

Abstract
A chemoenzymatic glycosylation remodeling method for the synthesis of selectively fluorinated glycoproteins is described. The method consists of chemical synthesis of a fluoroglycan oxazoline and its use as donor substrate for endoglycosidase (ENGase)-catalyzed transglycosylation to a GlcNAc-protein to form a homogeneous fluoroglycoprotein. The approach was exemplified by the synthesis of fluorinated glycoforms of ribonuclease B (RNase B). an interesting finding was that fluorination at the C-6 of the 6-branched mannose moiety in the Man3GlcNAc core resulted in significantly enhanced reactivity of the substrate in enzymatic transglycosylation. A structural analysis suggests that the enhancement in reactivity may come from favorable hydrophobic interactions between the fluorine and a tyrosine residue in the catalytic site of the enzyme (Endo-A). SPR analysis of the binding of the fluorinated glycoproteins with lectin concanavalin A (con A) revealed the importance of the 6-hydroxyl group on the α-1,6-branched mannose moiety in con A recognition. The present study establishes a facile method for preparation of selectively fluorinated glycoproteins that can serve as valuable probes for elucidating specific carbohydrate-protein interactions.
Keywords: fluorine, fluorosugar, glycoprotein, lectin, endoglycosidase, transglycosylation
1. Introduction
Glycans are involved in many important biological recognition processes: intracellular signaling, cell adhesion, host-pathogen interaction, and immune responses, to name a few 1–5. Such molecular recognition processes are, in many cases, mediated by specific interactions between glycans and glycan-binding proteins (GBPs) such as lectins, glycan-specific antibodies, and enzymes. A detailed understanding of the molecular mechanism behind these binding events requires well-defined oligosaccharides and glycoconjugates as molecular probes 6–8. Deoxyfluoro sugars represent an important class of selectively modified carbohydrates that have been successfully used for mapping antigen-antibody interactions 9, for probing lectin binding 10–12, and for deciphering enzyme mechanisms 13–15. Fluorine is highly electronegative and inherently hydrophobic. It is isosteric to a hydroxyl group, as the bond length and the van der Waals radius (1.39 and 1.37 Å, respectively) of a typical C-F bond are well comparable to those of a C-OH bond (1.43 and 1.40 Å, respectively) 16. Thus, replacement of the OH in a sugar moiety with a fluorine atom would not have significant steric effects. Fluorine can also serve as an acceptor in hydrogen-bond 17. Moreover, 19F NMR is highly sensitive to changes of environment and provides clear signals without background in biological system. These notable properties make fluorine a powerful probe for imaging and for elucidating carbohydrate-protein and protein-protein interactions in vitro and in vivo 18–22.
Despite the great potential of fluorinated compounds for imaging and biochemical studies, few methods are available for site-controlled incorporation of fluorine into proteins or glycoproteins 19–25. Recently, Davis and co-workers has reported an elegant method for site-selective incorporation of fluorosugars into proteins to make fluoroglycoproteins 25. The method applies site-directed mutagenesis to introduce L-homopropargylglycine at a pre-determined site into a protein, followed by conjugation with a fluorine-labeled glycosyl azide via Copper(I)-catalyzed alkyne-azide cycloaddition reaction to yield a homogeneous fluoroglycoprotein. The method should be generally useful for labeling proteins with various fluorosugars, albeit with the drawback of introduction of an unnatural triazole linkage into the protein. In this paper, we report a chemoenzymatic method for the synthesis of selectively fluorinated homogeneous glycoproteins, which takes advantage of the transglycosylation activity of endo-β-N-acetylglucosaminidase (ENGase) that was originally applied for glycopeptide synthesis 26,27 and more recently explored for glycoprotein synthesis and glycosylation remodeling with sugar oxazolines as the activated donor substrates 28–46. Using ribonuclease B (RNase B) as a model system, we found that synthetic fluorinated glycan oxazolines were able to serve as excellent substrates for ENGase-catalyzed transglycosylation to afford selectively fluorinated glycoforms of RNase B (Fig. 1). We also observed that a selectively fluorinated N-glycan oxazoline demonstrated significantly enhanced reactivity in enzymatic transglycosylation in comparison with the non-fluorinated glycan oxazoline. Notably, this method allows site-specific fluorine-labeling of the N-glycans in a glycoprotein while keeping the native N-glycan core structure intact. The binding of the synthetic fluorinated glycoforms of RNase B with concanavalin A (con A) was studied by SPR technology.
Figure 1.
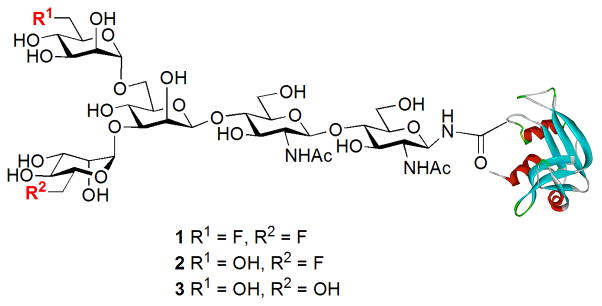
Structures of the synthetic fluoroglycoproteins.
2. Results and Discussion
2.1. Synthesis of Selectively Fluorinated Tetrasaccharide Oxazolines
The chemoenzymatic synthesis of the designed fluorinated glycoproteins (Fig. 1) via an ENGase-catalyzed transglycosylation requires the preparation of a fluorinated N-glycan oxazoline as the donor substrate and a GlcNAc-protein as the acceptor. A chemical synthesis of the di-fluoro-Man3GlcNAc oxazoline (10) was shown in Scheme 1. Briefly, Cu (OTf)2-catalyzed region-selective reductive ring opening of the 4′,6′-O-benzylidene group of disaccharide 4, which was synthesized according to the previously reported procedure 29, gave the diol (5) in 85% yield. TMSOTf-catalyzed double glycosylation of diol 5 with the 6-deoxy-6-fluoro-mannosyl trichloroacetimidate (6) 47 led to the formation of the fully protected tetrasaccharide derivative (7) in 86% yield, in which the two mannosyl moieties were introduced in α-glycosidic linkages. The α-selectivity of the newly formed glycosidic bonds was confirmed by the relatively large C-1/H-1 coupling constant 48 for the α-mannosidic linkages revealed by the 1H/13C HSQC NMR of 7 (GlcN, 1JC-1/H-1 = 160 Hz, β-linkage; Man1, 1JC-1/H-1 = 161 Hz, β-linkage; Man2, 1JC-1/H-1 = 175 Hz, assigned as α-linkage; and Man2′, 1JC-1/H-1 = 174 Hz, assigned as α-linkage). Removal of the O-acyl and N-phthaloyl protecting groups by treatment of 7 with hydrazine hydrate, followed by selective N-acetylation at the unmasked amino group with Ac2O in an aqueous MeOH-NaHCO3 solution gave the partially deprotected tetrasaccharide (8). Hydrogenolysis of 8 provided the free tetrasaccharide (9) in quantitative yield. Finally, treatment of 9 with excess 2-chloro-1,3-dimethylimidazolinium chloride under aqueous conditions in the presence of triethylamine afforded the difluoro-tetrasaccharide oxazoline (10) in 88% yield after purification by size-exclusion chromatography with a Sephadex G-10 column (Scheme 1). The difluoro-Man3GlcNAc oxazoline (10) was characterized by MS and NMR (1H, 19F, and 13C) analysis (see experimental sections for details).
Scheme 1. Synthesis of difluorinated tetrasaccharide oxazoline.
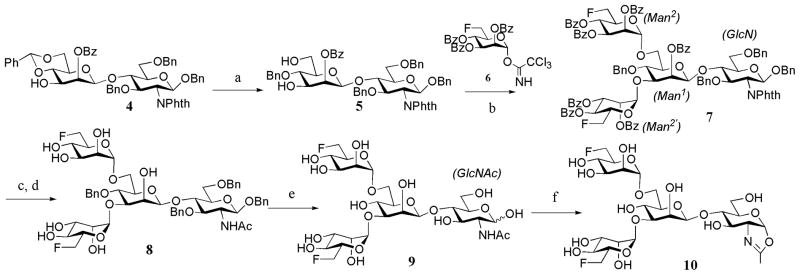
a) BH3.THF, Cu(OTf)2, CH2Cl2, 85%; b) TMSOTf, MS4A, 86%; c) EtOH, H2NNH2.H2O, H2O, 80 °C; d) 0.1 M NaHCO3, Ac2O, MeOH, 60% (2 steps); e) H2, Pd-C, MeOH, quant., f) 2-chloro-1, 3-dimethylimidazolinium chloride, Et3N, 4°C, 88%.
For the synthesis of the monofluoro-Man3GlcNAc oxazoline (17) carrying a single fluorine atom at the 6-position of the α-1,3-branched mannose, the disaccharide (4) was first glycosylated with the 6-deoxy-6-fluoro-mannosyl tricholoroacetimidate (6) to give the trisaccharide (11) (Scheme 2). The relatively large H-1/C-1 coupling constant found for the newly added mannose residue (1JC-1/H-1 = 172 Hz) indicated that the newly formed glycosidic bond was in α-linkage. Selective reductive ring opening of the 4,6-benzylidene group in 11 gave intermediate 12 with a free hydroxyl group at the C-6 position. Glycosylation at the free hydroxyl group of 12 using the known mannosyl trichloroacetimidate 13 47 furnished the protected tetrasaccharide 14. Removal of the N-phthalimido group of 11 using hydrazine hydrate, with concurrent de-O-acylation, followed by selective N-acetylation with Ac2O in an aqueous NaHCO3 solution, afforded the tetrasaccharide (15) in 60% yield for two steps. Hydrogenolysis of 15 gave free tetrasaccharide 16, which was finally converted into the corresponding monofluoro-Man3GlcNAc oxazoline (17) in a single step by treatment with excess 2-chloro-1, 3-dimethylimidazolinium chloride in an aqueous solution (Scheme 2).
Scheme 2. Synthesis of mono-fluorinated tetrasaccharide oxazoline.

Reagents and yields: a) TMSOTf, MS4A, CH2Cl2, 70%; b) Cu(OTf)2, BH3 THF, 60%; c) TMSOTf, MS4A, CH2Cl2, 73%; d) EtOH, H2NNH2.H2O, H2O, 80 °C, 60%; e) 0.1 M NaHCO3, Ac2O, MeOH, 66% (2 steps); f) H2, Pd-C, MeOH, 91%; g) 2-chloro-1,3-dimethylimidazolinium chloride, Et3N, 4°C, quant.
