

Abstract
Accumulating evidence indicates that activation of spinal cord astrocytes contributes importantly to nerve injury and inflammation-induced persistent pain and chronic opioid-induced antinociceptive tolerance. Phosphorylation of extracellular signal-regulated kinase (pERK) and induction of interleukin-1 beta (IL-1β) in spinal astrocytes have been implicated in astrocytes-mediated pain. Tissue plasminogen activator (tPA) is a serine protease that has been extensively used to treat stroke. We examined the potential involvement of tPA in chronic opioid-induced antinociceptive tolerance and activation of spinal astrocytes using tPA knockout (tPA−/−) mice and astrocyte cultures. tPA−/− mice exhibited unaltered nociceptive pain and morphine-induced acute analgesia. However, the antinociceptive tolerance, induced by chronic morphine (10 mg/kg/day, s.c.), is abrogated in tPA−/− mice. Chronic morphine induces tPA expression in GFAP-expressing spinal cord astrocytes. Chronic morphine also increases IL-1β expression in GFAP-expressing astrocytes, which is abolished in tPA-deficient mice. In cultured astrocytes, morphine treatment increases tPA, IL-1β, and pERK expression, and the increased IL-1β and pERK expression is abolished in tPA-deficient astrocytes. tPA is also sufficient to induce IL-1β and pERK expression in astrocyte cultures. Intrathecal injection of tPA results in up-regulation of GFAP and pERK in spinal astrocytes but not up-regulation of IBA-1 in spinal microglia. Finally, intrathecal tPA elicits persistent mechanical allodynia, which is inhibited by the astroglial toxin alpha-amino adipate and the MEK (ERK kinase) inhibitor U0126. Collectively, these data suggest an important role of tPA in regulating astrocytic signaling, pain hypersensitivity, and morphine tolerance.
Keywords: acute opioid analgesia, chronic morphine exposure, extracellular signal-regulated kinase (ERK), interleukin-1 beta (IL-1β), protease, tPA knockout mice
INTRODUCTION
Mounting evidence suggests that spinal cord astrocytes play an important role in the genesis of persistent pain (Gao and Ji, 2010b; Gwak et al., 2012; Milligan and Watkins, 2009; Ren and Dubner, 2010; Svensson and Brodin, 2010). Spinal cord astrocytes are persistently activated in neuropathic and inflammatory pain conditions (Garrison et al., 1994; Garrison et al., 1991; Hulsebosch et al., 2009; Raghavendra et al., 2004; Shi et al., 2012; Zhang and De Koninck, 2006). Upon activation, spinal astrocytes produce proinflammatory cytokines (e.g., IL-1β) and chemokines (e.g., CCL2), leading to central sensitization and enhanced pain states (Gao et al., 2009; Kawasaki et al., 2008b; Ren and Dubner, 2010). Of interest, spinal cord astrocytes are also activated after chronic opioid treatment, and this activation is associated with the development of anti-nociceptive tolerance, one of the side-effects of opioid therapy (Chen et al., 2012; DeLeo et al., 2004; Raghavendra et al., 2002; Song and Zhao, 2001; Watkins et al., 2005). Chronic but not acute morphine treatment increases IL-1β levels in the spinal cord (Berta et al., 2012; Johnston et al., 2004). Further, IL-1β has been shown to antagonize acute morphine-induced analgesia and promote chronic morphine-induced antinociceptive tolerance (Berta et al., 2012; Hutchinson et al., 2008).
