectroscopy and imaging, combined with other spectroscopic methods, will lead to an improved understanding, diagnosis, and treatment of protein-folding diseases.
Highlights.
We review FTIR spectroscopy for evaluating the secondary structure of proteins.
We describe its use for investigating protein misfolding and aggregation in vitro.
New technology has enabled determination of aggregate structure in cells and tissue.
Time-lapsed FTIR imaging of protein aggregation within living cells is demonstrated.
Results can reveal the structural intermediates and mechanisms of cell toxicity.
Acknowledgments
We thank Dr. David Borchelt (University of Florida College of Medicine) for providing the SOD1 cDNA used for the time-lapsed FTIR imaging study of G37R-SOD1 aggregation. The National Synchrotron Light Source is supported by the US Department of Energy under Contract No. DE-AC02-98CH10886. Beamline U10B at the NSLS was supported by the National Institutes of Health grant RR23782.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Ramirez-Alvarado M, Kelly JW, Dobson CM. Protein misfolding diseases: current and emerging principles and therapies. Wiley Publishers; 2010. [Google Scholar]
- 2.Ross CA, Poirier MA. Protein aggregation and neurodegenerative disease. Nature medicine. 2004;10(Suppl):S10–7. doi: 10.1038/nm1066. [DOI] [PubMed] [Google Scholar]
- 3.Ross CA, Poirier MA. Opinion: What is the role of protein aggregation in neurodegeneration? Nature reviews Molecular cell biology. 2005;6:891–8. doi: 10.1038/nrm1742. [DOI] [PubMed] [Google Scholar]
- 4.Cohen FE, Kelly JW. Therapeutic approaches to protein-misfolding diseases. Nature. 2003;426:905–9. doi: 10.1038/nature02265. [DOI] [PubMed] [Google Scholar]
- 5.Haris PI, Severcan F. FTIR spectroscopic characterization of protein structure in aqueous and non-aqueous media. Journal of Molecular Catalysis B-Enzymatic. 1999;7:207–221. [Google Scholar]
- 6.Byler DM, Susi H. Examination of the secondary structure of proteins by deconvolved FTIR spectra. Biopolymers. 1986;25:469–87. doi: 10.1002/bip.360250307. [DOI] [PubMed] [Google Scholar]
- 7.Goormaghtigh E, Ruysschaert JM, Raussens V. Evaluation of the information content in infrared spectra for protein secondary structure determination. Biophys J. 2006;90:2946–57. doi: 10.1529/biophysj.105.072017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Miller LM, Wang Q, Telivala TP, Smith RJ, Lanzirotti A, Miklossy J. Synchrotron-based infrared and X-ray imaging shows focalized accumulation of Cu and Zn co-localized with beta-amyloid deposits in Alzheimer’s disease. J Struct Biol. 2006;155:30–7. doi: 10.1016/j.jsb.2005.09.004. [DOI] [PubMed] [Google Scholar]
- 9.Sarver RW, Krueger WC. Protein Secondary Structure from Fourier-Transform Infrared-Spectroscopy - a Data-Base Analysis. Analytical Biochemistry. 1991;194:89–100. doi: 10.1016/0003-2697(91)90155-m. [DOI] [PubMed] [Google Scholar]
- 10.Choo LP, Wetzel DL, Halliday WC, Jackson M, LeVine SM, Mantsch HH. In situ characterization of beta-amyloid in Alzheimer’s diseased tissue by synchrotron Fourier transform infrared microspectroscopy. Biophys J. 1996;71:1672–9. doi: 10.1016/S0006-3495(96)79411-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rak M, Del Bigio MR, Mai S, Westaway D, Gough K. Dense-core and diffuse Abeta plaques in TgCRND8 mice studied with synchrotron FTIR microspectroscopy. Biopolymers. 2007;87:207–17. doi: 10.1002/bip.20820. [DOI] [PubMed] [Google Scholar]