2.2. Synthesis of selectively fluorinated glycoproteins by enzymatic transglycosylation
Using bovine ribonuclease B (RNase B), a small natural glycoprotein that has 124 amino acid residues and carries a heterogeneous high-mannose type N-glycan at the Asn-34 site, as a model system, we examined the feasibility of the synthetic fluoroglycan oxazolines (10 and 17) as substrates for enzymatic transglycosylation for making fluoroglycoproteins. To make the GlcNAc-protein as the acceptor, RNase B was deglycosylated with Endo-H to give the glycosylation acceptor, GlcNAc-RNase (18), following our previously reported procedures 30,33. The transglycosylation was performed by incubation of fluoroglycan oxazoline (10) and GlcNAc-RNase (18) (donor/acceptor, 5:1, molar ratio) in a phosphate buffer (0.1 M, pH 7) at 30 °C in the presence of catalytic amount of the endoglycosidase from Arthrobacter protophormiae (Endo-A). The reaction was monitored by RP-HPLC. It was found that the difluoroglycan oxazoline (10) could serve as an excellent donor substrate to give a transglycosylation product that appeared slightly earlier than the acceptor (18) under the RP-HPLC conditions (see Figure S1 in the supporting information). At 1 h, more than 85% of the GlcNAc-RNase (18) was converted to the product, glycoprotein 1. After 2 h, a complete conversion was achieved, and the transglycosylation product was readily purified by HPLC in essentially quantitative yield. The identity of the glycoprotein (1) was confirmed by ESI-MS: calculated, M = 14579.24; found: 1823.30 [M + 8 H] 8+, 1620.95 [M + 9 H] 9+, 1459.07 [ M + 10 H] 10+, 1326.61 [ M + 11 H] 11+, 1216.18 [ M + 12 H] 12+, 1122.71 [M + 13 H] 13+, 1042.56[M + 14 H] 14+; deconvolution data, M = 14582 (Figure S1, supporting information). It was found that the monofluoro-glycan oxazoline (17) also acted as an efficient substrate for Endo-A catalyzed transglycosylation with GlcNAc-RNase (18), providing the corresponding monofluoro-glycoprotein (2) in 92% yield. These two fluoroglycoproteins represnt the first examples synthesized by the chemoenzymatic method, in which the fluorine atom is site-specifically incorporated on the natural N-glycan core. Similarly, the Endo-A catalyzed reaction of the Man3GlcNAc oxazoline (19) with GlcNAc-RNase (18) under the same reaction conditions gave the corresponding glycoprotein product (3), which was isolated in 88% yield (Scheme 3). Again, the identity of the glycoprotein products (2 and 3) was confirmed by ESI-MS analysis (see Figure S2 for the HPLC and ESI-MS profiles of glycoproteins 2 and 3; supporting information). These experimental results indicate that the ENGase-catalyzed transglycosylation can be efficiently used for transferring pre-assembled fluoroglycans to a GlcNAc-containing protein to form homogeneous fluoroglycoproteins carrying a native N-glycan core structure.
Scheme 3. Transglycosylation of GlcNAc-RNase with fluorinasted glycan oxazolines.

a) Endo-A, phosphate buffer (0.2 M, pH 7.0), quant., b) Endo-A, phosphate buffer (0.2 M, pH 7.0), 92%; c) Endo-A, phosphate buffer (0.2 M, pH 7.0), 88%.
2.3. Comparison of the reactivity of the fluorinated and non-fluorinated glycan oxazolines in enzymatic transglycosylation
During our initial monitoring of the enzymatic transglycosylation reactions, we observed that the difluoro-glycan oxazoline (10) seemed to react much faster than the monofluoro (17) and the non-fluorinated Man3GlcNAc oxazoline (19). This initial observation prompted us to probe the fluorination effect on the enzymatic reaction in more details with a competitive assay employing the Fmoc-protected GlcNAc-Asn (20) 49 as the acceptor (Scheme 4). The incorporation of Fmoc on the Asn facilitated HPLC monitoring by UV absorbance and provided the necessary hydrophobicity for RP-HPLC separation of the products. The reaction was performed using a mixture of equimolar concentrations of the glycan oxazolines (10 and 17, or 10 and 19) and an excess of the acceptor, GlcNAc-Asn-Fmoc (20). When incubated with Endo-A, the transglycosylation products were formed, which appeared as distinct peaks under an appropriate RP-HPLC condition, allowing quantification by integration of the peaks. It was found that the initial rate of the Endo-A catalyzed transglycosylation with the difluoro-Man3GlcNAc oxazoline 10, which was measured as 6.5 nmol/min/μg enzyme, was at least 3-fold higher than that of the monofluoro-Man3GlcNAc oxazoline (17) (1.87 nmol/min/μg enzyme), and about 2.3 fold higher than that of the non-fluorinated Man3GlcNAc oxazoline (19) (2.95 nmol/min/μg enzyme) (Figure 2). The similar reactivity of 17 and 19 suggests that the substitution of the 6-OH of the α-1,3-branched mannose by a fluorine atom does not have much effect, while the substitution at the 6-OH of the α-1,6-branched mannose have a profound positive contribution to the enzymatic reactivity of the glycan oxazoline.
Scheme 4.
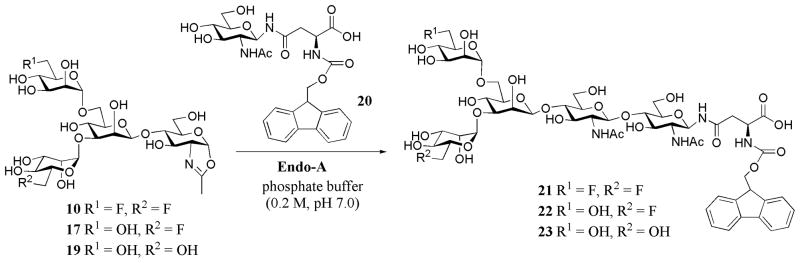
Competitive enzymatic transglycosylation with fluorinated and non-fluorinated glycan oxazolines.
Figure 2. Competitive enzymatic transglycosylation with the fluoroglycan oxazolines (10 and 17) and the non-fluorinated glycan oxazoline (19).
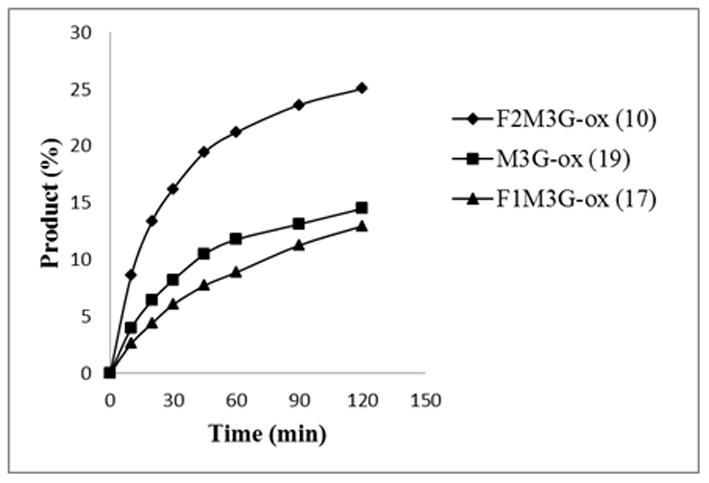
A mixture of the glycan oxazolines and the acceptor Fmoc-Asn(GlcNAc)-OH (20) in a phosphate buffer was incubated with Endo-A, and the respective transglycosylation products were separated on HPLC and quantified by integration of the peak areas to give the transglycosylation yields.
Fluorine has the highest electronic negativity and is inherently hydrophobic. The hydrophobicity has been exploited in designing higher affinity enzyme inhibitors 50. There are also examples showing that introduction of a fluorine atom at the C-2 or C-5 position of a glycoside significantly decreases the reactivity of the glycoside when it serves as a substrate for enzymatic hydrolysis or glycosylation, due to the electron-withdrawing effect of the fluorine13,14,51. To understand the “unexpected” enhancement in reactivity of the difluoro-glycan oxazoline (10) in the Endo-A catalyzed transglycosylation, we examined the recently reported structure of Endo-A 52 in complex with synthetic Man3GlcNAc-thiazoline 53, which can be viewed as a stable Man3GlcNAc-oxazoline mimic. Analysis of the binding pocket with a bound Man3GlcNAc-thiazoline revealed the positioning of the C-6 hydroxyl group of the α-1,6-branched mannose to be in close proximity with the tyrosine 131 residue in a relatively hydrophobic cleft. Thus, modeling of the difluoro-Man3GlcNAc-oxazoline (10) into the Endo-A binding pocket (Figure S3, supporting information) revealed that the fluorine introduced at the C-6 of the 6-branched mannose moiety was proximal to the Tyr-131 residue, which might result in enhanced hydrophobic interactions between the fluorine atom and the aromatic ring of the Tyr-131 in Endo-A, thus leading to much enhanced affinity of the substrate to the enzyme. This may explain the significantly increased reactivity of oxazoline 10 in the enzymatic reaction in comparison with the non-fluorinated sugar oxazoline (19). On the other hand, the 6-fluorine group on the α-1,3-branched mannose (F1Man3GlcNAc oxazoline, 17) does not seem to have interactions with any hydrophobic residues in the enzyme pocket (Figure S3, supporting information). This may partially explain our experimental observation that substitution of the 6-OH on the α-1,3-branched mannose did not have significant effect on the reactivity of the monofluoroglycan oxazoline (17). In fact, monofluoro-Man3GlcNAc oxazoline 17 showed a slightly decreased activity in comparison with the non-fluorinated Man3GlcNAc-oxazoline (19), probably due to a remote electron-withdrawing effect of the fluorination. However, this electron-withdrawing effect, if any, could be relatively weak as the fluorination site is far from the oxazoline reactive center.