The upstream mechanisms controlling the astrocyte activation and IL-1β expression are not fully understood. Proinflammatory cytokines (e.g., TNF-α, IL-18) (Chen et al., 2012; Gao et al., 2010b; Miyoshi et al., 2008), growth factors (e.g., FGF-2) (Ji et al., 2006), calcitonin gene-related peptide (Wang et al., 2009), and protein kinases [e.g., ERK and c-Jun N-terminal kinase (JNK)] (Gao et al., 2010a; Zhuang et al., 2005; Zhuang et al., 2006) were implicated in the activation of spinal cord astrocytes. Accumulating evidence suggests that proteases, such as matrix metalloprotease-2 (MMP-2) are also critical for glial activation and neuropathic pain sensitization (Clark et al., 2007; Ji et al., 2009). Tissue plasminogen activator (tPA) is an extracellular serine protease that has been widely used to treat stroke by breaking down the blood clot (Wang et al., 2003). tPA is induced as an immediate-early gene during seizure, kindling, and long-term potentiation (Qian et al., 1993) and modulates long-term potentiation (LTP) and synaptic growth in the hippocampus (Baranes et al., 1998). tPA promotes axon regrowth after spinal cord injury (Bukhari et al., 2011), regulates morphine-induced hyperlocomotion (Nagai et al., 2005), reinforcing (Yan et al., 2007), and rewarding actions (Bahi and Dreyer, 2008; Nagai et al., 2004), and enhances nerve injury-induced neuropathic pain (Kozai et al., 2007).
In this study, we investigated the role of tPA in chronic morphine-induced antinociceptive tolerance and spinal cord astrocyte activation. Our data demonstrated that morphine tolerance and IL-1β induction in spinal cord astrocytes were diminished in tPA knockout mice, although nociceptive pain and acute morphine analgesia were intact in these mice. Our results also showed that intrathecal injection of tPA resulted in ERK phosphorylation in astrocytes and ERK-dependent mechanical allodynia.
EXPERIMENTAL PROCEDURES
Animals
Adult male mice (25–35 g, 8–12 weeks old) were used for behavioral and biochemical studies. tPA−/− mice and the same background C57B/6 wild-type mice were obtained from Jackson Laboratories. These mice are viable, have normal sizes, and show no obvious developmental deficits. All experiments were performed according to the guidelines of the National Institutes of Health and the International Association for the Study of Pain. All animal procedures were approved by the Institutional Animal Care and Use Committee of Harvard Medical School and Duke University. All mice were housed in cages with food and water ad libitum in a temperature and light-controlled (12 hour light-dark cycle) room.
Drugs and administration
Morphine was purchased from Hospira, freshly prepared in saline, and subcutaneously administered at the dose of 10 mg/kg for 3 to 10 days. We also purchased tPA from Feldan, the astrocyte toxin L-α-aminoadipate (L-2-AA) and the MEK inhibitor U0126 from Sigma. tPA, α-aminoadipate, and U0126 were administered via intrathecal route to cerebral-spinal fluid. For intrathecal injection, a lumbar puncture was made at L5–L6 level with a 30 gauge needle under a brief isoflurane anesthesia (Hylden and Wilcox, 1980).
Behavior testing
Animals were habituated to the testing environment daily for at least two days before baseline testing. For testing mechanical sensitivity, animals were put in boxes on an elevated metal mesh floor and allowed 30 min for habituation before examination. The plantar surface of each hindpaw was stimulated with a series of von Frey hairs with logarithmically incrementing stiffness (0.02–2.56 grams, Stoelting), presented perpendicular to the plantar surface. The 50% paw withdrawal threshold was determined using Dixon's up-down method (Chaplan et al., 1994). For testing heat sensitivity, animals were put in plastic boxes and allowed 30 min for habituation before examination. Heat sensitivity was tested by radiant heat using Hargreaves apparatus (IITC Life Science Inc.) (Hargreaves et al., 1988) and expressed as paw withdrawal latency (PWL). The radiant heat intensity was adjusted so that PWL is between 9–12 seconds, with a cut-off of 20 seconds to prevent tissue damage.
Morphine analgesia was evaluated by tail-flick in hot water (Stone et al., 1997). Briefly, tail-flick test was performed by gently holding the mouse wrapped with a terry towel and kept tail exposed. Then one third of the length of the tail was immersed into the 52°C hot water, and the response latency was recorded after removal of the whole tail from the water. A maximum cut-off value of 10 seconds was set to avoid thermal injury. The observers were unaware of the genotype.