- 12.Leskovjan AC, Lanzirotti A, Miller LM. Amyloid plaques in PSAPP mice bind less metal than plaques in human Alzheimer’s disease. Neuroimage. 2009;47:1215–20. doi: 10.1016/j.neuroimage.2009.05.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Szczerbowska-Boruchowska M, Dumas P, Kastyak MZ, Chwiej J, Lankosz M, Adamek D, Krygowska-Wajs A. Biomolecular investigation of human substantia nigra in Parkinson’s disease by synchrotron radiation Fourier transform infrared microspectroscopy. Arch Biochem Biophys. 2007;459:241–8. doi: 10.1016/j.abb.2006.12.027. [DOI] [PubMed] [Google Scholar]
- 14.Bonda M, Perrin V, Vileno B, Runne H, Kretlow A, Forro L, Luthi-Carter R, Miller LM, Jeney S. Synchrotron infrared microspectroscopy detecting the evolution of Huntington’s disease neuropathology and suggesting unique correlates of dysfunction in white versus gray brain matter. Analytical Chemistry. 2011;83:7712–20. doi: 10.1021/ac201102p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bourassa MW. Chemistry. Stony Brook University; Stony Brook: 2012. The Involvement of Metal Ions in Copper-Zinc Superoxide Dismutase Related Amyotrophic Lateral Sclerosis. [Google Scholar]
- 16.Kneipp J, Miller LM, Spassov S, Sokolowski F, Lasch P, Beekes M, Naumann D. Scrapie-infected cells, isolated prions, and recombinant prion protein: a comparative study. Biopolymers. 2004;74:163–7. doi: 10.1002/bip.20064. [DOI] [PubMed] [Google Scholar]
- 17.Kretlow A, Wang Q, Beekes M, Naumann D, Miller LM. Changes in protein structure and distribution observed at pre-clinical stages of scrapie pathogenesis. Biochim Biophys Acta. 2008;1782:559–65. doi: 10.1016/j.bbadis.2008.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kretlow A, Wang Q, Kneipp J, Lasch P, Beekes M, Miller L, Naumann D. FTIR-microspectroscopy of prion-infected nervous tissue. Biochim Biophys Acta. 2006;1758:948–59. doi: 10.1016/j.bbamem.2006.05.026. [DOI] [PubMed] [Google Scholar]
- 19.Susi H, Byler DM. Protein structure by Fourier transform infrared spectroscopy: second derivative spectra. Biochem Biophys Res Commun. 1983;115:391–7. doi: 10.1016/0006-291x(83)91016-1. [DOI] [PubMed] [Google Scholar]
- 20.Arrondo JL, Muga A, Castresana J, Goni FM. Quantitative studies of the structure of proteins in solution by Fourier-transform infrared spectroscopy. Prog Biophys Mol Biol. 1993;59:23–56. doi: 10.1016/0079-6107(93)90006-6. [DOI] [PubMed] [Google Scholar]
- 21.Hering JA, Innocent PR, Haris PI. Neuro-fuzzy structural classification of proteins for improved protein secondary structure prediction. Proteomics. 2003;3:1464–75. doi: 10.1002/pmic.200300457. [DOI] [PubMed] [Google Scholar]
- 22.Hering JA, Innocent PR, Haris PI. Towards developing a protein infrared spectra databank (PISD) for proteomics research. Proteomics. 2004;4:2310–9. doi: 10.1002/pmic.200300808. [DOI] [PubMed] [Google Scholar]
- 23.Severcan M, Haris PI, Severcan F. Using artificially generated spectral data to improve protein secondary structure prediction from Fourier transform infrared spectra of proteins. Analytical Biochemistry. 2004;332:238–244. doi: 10.1016/j.ab.2004.06.030. [DOI] [PubMed] [Google Scholar]
- 24.Juszczyk P, Kolodziejczyk AS, Grzonka Z. FTIR spectroscopic studies on aggregation process of the beta-amyloid 11–28 fragment and its variants. J Pept Sci. 2009;15:23–9. doi: 10.1002/psc.1085. [DOI] [PubMed] [Google Scholar]
- 25.Ii K. The role of beta-amyloid in the development of Alzheimer’s disease. Drugs Aging. 1995;7:97–109. doi: 10.2165/00002512-199507020-00004. [DOI] [PubMed] [Google Scholar]
- 26.Barrow CJ, Yasuda A, Kenny PT, Zagorski MG. Solution conformations and aggregational properties of synthetic amyloid beta-peptides of Alzheimer’s disease. Analysis of circular dichroism spectra. J Mol Biol. 1992;225:1075–93. doi: 10.1016/0022-2836(92)90106-t. [DOI] [PubMed] [Google Scholar]