2.4. SPR analysis of the binding between the fluoroglycoproteins and concanavalin A
The nature and binding specificity of the interactions between lectin concanavalin A (con A) and various carbohydrate ligands have been well documented 54–56. Con A has preference to mannose and GlcNAc monosaccharides and has high affinity for high-mannose or hybrid N-glycans and related glycopeptides/glycoproteins. Using SPR technology, we investigated the interaction between the synthetic fluoroglycoproteins (1 and 2) and con A. The affinity of con A for three non-fluorinated glycoforms of RNase was measured, including the native RNase B carrying Man5–9 N-glycans, the synthetic Man3GlcNAc2 glycoform (3), and the deglycosylated glycoform (18) carrying only the innermost GlcNAc residue. The lectin was immobilized on the surface of the sensor chip and the interactions were analyzed by injecting the respective glycoproteins at serial dilutions. The binding profiles, as shown in Fig. 3, were subject to data analysis by Biacore evaluation software to obtain the dissociation constant (KD). It was found that the native RNase B had a relatively high affinity for con A with a KD of 1.2 μM. The monofluoro-Man3GlcNAc2 glycoform (2) and the glycoform carrying the Man3GlcNAc2 glycan core (3) had comparable affinity for con A with a KD of 2.4 and 2.5 μM, respectively. Interestingly, the difluoro-Man3GlcNAc2 glycoform (1) showed only very weak affinity for conA, similar to the deglycosylated RNase (18), for which the dissociation constant was not deduced (Fig. 3).
Figure 3. Typical SPR sensorgrams of the binding of con A with respective glycoforms of RNase.

Lectin con A was immobilized on the chip and the binding was analyzed by injecting the respective RNase glycoforms at serial 2-fold dilutions starting at 8 μM.
Glycoproteins 1, 2, and 3 differ only in the extent and pattern of fluorination. The binding affinity of the monofluoro-Man3GlcNAc2 glycoform (2) for con A is almost the same as that of the non-fluorinated Man3GlcNAc2 glycoform (3), suggesting that the C-6 hydroxyl group at the α-1,3-branched mannose is not involved in con A recognition. However, the dramatically decreased affinity of the difluoro-Man3GlcNAc2 glycoform (1) for con A clearly indicates that the C-6 hydroxyl group of the α-1,6-branched mannose is essential for con A recognition. These results are consistent with previously reported structure-activity relationship studies using deoxysugar derivatives of the trimannoside 57. The study showed that 6-deoxygenation of the α-1,6-branched mannose in the trimannoside led to dramatic decrease in its affinity for con A, while the 6-deoxygenation of the α-1,3-branched mannose had only little effect on affinity 57. In addition, our binding results are also in agreement with the findings from a crystal structural study on the complex of con A with a trimannoside 56. The structure of the complex indicated that the α-1,6 branched mannose occupied the con A high-affinity monosaccharide binding site involving several hydrogen bonds with con A amino acid residues, where the C-6 hydroxyl group forms a hydrogen bond with an aspartic acid residue at 208 position (Asp-208). Thus, substitution of this key hydroxyl group by a fluorine atom, as in the case of the difluoro-Man3GlcNAc2 glycoform (1), would break this hydrogen bond, resulting in diminished binding affinity. Moreover, the significant decrease in affinity of difluoroglycoprotein 1 for con A (dropped to the level of the GlcNAc-RNase) also suggests that the C-6 hydroxyl group is likely to act as the H-donor in the H-bond formation, rather than an H-acceptor. Otherwise, the C-F can substitute the OH to play the role of a H-acceptor at least to some extent 12. As also revealed in the crystal structure of the con A and trimannoside complex, the C-6 hydroxyl group of the α-1,3 branched mannose was not in contact with other amino acid residues of con A, explaining the comparable affinity of the monofluoro-Man3GlcNAc2 glycoform (2) and the non-fluorinated Man3GlcNAc2 glycoform (3) for con A. Taken together, our experimental results suggest that selectively fluorinated glycoproteins can serve as a valuable tool to probe specific carbohydrate-protein interactions.
3. Conclusion
A chemoenzymatic glycosylation remodeling method for the synthesis of selectively fluorinated homogeneous glycoproteins is described. We have shown that, for the first time, fluoroglycan oxazolines can serve as excellent donor substrates for ENGase-catalyzed transglycosylation for making 19F-labeled homogeneous glycoproteins. An interesting observation is that certain selectively fluorinated glycan oxazolines can have significantly enhanced reactivity in comparison with the corresponding non-fluorinated glycan oxazoline in the Endo-A catalyzed transglycosylation, probably due to favorable hydrophobic interactions of the introduced fluorine atom(s) with aromatic residues in the enzyme. Our binding studies on the recognition of con A and the synthetic fluoroglycan glycoforms of RNase B suggest that selectively fluorinated glycoproteins can serve as valuable probes for deciphering specific carbohydrate-protein interactions. The described chemoenzymatic method should be useful for the synthesis of other fluorine-labeled glycoproteins for various applications.
4. Experimental
4.1. General Procedures
Thin-layer chromatography was performed on glass plates coated with silica gel (Merck 60F254) and stained by spraying sulfuric acid (10% in ethanol) and heating at 120 °C. NMR spectra were recorded on Bruker DRX 500 (500 MHz) and JEOL ECX 400 (400 MHz) spectrometers in CDCl3 or D2O as noted. Chemical shifts were reported in parts per million (ppm). ESI-MS spectra were measured on a Micromass ZQ-4000 single-quadrupole mass spectrometer and MALDI-TOF MS spectra were acquired on a Bruker Autoflex Speed MALDI-TOF MS instrument. Analytical reverse-phase HPLC was carried on a Waters 626 HPLC instrument equipped with a C18 column (Symmetry 300, 4.6 X 250 mm, 5.0 μm) at 40 °C eluting with a suitable gradient at a flow rate of 1 mL/min. Preparative RP-HPLC was carried out on a C18 column (Symmetry 300A, 19X 300 mm, 7 μm) eluting with a suitable gradient at a flow rate of 12 mL/min.
4.2. Synthesis of benzyl 2-O-benzoyl-4-O-benzyl-β-D-mannopyranosyl-(1→4)-3,6-di-O-benzyl-2-deoxy-2-phthalimido-β-D-glucopyranoside (5)
To a solution of compound 4 (50 mg, 53.5 μmol) in CH2Cl2 was added BH3·THF (1.0 M in THF, 270 μl, 0.27 mmol). The mixture was stirred at room temperature under argon for 5 min. Then Cu(OTf)2 (3.87 mg, 10.7 μmol) was added and the residue was stirred at room temperature (r.t.) under argon for 2 h. The reaction was quenched by addition of triethylamine (100 μl) and MeOH (50 μl) at 0° C. After concentration, the resulting syrup was subject to silica gel column chromatography (hexane/EtOAc, 3:1 then 1:1, v/v) to afford diol 5 as a white solid (43 mg, 85%) 1H NMR (500 MHz, CDCl3) δ 8.18-6.83 (m, 29 H, Ar), 5.54 (s, 1 H), 5.16 (d, 1 H, J = 8.5 Hz), 4.91-4.86 (m, 6 H), 4.77 (d, 1 H, J = 11.5 Hz), 4.64 (d, 1 H, J = 12 Hz), 4.57 (d, 1 H, J = 12.5 Hz), 4.39 (d, 1 H, J = 12.5 Hz), 4.30 (m,1H), 4.25-4.18 (m, 2H), 3.98-3.51 (m, 6H), 3.36 (s,1 H); 1H-13C HSQC NMR (1H at 500 MHz, 13C at 125 MHz, CDCl3) δ 8.18/130.01 (Ar), 7.75/133.53 (Ar), 7.64/133.50 (Ar), 7.53/128.42 (Ar), 7.44/128.29 (Ar), 7.19/127.54 (Ar), 7.15/127.87 (Ar), 6.95/127.04 (Ar), 6.90/127.83 (Ar), 6.85/127.21 (Ar), 5.54/72.20 (Man-2), 5.16/97.28 (GlcN-1, 1JC-1/H-1 = 163 Hz, β), 4.91/98.61(Man-1, 1JC-1/H-1 = 162 Hz, β), 4.91/73.70 (Bn-CH2), 4.90/70.91 (Bn-CH2), 4.90/74.29 (Bn-CH2), 4.89/74.85 (Bn-CH2), 4.77/74.85 (Bn-CH2), 4.64/73.70 (Bn-CH2), 4.57/70.91 (Bn-CH2), 4.39/74.293 (Bn-CH2), 4.30/55.73 (GlcNAc-2), 4.24/78.87 (GlcNAc-5), 4.21/76.37 (GlcNAc-3), 3.98/68.44 (GlcNAc-6a), 3.97/62.26 (Man-6a), 3.89/68.44 (GlcNAc-6b), 3.82/75.55 (Man-4), 3.81/73.38 (Man-3), 3.76/62.26 (Man-6b), 3.46/70.93 (GlcNAc-4), 3.36/75.55 (Man-5); ESI-MS: calcd for C47H49O11, M = 789.33; found (m/z), 790.36 [M + H]+.