Quantitative RT-PCR (qPCR)
Two hours after the 3rd, 5th, and 7th daily morphine or vehicle (saline) injection, animals were terminally anesthetized with isoflurane. The spinal cord segments (L4–L5, dorsal part) were rapidly removed. Total RNA was extracted using RNeasy Plus Mini kit (Qiagen). Quantity and quality of the eluted RNA samples were verified by NanoDrop spectrophotometer (ThermoFisherScientific). A total of 1 μg of RNA was reverse-transcribed using QuantiTect Reverse Transcription Kit according to the protocol of the manufacturer (Qiagen). Specific primers for tPA, GFAP and IL-1β as well as glyceraldehyde3-phosphate dehydrogenase (GAPDH, housekeeping gene) were obtained from Sigma and the sequences of the primers were described in Table 1. We performed mRNA analyses using the Mini Opticon Real-Time PCR system (Bio-Rad,Hercules,CA) according to the protocol described in our previous publications (Liu et al., 2012). The relative level of the target mRNA was quantified and normalized to GAPDH and expressed as fold changes.
Table-1.
Sequences of primers for qRT-PCR.
Western blotting
Two hours after the 5th daily morphine or vehicle (saline) injection, animals were terminally anesthetized with isoflurane and transcardially perfused with phosphate balanced saline (PBS). DRGs and spinal cord segments (L4-L5, dorsal part) were rapidly removed. Tissues and cell cultures were homogenized in a lysis buffer containing a cocktail of proteinase inhibitors and phosphatase inhibitors. The protein concentrations were determined by BCA Protein Assay (Pierce). For western blot analysis 30 μg of proteins from tissues or cell cultures were loaded for each lane and separated on SDS-PAGE gel (4–15%, Bio-Rad). After the transfer, the blots were incubated overnight at 4°C with polyclonal antibody against tPA (rabbit, 1:10000, Molecular Innovations), IL-1β (rabbit, 1:500, Chemicon), or phosphoERK (pERK, rabbit, 1:500, Cell Signaling). For loading control, the blots were probed with GAPDH antibody (rabbit, 1:20000, Sigma).
Immunohistochemistry
Two hours after the 5th daily morphine or vehicle injection, animals were terminally anesthetized with isoflurane and perfused through the ascending aorta with saline followed by 4% paraformaldehyde with 1.5% picric acid in 0.16 M PB, and the spinal cords were removed and postfixed in the same fixative overnight. Spinal cord sections (free-floating, L4-L5, 30 μm) were cut in a cryostat and processed for immunofluorescence. All the sections were blocked with 2% goat serum, and incubated over night at 4°C with the following primary antibodies (prepared in 2% goat serum): anti-tPA (rabbit, 1:10000, Molecular Innovations), anti-GFAP (glial fibrillary acidic protein, mouse, 1:1000, Chemicon), anti-IBA-1 (ionized calcium binding adapter molecule 1, rabbit, 1:1000, Wako), anti-NeuN (mouse, 1:2000, Chemicon), anti-IL-1β (rabbit, 1:500, Chemicon), or anti-phosphoERK (pERK, rabbit, 1:500, Cell Signaling). The sections were then incubated for 1 h at room temperature with Cy3- or FITC-conjugated (1:400, Jackson immunolab) or Alexa488-conjugated (1:400, Molecular probe) secondary antibody in 1% goat serum. For double immunofluorescence, sections were incubated with a mixture of polyclonal and monoclonal primary antibodies followed by a mixture of FITC- and CY3-congugated secondary antibodies (Zhuang et al., 2005). The stained sections were examined with a Nikon fluorescence microscope, and images were captured with a CCD Spot camera and analyzed with NIH Image software or Adobe PhotoShop. Some spinal cord sections were also examined under a Zeiss LSM 510 inverted confocal microscope.