- 27.Barrow CJ, Zagorski MG. Solution structures of beta peptide and its constituent fragments: relation to amyloid deposition. Science. 1991;253:179–82. doi: 10.1126/science.1853202. [DOI] [PubMed] [Google Scholar]
- 28.Zhang S, Iwata K, Lachenmann MJ, Peng JW, Li S, Stimson ER, Lu Y, Felix AM, Maggio JE, Lee JP. The Alzheimer’s peptide a beta adopts a collapsed coil structure in water. J Struct Biol. 2000;130:130–41. doi: 10.1006/jsbi.2000.4288. [DOI] [PubMed] [Google Scholar]
- 29.Zhang S, Rich A. Direct conversion of an oligopeptide from a beta-sheet to an alpha- helix: a model for amyloid formation. Proc Natl Acad Sci U S A. 1997;94:23–8. doi: 10.1073/pnas.94.1.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Petty SA, Decatur SM. Experimental evidence for the reorganization of beta-strands within aggregates of the Abeta(16–22) peptide. J Am Chem Soc. 2005;127:13488–9. doi: 10.1021/ja054663y. [DOI] [PubMed] [Google Scholar]
- 31.Cerf E, Sarroukh R, Tamamizu-Kato S, Breydo L, Derclaye S, Dufrene YF, Narayanaswami V, Goormaghtigh E, Ruysschaert JM, Raussens V. Antiparallel beta-sheet: a signature structure of the oligomeric amyloid beta-peptide. Biochem J. 2009;421:415–23. doi: 10.1042/BJ20090379. [DOI] [PubMed] [Google Scholar]
- 32.Komatsu H, Liu L, Murray IV, Axelsen PH. A mechanistic link between oxidative stress and membrane mediated amyloidogenesis revealed by infrared spectroscopy. Biochim Biophys Acta. 2007;1768:1913–22. doi: 10.1016/j.bbamem.2007.05.026. [DOI] [PubMed] [Google Scholar]
- 33.Koppaka V, Axelsen PH. Accelerated accumulation of amyloid beta proteins on oxidatively damaged lipid membranes. Biochemistry. 2000;39:10011–6. doi: 10.1021/bi000619d. [DOI] [PubMed] [Google Scholar]
- 34.Berriman J, Serpell LC, Oberg KA, Fink AL, Goedert M, Crowther RA. Tau filaments from human brain and from in vitro assembly of recombinant protein show cross-beta structure. Proc Natl Acad Sci U S A. 2003;100:9034–8. doi: 10.1073/pnas.1530287100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Barghorn S, Davies P, Mandelkow E. Tau paired helical filaments from Alzheimer’s disease brain and assembled in vitro are based on beta-structure in the core domain. Biochemistry. 2004;43:1694–703. doi: 10.1021/bi0357006. [DOI] [PubMed] [Google Scholar]
- 36.Schmitt J, Beekes M, Brauer A, Udelhoven T, Lasch P, Naumann D. Identification of scrapie infection from blood serum by Fourier transform infrared spectroscopy. Anal Chem. 2002;74:3865–8. doi: 10.1021/ac015688s. [DOI] [PubMed] [Google Scholar]
- 37.Lasch P, Schmitt J, Beekes M, Udelhoven T, Eiden M, Fabian H, Petrich W, Naumann D. Antemortem identification of bovine spongiform encephalopathy from serum using infrared spectroscopy. Anal Chem. 2003;75:6673–8. doi: 10.1021/ac030259a. [DOI] [PubMed] [Google Scholar]
- 38.Lasch P, Beekes M, Schmitt J, Naumann D. Detection of preclinical scrapie from serum by infrared spectroscopy and chemometrics. Anal Bioanal Chem. 2007;387:1791–800. doi: 10.1007/s00216-006-0764-z. [DOI] [PubMed] [Google Scholar]
- 39.Thomzig A, Spassov S, Friedrich M, Naumann D, Beekes M. Discriminating scrapie and bovine spongiform encephalopathy isolates by infrared spectroscopy of pathological prion protein. J Biol Chem. 2004;279:33847–54. doi: 10.1074/jbc.M403730200. [DOI] [PubMed] [Google Scholar]
- 40.Conway KA, Harper JD, Lansbury PT., Jr Fibrils formed in vitro from alpha-synuclein and two mutant forms linked to Parkinson’s disease are typical amyloid. Biochemistry. 2000;39:2552–63. doi: 10.1021/bi991447r. [DOI] [PubMed] [Google Scholar]