4.3. Synthesis of benzyl 2,3,4-tri-O-benzoyl-6-deoxy-6-fluoro-α-D-mannopyranosyl-(1→3)-[2,3,4-tri-O-benzoyl-6-deoxy-6-fluoro-α-D-mannopyranosyl-(1→6)]-2-O-benzoyl-4-O-benzyl-β-D-mannopyranosyl-(1→4)-3,6-di-O-benzyl-2-deoxy-2-phthalimido-β-D-glucopyranoside (7)
A mixture of diol 5 (31 mg, 33 μmol), trichloroacetimidate 6 (70 mg, 110 μmol), and activated 4 Å molecular sieves (145 mg) in CH2Cl2 (3 ml) was stirred for 15 min at r.t. under argon, then it was cooled to −40 °C and stirred for 15 min. TMSOTf (1.8 μl, 9.95 μmol) was added and the mixture was stirred at −40 °C for 1h then slowly warmed up to r.t. After 2.5 h, the reaction was quenched by addition of triethylamine (80 μl). The residue was filtered through a Celite pad and the filtrate was concentrated and subject to silica gel column purification (hexane/EtOAc, 3:1 to 1:1) to give tetrasaccharide 7 as a solid (54 mg, 86%) 1H NMR (500 MHz, CDCl3) δ 8.4-6.5 (m, 59 H, Ar), 6.09-6.0 (m, 2H), 5.92-5.84 (m, 5 H), 5.51 (s,1 H), 5.34 (s,1 H), 5.28 (d, 1 H, J = 11.5 Hz), 5.11(d, 1 H, J = 8.5 Hz), 5.0-4.89 (m, 4 H), 4.76-4.58 (m, 4 H), 4.56-4.44 (m, 2 H), 4.36-4.25 (m, 3 H), 4.23-4.16 (m, 2 H), 4.13-4.07 (m, 2 H), 4.03-3.92 (m, 3 H), 3.63-3.61 (m, 1 H), 3.46-3.44 (m, 1 H); 1H-13C HSQC NMR (1H at 500 MHz, 13C at 125 MHz, CDCl3) δ 8.38/130.15 (Ar), 8.20/130.00 (Ar), 8.10/129.94 (Ar), 8.04/129.89 (Ar), 7.93/129.82 (Ar), 7.91/129.69 (Ar), 7.87/129.70 (Ar), 7.73/133.44 (Ar), 7.66/133.40 (Ar), 7.62/128.68 (Ar), 7.60/127.801 (Ar), 7.57/133.36 (Ar), 7.54/133.15 (Ar), 7.53/133.12 (Ar), 7.44/128.42 (Ar), 7.35/128.13 (Ar), 6.59/128.34 (Ar), 6.51/127.44 (Ar), 6.05/65.94, 6.0/70.18, 5.92/69.8, 5.90/69.84, 5.88/69.96 (Man2′-2), 5.87/71.89, 5.87/70.27, 5.51/99.7 (Man2-1, 1JC-1/H-1 = 175 Hz, α)), 5.34/98.12 (Man2′-1, 1JC-1/H-1 = 174 Hz, α)), 5.28/75.61 (Bn-CH2), 5.11/97.19 (GlcN-1, 1JC-1/H-1 = 160 Hz, β), 4.99/75.61 (Bn-CH2), 4.97/98.92 (Man1-1, 1JC-1/H-1 = 161 Hz, β), 4.95/73.54 (Bn-CH2), 4.86/81.70 (Man2-6a, 1JC-F = 190 Hz), 4.76/81.70 (Man2-6b), 4.83/74.39 (Bn-CH2), 4.82/70.78(Bn-CH2), 4.79/74.39 (Bn-CH2), 4.76/70.32, 4.65/81.48 (Man2′-6a, 1JC-F = 175 Hz), 4.65/73.54 (Bn-CH2), 4.56/81.48 (Man2′-6b), 4.51/70.78(Bn-CH2), 4.32/79.24, 4.30/69.93, 4.29/74.85, 4.20/66.72 (Man1-6a), 4.18/55.52 (GlcN-2), 4.10/66.72 (Man1-6b), 4.08/75.65, 4.0/80.41 (Man1-3), 3.99/68.44(GlcN-6a), 3.95/68.44 (GlcN-6b), 3.62/74.38, 3.45/74.85; 19F NMR (376 MHz, CDCl3) −232.2 (td, 2JF-H = 47 Hz, 3JF-H = 24 Hz), −232.4 (td, 2JF-H = 47 Hz, 3JF-H = 23 Hz). MALDI TOF-MS: calcd for C109H95F2NO27, M = 1888.91; found (m/z), 1911.10 [M + Na]+.
4.4. Synthesis of benzyl 6-deoxy-6-fluoro-α-D-mannopyranosyl-(1→3)-[6-deoxy-6-fluoro-α-D-mannopyranosyl-(1→6)]-4-O-benzyl-β-D-mannopyranosyl-(1→4)-2-deoxy-2-acetamido-3,6-di-O-benzyl-glucopyranoside (8)
Tetrasaccharide 7 (37 mg, 19.6 μmol) was dissolved in an 90% aqueous ethanol (4 ml) containing hydrazine monohydrate (800 μl). The mixture was stirred at 80 °C for 48 h, then the solvent was removed by evaporation. The residue was dissolved in MeOH (3 ml) and 0.1 M aqueous NaHCO3 (3 ml). Acetic anhydride (1.0 M in MeOH, 209 μl, 0.2 mmol) was added and the mixture was stirred at r.t. under argon for 1 h. The residue was subject to preparative HPLC purification using a gradient eluent of 25%–50% MeCN containing 0.1% TFA over 30 min. Compound 8 was obtained as a white solid (10 mg, 60% for 2 steps). 1H NMR (400 MHz, CDCl3) δ 7.4–7.24 (m, 20 H, Ar), 5.01 (s,1 H), 4.92 (d, 1 H, J = 11.9), 4.82-4.66 (m, 7 H), 4.61-4.58 (m, 2 H), 4.54-4.50 (m, 2 H), 4.47 (s, 1 H), 4.0-3.98 (m, 1 H), 3.92-3.90 (m, 1 H), 3.86 (dd, 1 H, J = 3.2, 9 Hz), 3.79-3.75 (m, 3 H), 3.72-3.67 (m, 3 H), 3.63 (t, 1 H, J = 9.6 Hz), 3.52 (s, 1 H), 3.50 (s, 1 H), 3.30-3.28 (m, 7 H), 1.78 (s, 3 H, CH3); 1H-13C HSQC NMR (1H at 500 MHz, 13C at 125 MHz CDCl3): δ 7.44/128.43 (Ar), 7.36/128.13 (Ar), 5.05/102.82 (Man2-1), 4.93/73.48 (Bn-CH2), 4.85/70.96 (Bn-CH2), 4.81/74.57 (Bn-CH2), 4.75/82.95(Man2-6a, 1JC-F = 180 Hz), 4.73/99.98 (Man2′-1), 4.72/73.21 (Bn-CH2), 4.66/82.95 (Man2-6b), 4.66/73.48(Bn-CH2), 4.62/70.76 (Bn-CH2), 4.59/74.57 (Bn-CH2), 4.56/100.3 (GlcNAc-1), 4.52/73.21 (Bn-CH2), 4.46/99.71 (Man1-1), 4.40/82.20 (Man2′-6a, 1JC-F = 160 Hz), 4.31/82.20 (Man2′-6b), 4.08/72.44, 4.03/72.19, 4.01/70.65 (Man1-2), 3.98/76.58, 3.96/0.64 (Man2-2), 3.91/70.80, 3.85/54.66 (GlcNAc-2), 3.82/70.22 (Man2′-2), 3.81/68.13(GlcNAc-6a), 3.78/68.13 (GlcNAc-6b), 3.73/73.73, 3.73/70.93, 3.72/65.97, 3.69/79.97, 3.67/65.67, 3.62/65.94 (Man1-6a), 3.56/74.12, 3.52/65.94 (Man1-6b), 3.49/82.30 (Man1-3), 3.18/74.02, 1.82/22.00 (CH3); 19F NMR (376 MHz, CDCl3), δ −232.5 (td, 2JF-H = 47.6 Hz, 3JF-H = 24.1 Hz), −234.28 (td, 2JF-H = 47.5 Hz, 3JF-H = 26.4 Hz); ESI-MS: calcd for C54H67F2NO19, M = 1071.43; found (m/z), 1072.56 [M + H]+.
4.5. Synthesis of 6-deoxy-6-fluoro-α-D-mannopyranosyl-(1→3)-[6-deoxy-6-fluoro-α-D-mannopyranosyl-(1→6)]-β-D-mannopyranosyl-(1→4)-2-deoxy-2-acetamido-D-glucopyranose (9)
Compound 8 (12 mg, 11.2 μmol) was dissolved in MeOH (3 ml) and palladium-carbon (10%, 66 mg) was added. The mixture was stirred under H2 atmosphere for 8 h, then it was filtered through a Celite pad. The filtrate was concentrated to afford tetrasaccharide 9 as a white powder (8.2 mg, quantitative). 1H NMR (400 MHz, D2O) δ 5.27 (d, 1 H, J = 3 Hz), 5.18 (s, 1 H), 4.99 (s, 1 H), 4.89-4.69 (m, 6 H), 4.24 (s, 1 H), 4.15 (s, 1H), 4.08-4.05 (m, 2 H), 4.02-3.89 (m, 7 H), 3.87-3.63 (m, 9 H), 2.11 (s, 3 H); 1H-13C HSQC NMR (1H at 500 MHz, 13C at 125 MHz, D2O) δ 5.27/90.59, 5.18/102.80, 4.99/99.98, 4.89/100.31, 4.83/82.5 (Man2-6a, 1JC-F = 180 Hz), 4.81/82.3 (Man2′-6a, 1JC-F = 160 Hz), 4.78/94.95, 4.74/82.5 (Man2-6b), 4.70/82.3 (Man2′-6b), 4.24/70.31, 4.15/69.99, 4.08/72.16, 4.05/69.74, 4.02/71.88, 3.99/70.182, 3.98/69.08, 3.97/69.10, 3.96/53.72, 3.94/53.72, 3.89/71.55, 3.87/80.94, 3.87/71.35, 3.87/65.62, 3.79/65.75, 3.78/80.03, 3.78/72.41, 3.78/56.06, 3.73/74.08, 3.63/74.44, 2.12/22.15 (CH3); 19F (376 MHz, D2O) −233.5 (td, 2JF-H = 47.1 Hz, 3JF-H = 25.7 Hz), −233.6 (td, 2JF-H = 47.2 Hz, 3JF-H = 25.9 Hz), −234.2 to −234.6 (m, unresolved). ESI-MS: calcd for C26H43F2NO19, M = 711.24; found (m/z), 734.24 [M + Na]+.