Astrocyte cultures
To get high quality and large quantity of astrocytes, we prepared astrocyte cultures from cerebral cortexes of neonatal (P2) WT and tPA−/− mice, as we previously described (Gao et al., 2010b). After dissection, the cerebral hemispheres were transferred to ice-cold Hank's buffer and the meninges were carefully removed. Tissues were then minced into ~1 mm pieces, triturated, filtered through a 100 μm nylon screen, and collected by centrifugation at ~3000g for 5 min. The cell pellets were broken with a pipette and resuspended in a medium containing 15% fetal bovine serum (FBS) in low glucose Dulbecco's Modified Eagle's Medium (DMEM). After trituration, the cells were filtered through a 10 μm screen and then plated onto 6-well plates at a density of 2.5 × 105 cells/ml, and cultured for 10–12 days. The medium was replaced twice a week, first with 15% FBS, then with 10% FBS. Once the cells were grown to about 95% confluence, 0.15 mM dibutyryl cAMP (Sigma) was added to induce differentiation. Three days later, the cells were treated with morphine (100 μM) for five days or tPA (50 ng/ml) for one day. Cells were then collected and processed for Western blotting.
Quantification and statistical analysis
The density of the specific bands from Western blot gels was measured with a computer-assisted imaging analysis system (Image J, NIH). To quantify immunoreactive cells in the spinal cord, 5 nonadjacent spinal cord sections were randomly selected from the L4-L5 spinal cord segment. The intensity of tPA, GFAP, IBA-1, and pERK staining in the superficial dorsal horn (laminae I–III) was also measured with Image J. To determine the degree of double labeling, the number of GFAP/IL-1β double-labeled cells in the dorsal horn (laminae I–III) was also counted.
All the data were expressed as mean ± SEM. Differences between groups were compared using student t-test or ANOVA, followed by Newman-Keuls test. The criterion for statistical significance was P<0.05.
RESULTS
Chronic morphine-induced tolerance but not acute morphine-induced analgesia is abrogated in mice lacking tPA
We first investigated whether baseline pain sensitivity is altered in tPA knockout mice (tPA−/−, C57B/6 background). Compared to the wild-type (WT, C57B/6) control mice, tPA−/− mice showed normal thermal pain sensitivity, as assessed by paw withdrawal latency (Hargreaves test, P>0.05, Student's t-test, n=5 mice, Fig. 1A), and unaltered mechanical pain sensitivity, as evaluated by paw withdrawal threshold (Von Frey hair test, P>0.05, Student's t-test, n=5 mice, Fig. 1A). Neither did tPA−/− mice exhibit any deficits in gross anatomy and motor function (data not shown).
Figure 1. tPA−/− mice display normal nociceptive pain, unaltered acute morphine analgesia, but diminished morphine tolerance.
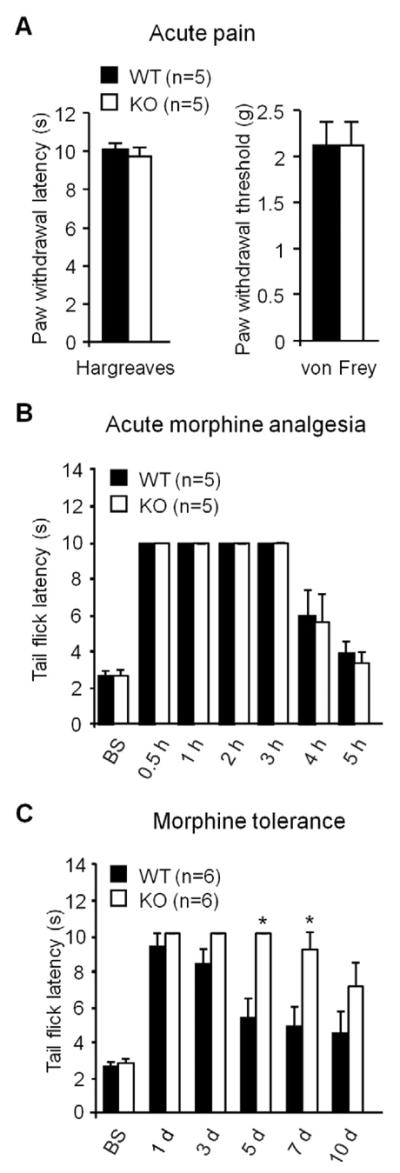
(A) Heat and mechanical sensitivity in WT and tPA−/− mice. n=5 mice. Heat and mechanical pain sensitivity was measured by radiant heat (Hargreaves) and von Frey hairs, respectively. (B) Acute morphine (10 mg/kg, s.c.)-induced analgesia in WT and tPA−/− mice. n=5 mice. Morphine analgesia was determined by tail flick latency in the hot water immersion test. (C) Chronic morphine (10 mg/kg, s.c., daily for 10 days)-induced antinociceptive tolerance in WT and tPA−/− mice. *P<0.05, compared with WT mice at the same time point, n=6 mice, t-test. Morphine analgesia was determined by tail flick latency at 30 min after the daily morphine injection.