- 41.Ulrih NP, Barry CH, Fink AL. Impact of Tyr to Ala mutations on alpha-synuclein fibrillation and structural properties. Biochim Biophys Acta. 2008;1782:581–5. doi: 10.1016/j.bbadis.2008.07.004. [DOI] [PubMed] [Google Scholar]
- 42.Yamin G, Munishkina LA, Karymov MA, Lyubchenko YL, Uversky VN, Fink AL. Forcing nonamyloidogenic beta-synuclein to fibrillate. Biochemistry. 2005;44:9096–107. doi: 10.1021/bi048778a. [DOI] [PubMed] [Google Scholar]
- 43.Uversky VN, Li J, Souillac P, Millett IS, Doniach S, Jakes R, Goedert M, Fink AL. Biophysical properties of the synucleins and their propensities to fibrillate: inhibition of alpha-synuclein assembly by beta- and gamma-synucleins. J Biol Chem. 2002;277:11970–8. doi: 10.1074/jbc.M109541200. [DOI] [PubMed] [Google Scholar]
- 44.Poirier MA, Li H, Macosko J, Cai S, Amzel M, Ross CA. Huntingtin spheroids and protofibrils as precursors in polyglutamine fibrilization. J Biol Chem. 2002;277:41032–7. doi: 10.1074/jbc.M205809200. [DOI] [PubMed] [Google Scholar]
- 45.Duncan W, Williams GP. Infrared Synchrotron Radiation from Electron Storage Rings. Applied Optics. 1983;22:2914–23. doi: 10.1364/ao.22.002914. [DOI] [PubMed] [Google Scholar]
- 46.Carr GL, Reffner JA, Williams GP. Performance of an infrared microspectrometer at the NSLS. Rev Sci Instr. 1995;66:1490–1492. [Google Scholar]
- 47.Reffner JA, Martoglio PA, Williams GP. Fourier Transform Infrared Microscopical Analysis with Synchrotron Radiation: The Microscope Optics and System Performance. Rev Sci Instr. 1995;66:1298. [Google Scholar]
- 48.Strodel B, Lee JW, Whittleston CS, Wales DJ. Transmembrane structures for Alzheimer’s Abeta(1–42) oligomers. Journal of the American Chemical Society. 2010;132:13300–12. doi: 10.1021/ja103725c. [DOI] [PubMed] [Google Scholar]
- 49.Kim HY, Cho MK, Kumar A, Maier E, Siebenhaar C, Becker S, Fernandez CO, Lashuel HA, Benz R, Lange A, Zweckstetter M. Structural properties of pore-forming oligomers of alpha-synuclein. Journal of the American Chemical Society. 2009;131:17482–9. doi: 10.1021/ja9077599. [DOI] [PubMed] [Google Scholar]
- 50.Peralvarez-Marin A, Barth A, Graslund A. Time-resolved infrared spectroscopy of pH-induced aggregation of the Alzheimer Abeta(1–28) peptide. Journal of molecular biology. 2008;379:589–96. doi: 10.1016/j.jmb.2008.04.014. [DOI] [PubMed] [Google Scholar]
- 51.Carr GL, Chubar O, Dumas P. Multichannel detection with a synchrotron light source: design and potential. In: Bhargava R, Levin IW, editors. Spectrochemical analysis using infrared detectors. Blackwell Publishing; 2006. pp. 56–84. [Google Scholar]
- 52.Nasse MJ, Walsh MJ, Mattson EC, Reininger R, Kajdacsy-Balla A, Macias V, Bhargava R, Hirschmugl CJ. High-resolution Fourier-transform infrared chemical imaging with multiple synchrotron beams. Nature Methods. 2011;8:413–U58. doi: 10.1038/nmeth.1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nasse MJ, Ratti S, Giordano M, Hirschmugl CJ. Demountable Liquid/Flow Cell for in Vivo Infrared Microspectroscopy of Biological Specimens. Applied Spectroscopy. 2009;63:1181–1186. doi: 10.1366/000370209789553101. [DOI] [PubMed] [Google Scholar]
- 54.Birarda G, Grenci G, Businaro L, Marmiroli B, Pacor S, Piccirilli F, Vaccari L. Infrared microspectroscopy of biochemical response of living cells in microfabricated devices. Vibrational Spectroscopy. 2010;53:6–11. [Google Scholar]
- 55.Holman HYN, Miles R, Hao Z, Wozei E, Anderson LM, Yang H. Real-Time Chemical Imaging of Bacterial Activity in Biofilms Using Open-Channel Microfluidics and Synchrotron FTIR Spectromicroscopy. Analytical Chemistry. 2009;81:8564–8570. doi: 10.1021/ac9015424. [DOI] [PubMed] [Google Scholar]