4.6. Synthesis of 6-deoxy-6-fluoro-α-D-mannopyranosyl-(1→3)-[6-deoxy-6-fluoro-α-D-mannopyranosyl-(1→6)]-β-D-mannopyranosyl-(1→4)-1,2-dideoxy-α-D-glucopyrano-[2,1-d]-2-oxazoline (10)
A mixture of 9 (8.2 mg, 11.5 μmol), 2-chloro-1,3-dimethylimidazolinium chloride (39 mg, 230 μmol), and triethylamine (72 μl, 520 μmol) in H2O (200 μl) was stirred on an ice bath for 1.5 h. The solution was concentrated and the residue was subjected to gel filtration on a Sephadex G-10 column, which was eluted with 0.05 % triethylamine in H2O. The saccharide fractions were combined and further purified using preparative HPLC eluting with a linear gradient of 0–10 % acetonitrile containing 0.05% triethylamine over 30 min. The product fractions were combined and lyophilized to give oxazoline 10 as a white powder (7 mg, 88%) 1H NMR (500 MHz, D2O), δ 6.18 (d, 1H, J = 7 Hz), 5.19 (s, 1 H), 5.05 (s, 1 H), 4.91-4.72 (m, 5 H), 4.45 (br S, 1 H), 4.29-4.26 (m, 1 H), 4.17-4.13 (m, 3 H), 4.07-3.79 (m, 12 H), 3.75-3.71 (m, 1 H), 3.68 (m, 1 H), 3.5 (m, 1 H), 2.15 (s, 3 H); 1H-13C HSQC NMR (1H at 500 MHz, 13C at 125 MHz, D2O) δ 6.18/99.91 (GlcNAc-1), 5.19/102.78 (Man2-1), 5.05/99.99 (Man2′-1), 4.89/82.5 (Man2-6a, 1JC-F = 155 Hz), 4.84/82.5 (Man2′-6a, 1JC-F = 160 Hz), 4.82/101.25 (Man1-1), 4.78/82.5 (Man2-6b), 4.73/82.5 (Man2′-6b), 4.45/69.07 (GlcNAc-3), 4.28/65.28 (GlcNAc-2), 4.17/70.03 (Man2-2), 4.16/70.32 (Man1-2), 4.13/69.84 (Man2′-2), 4.07/72.18 (Man2-5), 4.05/65.89 (Man1-6a), 4.03/72.0 (Man2-5), 4.01/70.29 (Man2-3), 3.97/69.05 (Man2′-3), 3.92/65.89 (Man1-6b), 3.92/65.29 (Man2-4), 3.89/65.59 (Man1-4), 3.89/71.56 (Man2′-4), 3.84/77.79 (GlcNAc-4), 3.84/61.87 (GlcNAc-6a), 3.79/81.25 (Man1-3), 3.73/61.87 (GlcNAc-6b), 3.68/74.39 (Man1-5), 3.50/70.96 (GlcNAc-5), 2.15/13.11 (CH3); 19F (376 MHz, D2O): −233.9 (td, 2JF-H = 47.2 Hz, 3JF-H = 26.5 Hz), −234.3 (td, 2JF-H = 47.3 Hz, 3JF-H = 27.3 Hz). ESI-MS: calcd for C26H41F2NO18, M = 693.24; found (m/z), 694.30 [M + H]+.
4.7. Synthesis of benzyl 2,3,4-tri-O-benzoyl-6-deoxy-6-fluoro-α-D-mannopyranosyl-(1→3)-2-O-benzoyl-4,6-O-benzylidene-β-D-mannopyranosyl-(1→4)-3,6-di-O-benzyl-2-deoxy-2-phthalimido-β-D-glucopyranoside (11)
A mixture of 4 (48 mg, 51.4 μmol), 6 (132 mg, 0.21 mmol), and 4 Å molecular sieves (150 mg) in CH2Cl2 (3 ml) was stirred at r.t. under argon for 15 min, then TMSOTf (4 μl, 21 μmol) was added, and the mixture was stirred at r.t. under argon for 24 h. The reaction was quenched by addition of triethylamine (100 μl) and the solution was filtered through a Celite pad. The filtrate was concentrated and the resulting syrup was subject to silica gel column chromatography (hexane/EtOAc, 3:1 to 2:1) to afford trisaccharide 11 as a white solid (50 mg, 70%). 1H NMR (500 MHz, CDCl3), δ 8.35-6.87 (m, 45 H, Ar), 5.97 (t, 1 H, J = 10 Hz), 5.86-5.76 (m, 4 H), 5.55 (s, 1 H), 5.18 (d, 1 H, J = 8.5 Hz), 4.98-4.89 (m, 4 H), 4.85 (d, 1 H, J = 3.0 Hz), 4.76 (s, 2 H), 4.65-4.58 (m, 2 H), 4.50-4.46 (m, 2 H), 4.36-4.31 (m, 2 H), 4.27-4.19 (m, 2 H), 4.13 (dd, 1 H, J = 3.2, 9.5 Hz), 4.01-3.93 (m, 2 H), 3.86 (t, 1 H, J = 10 Hz), 3.66 (d, 1 H, J = 10 Hz), 3.35-3.32 (m, 1 H); 1H-13C HSQC NMR (1H at 500 MHz, 13C at 125 MHz, CDCl3), δ 8.35/130.00 (Ar), 8.15/129.98 (Ar), 7.86/129.68 (Ar), 7.73/133.42 (Ar), 7.72/133.40 (Ar), 7.71/133.40 (Ar), 7.69/133.41 (Ar), 7.67/133.40 (Ar), 7.63/125.95 9Ar), 7.61/128.44 (Ar), 7.60/128.43 (Ar), 7.58/128.44 (Ar), 7.57/128.42 (Ar), 7.42/128.11 (Ar), 7.34/128.11 (Ar), 7.15/127.77 (Ar), 6.91/127.77 (Ar), 6.87/127.70 (Ar), 5.97/66.13, 5.86/71.432 (Man1-2), 5.81/70.00 (Man2′-2), 5.78/69.60, 5.76/71.27, 5.55/98.62 (Man2′-1, 1JC-1/H-1 = 172 Hz, α), 5.18/97.30 (GlcN-1, 1JC-1/H-1 = 165 Hz, β), 4.98/99.07 (Man1-1, 1JC-1/H-1 = 164 Hz, β), 4.96/73.60 (Bn-CH2), 4.93/4.50 (Bn-CH2), 4.89/70.81 (Bn-CH2), 4.85/81.87 (Man2′-6a, 1JC-F = 160 Hz), 4.80/70.29, 4.76/81.87 (Man2′-6b), 4.65/73.60 (Bn-CH2), 4.58/70.81 (Bn-CH2), 4.50/68.45 (Man1-6a), 4.46/74.50 (Bn-CH2), 4.36/78.95, 4.31/5.63 (GlcN-2), 4.27/76.30, 4.19/78.45, 4.13/74.68, 4.01/68.16 (GlcN-6a), 3.93/68.16 (GlcN-6b), 3.86/68.45 (Man1-6b), 3.66/74.31, 3.33/66.65; 19F (376 MHz, D2O): −234.9, 2JF-H = 47.3 Hz, 3JF-H = 26 Hz. MALDI TOF-MS: calcd for C82H72FNO20, M = 1409.46; found (m/z), 1432.94 [M + Na]+.
4.8. Synthesis of benzyl 2,3,4-tri-O-benzoyl-6-deoxy-6-fluoro-α-D-mannopyranosyl-(1→3)-2-O-benzoyl-4-O-benzyl-β-D-mannopyranosyl-(1→4)-3,6-di-O-benzyl-2-deoxy-2-phthalimido-β-D-glucopyranoside (12)
A solution of 11 (40 mg, 28.4 μmol) and BH3·THF (1.0 M in THF, 500 μl, 0.5 mmol) in CH2Cl2 (3 mL) was stirred at r.t. under argon for 10 min. Cu(OTf)2 (4 mg, 11 μmol) was then added and the mixture was stirred for 6 h at r.t. under argon. The reaction was quenched by addition of triethylamine (20 μl) and MeOH (100 μl). The solvent was removed and the syrup was subject to silica gel column chromatography (hexane/EtOAc, 3:2) to give the alcohol 12 as a solid (15 mg, 60%). 1H NMR (500 MHz, CDCl3), δ 8.17-6.71 (m, 44 H, Ar), 5.87 (t, 1 H, J = 10 Hz), 5.70-5.65 (m, 3 H), 5.36-5.31 (m, 1 H), 5.03 (d, 1 H, J = 8.5 Hz), 4.91 (d, 1 H, J = 10.5 Hz), 4.80-4.72 (m, 5 H), 4.66-4.63 (m, 2 H), 4.53-4.45 (m, 2 H), 4.25 (d, 1 H, J = 12.5 Hz), 4.20-4.09 (m, 3 H), 3.93 (t, 1 H, J = 9.5 Hz), 3.85-3.79 (m, 4 H), 3.61-3.57 (m, 1 H), 3.52 (d, 1 H, J = 10 Hz), 3.43 (m, 1 H), 3.21 (m, 1 H); 1H-13C HSQC NMR (1H at 500 MHz, 13C at 125 MHz, CDCl3) δ 8.17/130.11 (Ar), 8.05/130.04 (Ar), 7.92/130.00 (Ar), 7.77/129.71 (Ar), 7.63/133.44 (Ar), 7.61/133.43 (Ar), 7.60/133.44 (Ar), 7.58/133.43 (Ar), 7.56/133.41 (Ar), 7.54/133.43 (Ar), 7.53/133.56 (Ar), 7.50/128.60 (Ar), 7.47/128.68 (Ar), 7.41/128.11 (Ar), 7.40/133.12 (Ar), 7.40/128.10 (Ar), 7.39/128.44 (Ar), 7.37/128.35 (Ar), 7.36/128.13 (Ar), 7.27/128.43 (Ar), 7.23/128.13 (Ar), 7.06/127.51 (Ar), 7.01/127.82 (Ar), 6.83/127.08 (Ar), 6.78/127.82 (Ar), 6.71/127.21 (Ar), 5.87/65.94, 5.70/70.00 (Man2′-2), 5.69/65.94, 5.65/71.88 (Man1-2), 5.36/99.69 (Man2′-1), 5.03/97.34 (GlcN-1), 4.91/75.73 (½ Bn), 4.80/98.36 (Man1-1), 4.79/73.63 (Bn-CH2), 4.76/74.20 (Bn-CH2), 4.76/70.86 (Bn-CH2), 4.72/81.73 (C-6a, 1JC-F = 195 Hz), 4.66/75.73 (Bn-CH2), 4.63/81.73 (C-6b), 4.53/73.63 (Bn-CH2), 4.45/70.86 (Bn-CH2), 4.25/74.20 (Bn-CH2), 4.20/55.64 (GlcN-2), 4.17/78.75, 4.09/76.37, 3.93/74.70, 3.85/79.98, 3.82/68.17 (GlcN-6a), 3.82/61.97 (Man1-6a), 3.79/68.17 (GlcN-6b), 3.60/61.97 (Man1-6b), 3.52/74.38, 3.43/70.78, 3.21/75.52; 19F NMR (376 MHz, CDCl3), −232.2 (td, 2JF-H = 47.1 Hz, 3JF-H = 24.0 Hz). MALDI TOF-MS: calcd for C82H74FNO20, M = 1411.47; found (m/z), 1434.56 [M + Na]+.