We next tested morphine-induced acute analgesia in WT and tPA−/− mice. Subcutaneous morphine injection (10 mg/kg, s.c.) induced marked analgesia in WT mice, as determined by tail flick latency in the hot water tail immersion test (Fig. 1B). Notably, morphine-induced acute analgesia was unaltered in tPA−/− mice (Fig. 1B, P>0.05, Student's t-test, n=5 mice).
We further compared chronic morphine-induced antinociceptive tolerance in WT and tPA−/− mice. Subcutaneous morphine injections (10 mg/kg, s.c., once a day for 5 days) induced marked antinociceptive tolerance, which was evident on day 5 and maintained on day 10 (Fig. 1C). Of note, this tolerance on day 5 and 7 was significantly attenuated in tPA−/− mice (Fig. 1C, P<0.05, Student's t-test, n=6 mice). These data suggest that tPA is essential for the development of chronic morphine-induced antinociceptive tolerance.
Chronic morphine induces tPA expression in spinal cord astrocytes
We investigated the tPA expression in the dorsal root ganglia (DRGs) and spinal cords after chronic morphine exposure (5 daily injections, 10 mg/kg, s.c.). Western blotting analysis revealed no change in tPA expression in DRGs (Fig. 2A, P>0.05, t-test, n=5 mice) but a significant increase in tPA expression in the spinal cord dorsal horn (Fig. 2B, 1.47±0.14 fold of control; t-test P<0.05, n=5 mice). Immunohistochemistry also showed a significant tPA increase in the superficial dorsal horn (Fig. 2C, 1.29±0.07 fold of control, P<0.05, t-test, n=5).
Figure 2. Chronic morphine treatment induces tPA expression in spinal cord astrocytes.

(A, B) Western blotting showing tPA expression in the DRG and spinal cord dorsal horn following chronic morphine exposure (5 d). Low panels, intensity of tPA bands. *P<0.05, compared to control (Saline), t-test, n=5 mice. (C) Immunohistochemistry showing chronic morphine-induced tPA increase in the superficial dorsal horn. Low panel, intensity of tPA staining in the superficial dorsal horn. *P<0.05, t-test, compared to control (Saline), n=5 mice. Scale bar, 100 μm. (D) Confocal images showing colocalization of tPA and GFAP after chronic morphine. Scale bars, 100 μm (low magnification image) and 25 μm (high magnification images). (E) Real-time qRT-PCR analysis showing time courses of tPA, GFAP and IL-1β mRNA expression in the dorsal horn, before and 3, 5 and 7 days after chronic morphine exposure. *P<0.05, compared to control (Ctrl), t-test, n=4 mice. Spinal cord (dorsal part) and DRG tissues were collected 2 h after the 3th, 5th, or 7th daily morphine injection (10 mg/kg, s.c.).
Previous studies showed spinal tPA up-regulation in central terminals of primary afferents and astrocytes after peripheral nerve injury (Kozai et al., 2007; Yamanaka et al., 2004). To determine the cellular localization of tPA in the spinal cord after chronic morphine, we performed double staining for tPA and GFAP, a marker of astrocytes. We found that tPA was largely colocalized with GFAP (Fig. 2D). There are also some tPA single-labeled dots (GFAP-negative), possibly derived from primary afferents of DRG neurons (Fig. 2D).