- 56.Tobin M, Puskar L, Barber R, Harvey E, Heraud P, Wood B, Bambery K, Dillon C, Munro K. FTIR spectroscopy of single live cells in aqueous media by synchrotron IR microscopy using microfabricated sample holders. Vibrational Spectroscopy. 2010;53:34–38. [Google Scholar]
- 57.Kuimova MK, Chan KL, Kazarian SG. Chemical imaging of live cancer cells in the natural aqueous environment. Applied Spectroscopy. 2009;63:164–71. doi: 10.1366/000370209787391969. [DOI] [PubMed] [Google Scholar]
- 58.Yang WY, Xiao XL, Tan J, Cai QY. In situ evaluation of breast cancer cell growth with 3D ATR-FTIR spectroscopy. Vibrational Spectroscopy. 2009;49:64–67. [Google Scholar]
- 59.Mariangela CG, Seydou Y, Diego S, Sabine C, Augusto M, Cyril P. Experimental ATR device for real-time FTIR imaging of living cells using brilliant synchrotron radiation sources. Biotechnology advances. 2011 doi: 10.1016/j.biotechadv.2011.11.009. [DOI] [PubMed] [Google Scholar]
- 60.Abel O. ALSOD (3.0) 2012. ALS Online Genetic Database. [Google Scholar]
- 61.Lill CM, Abel O, Bertram L, Al-Chalabi A. Keeping up with genetic discoveries in amyotrophic lateral sclerosis: The ALSoD and ALSGene databases. Amyotrophic Lateral Sclerosis. 2011;12:238–249. doi: 10.3109/17482968.2011.584629. [DOI] [PubMed] [Google Scholar]
- 62.Lynch SM, Boswell SA, Colón W. Kinetic Stability of Cu/Zn Superoxide Dismutase Is Dependent on Its Metal Ligands:Implications for ALS†. Biochemistry. 2004;43:16525–16531. doi: 10.1021/bi048831v. [DOI] [PubMed] [Google Scholar]
- 63.Shaw BF, Lelie HL, Durazo A, Nersissian AM, Xu G, Chan PK, Gralla EB, Tiwari A, Hayward LJ, Borchelt DR. Detergent-insoluble aggregates associated with amyotrophic lateral sclerosis in transgenic mice contain primarily full-length, unmodified superoxide dismutase-1. Journal of Biological Chemistry. 2008;283:8340–8350. doi: 10.1074/jbc.M707751200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lelie HL, Liba A, Bourassa MW, Chattopadhyay M, Chan PK, Gralla EB, Miller LM, Borchelt DR, Valentine JS, Whitelegge JP. Copper and Zinc Metallation Status of Copper-Zinc Superoxide Dismutase from Amyotrophic Lateral Sclerosis Transgenic Mice. Journal of Biological Chemistry. 2011;286:2795–2806. doi: 10.1074/jbc.M110.186999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Oztug Durer ZA, Cohlberg JA, Dinh P, Padua S, Ehrenclou K, Downes S, Tan JK, Nakano Y, Bowman CJ, Hoskins JL, Kwon C, Mason AZ, Rodriguez JA, Doucette PA, Shaw BF, Valentine JS. Loss of Metal Ions, Disulfide Reduction and Mutations Related to Familial ALS Promote Formation of Amyloid-Like Aggregates from Superoxide Dismutase. PLoS ONE. 2009;4:e5004. doi: 10.1371/journal.pone.0005004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Chattopadhyay M, Durazo A, Sohn S, Strong C, Gralla E, Whitelegge J, Valentine J. Initiation and elongation in fibrillation of ALS-linked superoxide dismutase. Proceedings of the National Academy of Sciences. 2008;105:18663. doi: 10.1073/pnas.0807058105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Prudencio M, Hart PJ, Borchelt DR, Andersen PM. Variation in aggregation propensities among ALS-associated variants of SOD1: Correlation to human disease. Human Molecular Genetics. 2009;18:3217–3226. doi: 10.1093/hmg/ddp260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Prudencio M, Borchelt DR. Superoxide dismutase 1 encoding mutations linked to ALS adopts a spectrum of misfolded states. Molecular Neurodegeneration. 2011;6:1–19. doi: 10.1186/1750-1326-6-77. [DOI] [PMC free article] [PubMed] [Google Scholar]