4.9. Synthesis of benzyl 2,3,4-tri-O-benzoyl-6-deoxy-6-fluoro-α-D-mannopyranosyl-(1→3)-[2,3,4,6-tetra-O-benzoyl-α-D-mannopyranosyl-(1→6)]-2-O-benzoyl-4-O-benzyl-β-D-mannopyranosyl-(1→4)-3,6-di-O-benzyl-2-deoxy-2-phthalimido-β-D-glucopyranoside (14)
A mixture of 12 (15 mg, 10.6 μmol), 13 (30 mg, 40.5 μmol), and 4 Å molecular sieves (200 mg) in CH2Cl2 (2 ml) was stirred at r.t. under argon for 10 min, then TMSOTf (1.5 μl, 8 μmol) was added, and the resulting mixture was stirred at r.t. for 16 h. The reaction was quenched by addition of triethylamine (100 μl) and the solution was filtered through a Celite pad. The filtrate was concentrated and the residue was subject to silica gel column chromatography (hexanes/EtOAc, 2:1 to 1:1) to afford 14 as a white solid (16 mg, 73%). 1H NMR (500 MHz, CDCl3), δ 8.39-6.44 (m, 64 H Ar), 6.18 (t, 1 H, J = 10 Hz), 6.05-6.0 (m, 2 H), 5.92-5.86 (m, 4 H), 5.50 (s, 1 H), 5.32-5.30 (m, 2 H), 5.09 (d, 1 H, J = 8.5 Hz), 4.98-4.89 (m, 2 H), 4.94 (t, 1 H, J = 11 Hz), 4.85-4.76 (m, 4 H), 4.75-4.65 (m, 3 H), 4.51-4.46 (m, 3 H), 4.39-4.31 (m, 3 H), 4.23-4.20 (m, 2 H), 4.08-4.07 (m, 2 H), 4.01-3.98 (m, 2 H), 3.94-3.92 (m, 1 H), 3.60 (d, 1 H, J = 9.5 Hz), 3.42 (d, 1 H, J = 9.5Hz); 1H-13C HSQC NMR (1H at 500 MHz, 13C at 125 MHz, CDCl3) δ 8.39/130.21 (Ar), 8.20/129.94 (Ar), 8.11/129.99 (Ar), 8.08/128.88 (Ar), 8.0/129.77 (Ar), 7.86/129.70 (Ar), 7.74/133.44 (Ar), 7.73/122.98 (Ar), 7.67/133.12 (Ar), 7.61/128.73 (Ar), 7.60/127.68 (Ar), 7.58/133.13 (Ar), 7.54/128.03 (Ar), 7.52/133.11 (Ar), 7.51/128.44 (Ar), 7.40/128.43 (Ar), 7.37/122.89 (Ar), 7.31/128.14 (Ar), 7.14/127.49 (Ar), 7.08/128.03 (Ar), 7.07/127.74 (Ar), 6.57/126.57 (Ar), 6.56/128.20 (Ar), 6.44/127.44 (Ar), 6.18/66.57, 6.05/65.94, 6.0/70.32, 5.92/69.72, 5.91/69.82 (Man2-2), 5.87/71.88 (Man1-2), 5.86/70.30 (Man2′-2), 5.50/99.70 (Man2-1, 1JC-1/H-1 = 173 Hz, α), 5.32/98.28 (Man2′-1, 1JC-1/H-1 = 176 Hz, α).), 5.30/75.63 (Bn-CH2), 5.09/97.20 (GlcN-1, 1JC-1/H-1 = 163 Hz, β), 4.98/99.07 (Man1-1, 1JC-1/H-1 = 162 Hz, β), 4.99/75.63 (Bn-CH2), 4.94/73.43 (Bn-CH2), 4.85/81.72 (Man2-6a, 1JC-F = 195 Hz), 4.81/70.69 (Bn-CH2), 4.78/74.33 (Bn-CH2), 4.76/81.72 (Man2-6b), 4.75/70.32, 4.69/62.68 (Man2′-6a), 4.65/73.43 (Bn-CH2), 4.51/70.69 (Bn-CH2), 4.48/74.33 (Bn-CH2), 4.46/62.68 (Man2-6b), 4.39/69.07, 4.35/74.71, 4.31/79.38, 4.23/66.43 (Man1-6a), 4.20/55.63 (GlcN-2), 4.08/75.62, 4.07/66.43 (Man1-6b), 4.01/80, 3.98/68.44 (GlcN-6a), 3.94/68.44 (GlcN-6b), 3.60/74.84, 3.42/74.7; MALDI TOF-MS: calcd for C116H100FNO29, M = 1989.64; found (m/z), 2012.26 [M + Na]+.
4.10. Synthesis of benzyl 6-deoxy-6-fluoro-α-D-mannopyranosyl-(1→3)-[α-D-mannopyranosyl-(1→6)]-4-O-benzyl-β-D-mannopyranosyl-(1→4)-2-deoxy-2-acetamido-3,6-di-O-benzyl-glucopyranoside (15)
A solurion of 14 (15 mg, 7.5 μmol) in 90% aqueous EtOH (4 ml) containing hydrazine monohydrate (600 μl) was stirred at 80 ° C for 7 h. The solvent was then removed and the residue was dissolved in MeOH (5 ml) and 1.0 M aqueous NaHCO3 (5 ml). Acetic anhydride (30 μl) was added and the mixture was stirred at r.t. for 1 h. The solution was concentrated and the residue was subject to preparative HPLC purification to give 15 as a white powder (5 mg, 60% for two steps). 1H NMR (500 MHz, D2O) δ 7.53-7.34 (m, 20 H, Ar), 5.14 (s, 1 H), 5.08 (s, 1 H), 4.94-4.89 (m, 3 H), 4.84-4.79 (m, 3 H), 4.75-4.63 (m, 6 H), 4.18-4.1 (m, 4 H), 4.0 (br S, 1 H), 3.97-3.9 (m, 5 H), 3.88-3.85 (m, 3 H), 3.82-3.76 (m, 3 H), 3.74-3.71 (m, 2 H), 3.66-3.61 (m, 4 H), 1.9 (s, 3 H, CH3); 1H-13C HSQC NMR (1H at 500 MHz, 13C at 125 MHz, D2O) δ 7.53/127.82 (Ar), 7.49/128.21 (Ar), 7.48/128.14 (Ar), 7.47/127.92 (Ar), 7.42/127.84 (Ar), 7.41/127.72, 7.36/127.19, 5.14/103.13 (Man2-1), 5.08/73.61 (Bn-CH2), 4.94/70.47 (Bn-CH2), 4.91/100.00 (Man2′-1), 4.90/74.59 (Bn-CH2), 4.84/73.61 (Bn-CH2), 4.81/82.81 (Man2-6a, 1JC-F = 160 Hz), 4.79/73.13 (Bn-CH2), 4.74/82.81(Man2-6b), 4.74/74.59 (Bn-CH2), 4.69/99.93(Man1-1), 4.68/70.47 (Bn-CH2), 4.65/100.31 (GlcNAc-1), 4.63/73.13 (Bn-CH2), 4.19/72.53, 4.16/70.84, 4.15/72.49 (Man1-2), 4.11/77.10, 4.0/70.95 (Man2-2), 3.96/71.09, 3.93/54.99 (GlcNAc-2), 3.93/68.45 (GlcNAc-6a), 3.92/70.63 (Man2′-2), 3.90/68.45 (GlcNAc-6b), 3.87/74.06, 3.85/71.27, 3.85/65.83 (Man-6a), 3.81/79.69, 3.79/66.24, 3.75/67.18, 3.73/61.24 (Man-6a), 3.71/65.83 (Man-6b), 3.68/61.24 (Man-6b), 3.64/82.82, 3.62/73.108, 3.61/74.65, 1.90/21.67 (CH3); 19F NMR (376 MHz, D2O), δ −233.4 (td, 2JF-H = 47.6 Hz, 3JF-H = 24 Hz). ESI-MS: calcd for C54H68FNO20, M = 1069.43; found (m/z), 1070.74 [M + H]+.
4.11. Synthesis of 6-deoxy-6-fluoro-α-D-mannopyranosyl-(1→3)-[α-D-mannopyranosyl-(1→6)]-β-D-mannopyranosyl-(1→4)-2-deoxy-2-acetamido-D-glucopyranose (16)
A mixture of 15 (5 mg, 4.7 μmol) and palladium-carbon (5%, 25 mg) in MeOH (2 ml) was stirred for 16 h under a H2 atmosphere. Then the mixture was filtered through a Celite pad and the filtrate was concentrated to dryness. The residue was dissolved in water and lyophilized to give free tetrasaccharide 16 as a white solid (3 mg, 91%). 1H NMR (500 MHz, D2O) δ 5.27 (d, 1 H, J = 3 Hz), 5.18 (s, 1 H), 4.96 (s, 1 H), 4.88-4.78 (br S, 3 H), 4.71-4.74 (m, 1H), 4.27-4.24 (m, 2 H), 4.16-4.14 (m, 2 H), 4.07-4.06 (m, 2 H), 4.03-4.0 (m, 3 H), 3.98-3.88 (m, 10 H), 3.85-3.77 (m, 8 H), 3.72-3.67 (m, 4 H), 3.63-3.58 (m, 1 H), 2.11 (s, 3 H, CH3); 1H-13C HSQC NMR (1H at 500 MHz, 13C at 125 MHz, D2O) δ 5.27/90.53, 5.18/102.68 (Man2-1), 4.96/99.66 (Man2′-1), 4.85/100.34 (Man1-1), 4.78/94.98 (GlcNAc-1), 4.83/82.6 (Man2-6a, 1JC-F = 168 Hz), 4.73/82.6 (Man2-6b), 4.28/70.35, 4.24/70.31(Man1-2), 4.16/68.48, 4.15/69.98 (Man2-2), 4.07/72.18, 4.06/70.0, 4.03/69.98 (Man2′-2), 4.02/71.90, 4.0/69.12, 3.99/69.12, 3.98/70.1, 3.98/65.93 (Man1-6a), 3.97/69.13, 3.96/70.31, 3.95/60.97 (6a), 3.95/53.68, 3.94/70.34, 3.91/60.31 (6a), 3.88/65.93(Man1-6b), 3.85/60.31(6b), 3.84/72.15, 3.83/80.94, 3.82/65.63, 3.81/60.97 (6b), 3.78/72.48, 3.78/55.95 (GlcNAc-2), 3.77/80.03, 3.72/74.10, 3.71/72.79, 3.70/66.87, 3.68/66.88, 3.61/74.38, 2.12/22.12 (CH3); 19F NMR (376 MHz, D2O), δ −233.6 (td, 2JF-H = 46.8 Hz, 3JF-H =25.6 Hz), −233.6 (td, 2JF-H = 47.5 Hz, 3JF-H = 25.7 Hz). ESI-MS: calcd for C26H44FNO20, M = 709.24; found (m/z), 710.34 [M + H]+.