To determine the correlation of tPA expression and astrocyte activation (e.g., GFAP and IL-1β expression) to the development of morphine-induced tolerance, we performed qPCR from the spinal cord samples collected 3, 5, and 7 days after daily morphine treatment. Of note, morphine treatment significantly increased tPA, GFAP, and IL-1β mRNA expression on day 5 when morphine tolerance began to develop (Fig. 1C, Fig. 2E). Like antinociceptive tolerance, morphine-induced up-regulation of tPA, GFAP, and IL-1β mRNA expression was maintained on day 7 (Fig. 2E).
Chronic morphine-induced IL-1β expression in spinal astrocytes is abolished in tPA−/− mice
Chronic morphine treatment increased GFAP expression in the dorsal horn of WT mice (Fig. 3A, B). However, this increase was not reduced in tPA−/− mice (Fig. 3A, B). Chronic morphine also increased GFAP immunofluorescence in the dorsal horn of both WT and tPA−/− mice (Fig. 3C, D, E). Thus, GFAP expression is not well correlated with morphine-induced antinociceptive tolerance.
Figure 3. Chronic morphine induces IL-1β expression in spinal cord astrocytes via tPA.

(A) Western blot showing GFAP expression in the spinal cord dorsal horn of WT and tPA−/− mice following chronic morphine exposure (5 d). (B) Quantification of the GFAP bands. *P<0.05, compared to control (Saline), t-test, n=5 mice. (C) GFAP immunostaining in the dorsal horn of WT and tPA−/− mice after chronic morphine treatment. The boxes are enlarged in D. Scale, 100 μM. (D) GFAP and IL-1β double staining in the dorsal horn of WT and tPA−/− mice after chronic morphine treatment. Arrows indicate the double-labeled cells. The cell indicated by blue arrow is enlarged in the small insert. Note that chronic morphine induces IL-1β expression in astrocytes. Scale, 25 μM. (E) Intensity of GFAP staining in the superficial dorsal horn (laminae I–III). *P<0.05, compared to WT control, ANOVA, n=5 mice. (F) Number of IL-1β/GFAP double-labeled cells in the superficial dorsal horn (laminae I–III). *P<0.05, compared to WT control, #P<0.05, compared to morphine WT group, ANOVA, n=5 mice.
We next examined morphine-induced IL-1β expression in the spinal cord. Double staining revealed a significant increase of IL-1β in GFAP-expressing astrocytes after chronic morphine (Fig. 3D and F, 1.89±0.07 fold of control, P<0.05). Importantly, chronic morphine-induced IL-1β expression was eliminated in tPA−/− mice (Fig. 3D and F, 1.12±0.12 fold of control, P<0.05). Thus, tPA is required for chronic opioid-induced IL-1β expression in spinal astrocytes.
tPA induces IL-1β and pERK expression in astrocyte cultures following chronic morphine
We investigated whether chronic morphine would directly impact astrocyte signaling in vitro. Incubation of astrocyte cultures with morphine (100 μM, 5 days) significantly increased tPA expression (P<0.05, t-test, Fig. 4A). The morphine treatment also increased the expression of pERK and IL-1β (Fig. 4B, C) in astrocyte cultures. Interestingly, both increases in pERK and IL-1β expression by morphine were abolished in tPA-deficient astrocytes (Fig. 4B, C). Furthermore, incubation of astrocyte cultures with recombinant tPA (50 ng/ml, 1 day) significantly increased the expression of pERK and IL-1β (Fig. 4D). Together, these in vitro data suggest that tPA is both sufficient and necessary for chronic morphine-induced pERK and IL-1β expression in astrocytes.
Figure 4. Chronic morphine induces IL-1β and pERK expression in astrocyte cultures via tPA.

(A) Western blot showing tPA expression following chronic morphine treatment (100 μM, 5 days) in astrocyte cultures from WT mice. The line under the gel indicates the two parts are from the same gel but not adjacent. Right panel, intensity of tPA bands. *P<0.05, compared to Control (saline), t-test, n=4 cultures. (B) Western blots showing pERK and IL-1β expression in astrocytes from WT (left blot) and tPA−/− (right blot) mice before and after morphine treatment. (C) Intensity of pERK (p42/44) and IL-1β bands in astrocytes from WT (left graph) and tPA−/− (right graph) mice before and after morphine treatment. Morphine increases pERK and IL-1β expression in wild-type but not tPA-deficient astrocytes. *P<0.05, compared to control; #P<0.05; n=4 cultures. (D) tPA (50 ng/ml, 1 day) induces pERK and IL-1β expression in WT astrocytes. *P<0.05, compared with control, n=4 cultures.