4.12. Synthesis of 6-deoxy-6-fluoro-α-D-mannopyranosyl-(1→3)-[α-D-mannopyranosyl-(1→6)]-β-D-mannopyranosyl-(1→4)-1,2-dideoxy-α-D-glucopyrano-[2,1-d]-2-oxazoline (17)
A solution of 16 (3 mg, 4.23 μmol), 2-chloro-1,3-dimethylimidazolinium chloride (15 mg, 88.7 μmol), and triethylamine (26 μl) in H2O (500 μl) was stirred at 0°C for 1 h. The residue was subject to preparative HPLC purification with a linear gradient eluent of 0–10% MeCN containing 0.05 % triethylamine over 30 min. The pooled fractions were lyophilized to give the oxazoline 17 as a white powder (2 mg, quantitative). 1H NMR (500 MHz, D2O) δ 6.15 (d, 1 H, J = 7 Hz), 5.16 (s, 1H), 5.0 (s, 1 H), 4.90-4.80 (m, 2 H), 4.79 (s, 1 H), 4.44 (s, 1 H), 4.26 (m,1 H), 4.14 (s, 1 H), 4.07 (s, 1 H), 4.04-3.64 (m, 17 H), 2.12 (s, 3 H); 1H-13C HSQC NMR NMR (1H at 500 MHz, 13C at 125 MHz, D2O) δ 6.15/99.94 (GlcNAc-1), 5.16/102.79 (Man2-1), 5.0/99.65 (Man2′-1), 4.81/82.53 (Man2-6a, 1JC-F = 163 Hz), 4.79/101.26 (Man1-1), 4.72/82.53 (Man2-6b), 4.44/60.07, 4.26/65.10 (GlcNAc-2), 4.14/70.19 (Man2-2), 4.13/73.64 (Man1-2), 4.07/70.00 (Man2′-2), 4.04/72.19, 4.00/81.24, 4.02/65.63 (Man1-6a), 3.98/70.32, 3.97/60.94 (Man2′-6a), 3.91/70.62, 3.89/65.63 (Man1-6b), 3.83/65.63, 3.83/60.94 (Man2′-6b), 3.81/77.71, 3.81/61.88 (GlcNAc-6a), 3.78/81.23, 3.76/72.80, 3.72/66.87, 3.70/61.88 (GlcNAc-6b), 3.64/74.38, 3.47/70.95 (GlcNAc-5), 2.12/13.12 (CH3); 19F NMR (376 MHz, D2O); δ −233.9 (td, 2JF-H = 47.5 Hz, 3JF-H = 25.8 Hz). ESI-MS: calcd for C26H42FNO19, M = 691.23; found (m/z), 691.81[M + H]+.
4.13. Synthesis of the difluoro-glycoprotein (1) through Endo A catalyzed transglycosylation
A solution of difluoroglycan oxazoline 10 (1.6 mg, 2.3 μmol), GlcNAc-RNase 18 30 (5.7 mg, 0.41 μmol) in phosphate buffer (0.1 M, 100 μL, pH 7) was incubated at 30 °C with Endo-A (25 μg). The reaction was monitored by analytical HPLC and the transglycosylation product appeared slightly earlier than the acceptor under the analytical HPLC conditions. After 2 h, a complete conversion of GlcNAc-RNase into the transglycosylation product was achieved. The product was purified by preparative HPLC using a linear gradient of 24–33% MeCN containing 0.1% TFA over 30 min. The fractions containing the product were pololed and lyophilized to give the difluoroglycoprotein (1) as a white powder (6 mg, quantitative). Product quantification was carried out on the basis of UV absorbance at 280 nm with a reference standard solution of GlcNAc-RNase (18). ESI-MS: calcd for glycoprotein 1, M = 14579.24; found (m/z): 1823.30 [M + 8 H]8+, 1620.95 [M + 9 H]9+, 1459.07 [M + 10 H]10+, 1326.61 [M + 11 H]11+, 1216.18 [M + 12 H]12+, 1122.71 [M + 13 H]13+, 1042.56 [M + 14 H]14+; deconvolution data, M = 14582.
4.14. Synthesis of the monofluoro-glycoprotein (2) through Endo A catalyzed transglycosylation
A solution of tetrasaccharide oxazoline 17 (1 mg, 1.5 μmol), GlcNAc-RNase 18 (4 mg, 0.29 μmol) in phosphate buffer (0.1 M, 60 μL, pH 7) was incubated at 30 °C with Endo-A (2 μg). The reaction proceeded for 2 h and the product was purified in the same way as described for the preparation of glycoprotein 1 to afford the monofluoro-glycoprotein (2) as a white powder (3.8 mg, 92%). ESI-MS: calcd for glycoprotein 2, M = 14578.25; found, 1823.10[M + 8 H]8+, 1620.79 [M + 9 H] 9+, 1458.92 [M + 10 H] 10+, 1326.49 [M + 11 H] 11+, 1216.07 [M + 12 H] 12+, 1122.59 [M + 13 H]13+, 1042.45 [M + 14 H]14+; deconvolution data, M = 14580.
4.15. Synthesis of the glycoprotein (3) through Endo A catalyzed transglycosylation
A solution of tetrasaccharide oxazoline 19 (2 mg, 3 μmol), GlcNAc-RNase 18 (8 mg, 0.58 μmol) in phosphate buffer (0.1 M, 30 μL, pH 7) was incubated at 30 °C with Endo-A (4 μg) for 2 h. The transglycosylation product was purified in the same way as described for the preparation of glycoprotein 1 to afford the Man3GlcNAc2-RNase (3) as a white powder (7.0 mg, 88%). ESI-MS: calcd for glycoprotein 3, M = 14576.25; found, 1822.94 [M + 8 H]8+, 1620.59 [M + 9 H]9+, 1458.70 [M + 10 H] 10+, 1326.31 [M + 11 H] 11+, 1215.88 [M + 12 H] 12+, 1122.44 [M + 13 H]13+, 1042.33 [M + 14 H]14+; deconvolution data, M = 14578.
4.16. Analysis of the relative reactivity of the glycan oxazolines in the enzymatic transglycosylation
A mixture of difluoroglycan oxazoline 10 (3 mM), monofluoro-glycan oxazoline 17 (3 mM), and acceptor Fmoc-Asn(GlcNAc)-OH (20) (5 mM) in a phosphate buffer (50 mM, pH 7, 20 μL) was incubated at 30° C with Endo A enzyme (6.7 μg/mL) for competitive reaction. The reaction was monitored by analytical HPLC using a linear gradient of 25–34% MeCN containing 0.1% TFA over 30 min. Under this condition, the respective transglycosylation products (21 and 22) were eluted earlier than the acceptor (20) and the two were well separated. Aliquots were taken at intervals and the relative yields were calculated by integration of the peak areas for the respective products. Competitive transglycosylation reactions between the difluoro-glycan oxazoline (10) and the non-fluorinated glycan oxazoline (19) were performed in the same way and the relative reaction rate was calculated by inergration of the peaks corresponding to the transglycosylation products (21 and 23, respectively). ESI-MS of transglycosylation product 21, calcd for C53H72F2N4O28, M = 1250.43; found (m/z), 1251.63 [M + H]+. ESI-MS of transglycosylation product 22, calcd for C53H73FN4O29, M = 1248.43; found (m/z), 1249.71 [M + H]+. ESI-MS of transglycosylation product 23, calcd for C53H74N4O30, M = 1246.44; found (m/z), 1247.72 [M + H]+.
4.17. Surface Plasmon Resonance (SPR) Measurement
The interactions between the the synthetic glycoforms of RNase and ConA were evaluated by SPR using a BIAcore T100 system (GE Healthcare). Con A were immobilized on a CM5 sensor chip in an acetate buffer (10 mM, pH 5.0) by use of the amine coupling kit provided by the manufacturer. The immobilization was performed until 3000 response unit (RU) was reached. The chip was blocked by the addition of ethanolamine HCl (1 M, pH 8.5). A reference cell without the con A was prepared using similar procedures. Analyses were performed at 25 °C by injecting a solution of the respective RNase glycoforms in a running buffer of HBS-P (10 mM HEPES, 150 mM NaCl, and 0.05% surfactant P20, pH 7.4) containing 1 mM CaCl2 and 1 mM MnCl2 at a flow rate of 30 μL/min for 2 min, followed by wash for 6 min with the running buffer for dissociation. The injection was performed by a serial 2-fold dilutions starting at 8 μM. The sensor surface was regenerated through a two-step wash (first wash, 0.1 M glycine pH 1.7 for 1.5 min; second wash, running buffer for 1.5 min). Data processing was carried out using the BIAcore T100 evaluation software.
Supplementary Material
Acknowledgments
We thank Prof. C. Allen Bush for valuable discussions and members of Wang Lab for technical assistance. This work was supported by the National Institutes of Health (NIH grant R01 GM080374).
Footnotes
Dedicated to Professor Benjamin G. Davis on the occasion of his receiving the Tetrahedron Young Investigator Award
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Dwek RA. Chem Rev. 1996;96:683–720. doi: 10.1021/cr940283b. [DOI] [PubMed] [Google Scholar]
- 2.Helenius A, Aebi M. Science. 2001;291:2364–2369. doi: 10.1126/science.291.5512.2364. [DOI] [PubMed] [Google Scholar]
- 3.Hart GW, Copeland RJ. Cell. 2010;143:672–676. doi: 10.1016/j.cell.2010.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Marth JD, Grewal PK. Nat Rev Immunol. 2008;8:874–887. doi: 10.1038/nri2417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jefferis R. Nat Rev Drug Discov. 2009;8:226–234. doi: 10.1038/nrd2804. [DOI] [PubMed] [Google Scholar]
- 6.Gamblin DP, Scanlan EM, Davis BG. Chem Rev. 2009;109:131–163. doi: 10.1021/cr078291i. [DOI] [PubMed] [Google Scholar]
- 7.Schmaltz RM, Hanson SR, Wong CH. Chem Rev. 2011;111:4259–4307. doi: 10.1021/cr200113w. [DOI] [PubMed] [Google Scholar]
- 8.Wang LX, Lomino JV. ACS Chem Biol. 2012;7:110–122. doi: 10.1021/cb200429n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Glaudemans CPJ. Chem Rev. 1991;91:25–33. [Google Scholar]
- 10.Tatami A, Hon YS, Matsuo I, Takatani M, Koshino H, Ito Y. Biochem Biophys Res Commun. 2007;364:332–337. doi: 10.1016/j.bbrc.2007.10.008. [DOI] [PubMed] [Google Scholar]
- 11.Allman SA, Jensen HH, Vijayakrishnan B, Garnett JA, Leon E, Liu Y, Anthony DC, Sibson NR, Feizi T, Matthews S, Davis BG. ChemBioChem. 2009 doi: 10.1002/cbic.200900425. [DOI] [PubMed] [Google Scholar]
- 12.Garnett JA, Liu Y, Leon E, Allman SA, Friedrich N, Saouros S, Curry S, Soldati-Favre D, Davis BG, Feizi T, Matthews S. Protein Sci. 2009;18:1935–1947. doi: 10.1002/pro.204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Withers SG, Rupitz K, Street IP. J Biol Chem. 1988;263:7929–7932. [PubMed] [Google Scholar]
- 14.Lee SS, Greig IR, Vocadlo DJ, McCarter JD, Patrick BO, Withers SG. J Am Chem Soc. 2011;133:15826–15829. doi: 10.1021/ja204829r. [DOI] [PubMed] [Google Scholar]
- 15.Brown CD, Rusek MS, Kiessling LL. J Am Chem Soc. 2012;134:6552–6555. doi: 10.1021/ja301723p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Walsh C. Adv Enzymol Rel Areas Mol Biol. 1983;55:197–289. doi: 10.1002/9780470123010.ch3. [DOI] [PubMed] [Google Scholar]
- 17.Glaudemans CP, Kovac P, Rao AS. Carbohydr Res. 1989;190:267–277. doi: 10.1016/0008-6215(89)84130-8. [DOI] [PubMed] [Google Scholar]
- 18.Ametamey SM, Honer M, Schubiger PA. Chem Rev. 2008;108:1501–1516. doi: 10.1021/cr0782426. [DOI] [PubMed] [Google Scholar]
- 19.Sun Y, Takaoka Y, Tsukiji S, Narazaki M, Matsuda T, Hamachi I. Bioorg Med Chem Lett. 2011;21:4393–4396. doi: 10.1016/j.bmcl.2011.06.038. [DOI] [PubMed] [Google Scholar]
- 20.Takaoka Y, Kiminami K, Mizusawa K, Matsuo K, Narazaki M, Matsuda T, Hamachi I. J Am Chem Soc. 2011;133:11725–11731. doi: 10.1021/ja203996c. [DOI] [PubMed] [Google Scholar]
- 21.Takaoka Y, Sakamoto T, Tsukiji S, Narazaki M, Matsuda T, Tochio H, Shirakawa M, Hamachi I. Nat Chem. 2009;1:557–561. doi: 10.1038/nchem.365. [DOI] [PubMed] [Google Scholar]
- 22.Tsukiji S, Miyagawa M, Takaoka Y, Tamura T, Hamachi I. Nat Chem Biol. 2009;5:341–343. doi: 10.1038/nchembio.157. [DOI] [PubMed] [Google Scholar]
- 23.Flavell RR, Kothari P, Bar-Dagan M, Synan M, Vallabhajosula S, Friedman JM, Muir TW, Ceccarini G. J Am Chem Soc. 2008;130:9106–9112. doi: 10.1021/ja801666z. [DOI] [PubMed] [Google Scholar]
- 24.Wangler B, Quandt G, Iovkova L, Schirrmacher E, Wangler C, Boening G, Hacker M, Schmoeckel M, Jurkschat K, Bartenstein P, Schirrmacher R. Bioconjug Chem. 2009;20:317–321. doi: 10.1021/bc800413g. [DOI] [PubMed] [Google Scholar]
- 25.Boutureira O, D’Hooge F, Fernandez-Gonzalez M, Bernardes GJ, Sanchez-Navarro M, Koeppe JR, Davis BG. Chem Commun (Camb) 2010;46:8142–8144. doi: 10.1039/c0cc01576h. [DOI] [PubMed] [Google Scholar]
- 26.Haneda K, Inazu T, Yamamoto K, Kumagai H, Nakahara Y, Kobata A. Carbohydr Res. 1996;292:61–70. doi: 10.1016/s0008-6215(96)91025-3. [DOI] [PubMed] [Google Scholar]
- 27.Wang LX, Tang M, Suzuki T, Kitajima K, Inoue Y, Inoue S, Fan JQ, Lee YC. J Am Chem Soc. 1997;119:11137–11146. [Google Scholar]
- 28.Fujita M, Shoda S, Haneda K, Inazu T, Takegawa K, Yamamoto K. Biochim Biophys Acta. 2001;1528:9–14. doi: 10.1016/s0304-4165(01)00164-7. [DOI] [PubMed] [Google Scholar]
- 29.Li B, Zeng Y, Hauser S, Song H, Wang LX. J Am Chem Soc. 2005;127:9692–9693. doi: 10.1021/ja051715a. [DOI] [PubMed] [Google Scholar]
- 30.Li B, Song H, Hauser S, Wang LX. Org Lett. 2006;8:3081–3084. doi: 10.1021/ol061056m. [DOI] [PubMed] [Google Scholar]
- 31.Wei Y, Li C, Huang W, Li B, Strome S, Wang LX. Biochemistry. 2008;47:10294–10304. doi: 10.1021/bi800874y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Umekawa M, Huang W, Li B, Fujita K, Ashida H, Wang LX, Yamamoto K. J Biol Chem. 2008;283:4469–4479. doi: 10.1074/jbc.M707137200. [DOI] [PubMed] [Google Scholar]
- 33.Ochiai H, Huang W, Wang LX. J Am Chem Soc. 2008;130:13790–13803. doi: 10.1021/ja805044x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang LX, Huang W. Curr Opin Chem Biol. 2009;13:592–600. doi: 10.1016/j.cbpa.2009.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Huang W, Li C, Li B, Umekawa M, Yamamoto K, Zhang X, Wang LX. J Am Chem Soc. 2009;131:2214–2223. doi: 10.1021/ja8074677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Umekawa M, Li C, Higashiyama T, Huang W, Ashida H, Yamamoto K, Wang LX. J Biol Chem. 2010;285:511–521. doi: 10.1074/jbc.M109.059832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schwarz F, Huang W, Li C, Schulz BL, Lizak C, Palumbo A, Numao S, Neri D, Aebi M, Wang LX. Nat Chem Biol. 2010;6:264–266. doi: 10.1038/nchembio.314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Huang W, Yang Q, Umekawa M, Yamamoto K, Wang LX. ChemBioChem. 2010;11:1350–1355. doi: 10.1002/cbic.201000242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zou G, Ochiai H, Huang W, Yang Q, Li C, Wang LX. J Am Chem Soc. 2011;133:18975–18991. doi: 10.1021/ja208390n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Amin MN, Huang W, Mizanur RM, Wang LX. J Am Chem Soc. 2011;133:14404–14417. doi: 10.1021/ja204831z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fan SQ, Huang W, Wang LX. J Biol Chem. 2012;287:11272–11281. doi: 10.1074/jbc.M112.340497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Huang W, Giddens J, Fan SQ, Toonstra C, Wang LX. J Am Chem Soc. 2012;134:12308–12318. doi: 10.1021/ja3051266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rising TW, Claridge TD, Moir JW, Fairbanks AJ. ChemBioChem. 2006;7:1177–1180. doi: 10.1002/cbic.200600183. [DOI] [PubMed] [Google Scholar]
- 44.Rising TW, Heidecke CD, Moir JW, Ling Z, Fairbanks AJ. Chem Eur J. 2008;14:6444–6464. doi: 10.1002/chem.200800365. [DOI] [PubMed] [Google Scholar]
- 45.Heidecke CD, Ling Z, Bruce NC, Moir JW, Parsons TB, Fairbanks AJ. ChemBioChem. 2008;9:2045–2051. doi: 10.1002/cbic.200800214. [DOI] [PubMed] [Google Scholar]
- 46.Parsons TB, Moir JW, Fairbanks AJ. Org Biomol Chem. 2009;7:3128–3140. [Google Scholar]
- 47.Wang J, Li H, Zou G, Wang LX. Org Biomol Chem. 2007;5:1529–1540. doi: 10.1039/b702961f. [DOI] [PubMed] [Google Scholar]
- 48.Travoska I, Taravel FR. Adv Carbohydr Chem Biochem. 1995;51:15–61. doi: 10.1016/s0065-2318(08)60191-2. [DOI] [PubMed] [Google Scholar]
- 49.Huang W, Li J, Wang LX. ChemBioChem. 2011;12:932–941. doi: 10.1002/cbic.201000763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gao J, Qiao S, Whitesides GM. J Med Chem. 1995;38:2292–2301. doi: 10.1021/jm00013a005. [DOI] [PubMed] [Google Scholar]
- 51.Ly HD, Howard S, Shum K, He S, Zhu A, Withers SG. Carbohydr Res. 2000;329:539–547. doi: 10.1016/s0008-6215(00)00214-7. [DOI] [PubMed] [Google Scholar]
- 52.Yin J, Li L, Shaw N, Li Y, Song JK, Zhang W, Xia C, Zhang R, Joachimiak A, Zhang HC, Wang LX, Liu ZJ, Wang P. PLoS One. 2009;4:e4658. doi: 10.1371/journal.pone.0004658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Li B, Takegawa K, Suzuki T, Yamamoto K, Wang LX. Bioorg Med Chem. 2008;16:4670–4675. doi: 10.1016/j.bmc.2008.02.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bhattacharyya L, Brewer CF. Eur J Biochem. 1989;178:721–726. doi: 10.1111/j.1432-1033.1989.tb14503.x. [DOI] [PubMed] [Google Scholar]
- 55.Mandal DK, Bhattacharyya L, Koenig SH, Brown RD, 3rd, Oscarson S, Brewer CF. Biochemistry. 1994;33:1157–1162. doi: 10.1021/bi00171a015. [DOI] [PubMed] [Google Scholar]
- 56.Naismith JH, Field RA. J Biol Chem. 1996;271:972–976. doi: 10.1074/jbc.271.2.972. [DOI] [PubMed] [Google Scholar]
- 57.Gupta D, Dam TK, Oscarson S, Brewer CF. J Biol Chem. 1997;272:6388–6392. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.