Spinal injection of tPA induces persistent mechanical allodynia via ERK and astrocyte activation
Intrathecal injection of the recombinant tPA (10 μg, i.t.) elicited robust mechanical allodynia, a reduction in paw withdrawal threshold; this allodynia maintained on day 5 but recovered on day 9 after the tPA injection (Fig. 5A). In contrast, intrathecal tPA failed to induce heat hyperalgesia, a reduction in paw withdrawal latency, in all the time points we tested (Fig. 5B).
Figure 5. Intrathecal injection of tPA elicits persistent mechanical allodynia.
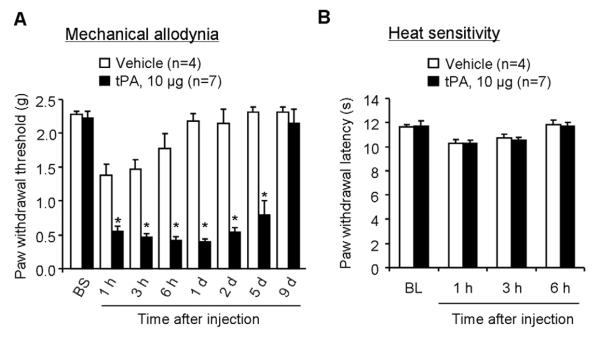
(A) A single intrathecal injection of tPA (10 μg) induces rapid and persistent mechanical allodynia, as assessed by von Frey hairs. *P<0.05, vs. PBS control, t-test. n=4–7 mice. (B) Intrathecal injection of tPA (10 μg) does not produce heat hyperalgesia, as measured by radiant heat test (Hargreaves). P>0.05, vs. vehicle control, t-test, n=4–7 mice.
Next, we tested whether spinal tPA injection also results in glial reaction, by examining the expression of IBA-1 (microglial marker), GFAP, and pERK in the spinal cord. Immunohistochemistry revealed significant increases in GFAP and pERK but not IBA-1 immunoreactivity in the dorsal horn (Fig. 6A–D). Moreover, confocal images demonstrated colocalization of pERK with GFAP in the superficial dorsal horn (Fig. 6E), indicating that tPA induces pERK in astrocytes.
Figure 6. Intrathecal injection of tPA induces pERK in astrocytes of the spinal cord dorsal horn.

(A) Immunofluorescence of IBA-1, GFAP, and pERK in the dorsal horn 2 days after intrathecal injection of tPA (10 μg) and saline. The white boxes are enlarged in the panels below. Scale bars, 100 μm. (B–D) Intensity of immunofluorescence of IBA-1 (B), GFAP (C) and pERK (D) in the superficial dorsal horn (laminae I-III) 2 days after intrathecal tPA (10 μg) and vehicle. *P<0.05, vs. vehicle control, t-test. n=5 mice. (E) Confocal images showing the double staining of pERK and GFAP in the superficial dorsal horn 2 days after intrathecal tPA (10 μg). Arrows indicate the double-labeled cells. Scale, 25 μm.
To test whether tPA induces mechanical allodynia via astrocyte signaling, we intrathecally injected an astroglial toxin, L-2-alpha aminoadipate (L-2-AA), which has been shown to inhibit inflammatory and neuropathic pain (Gao and Ji, 2010a; Zhuang et al., 2006). Intrathecal L-2-AA (50 nmol) reduced tPA-induced mechanical allodynia (Fig. 7A). tPA-induced mechanical allodynia was further suppressed by the ERK kinase (MEK) inhibitor U0126 (5 μg, i.t., Fig. 7B).
Figure 7. tPA-induced mechanical allodynia is attenuated by intrathecal inhibition of astrocytes and ERK.
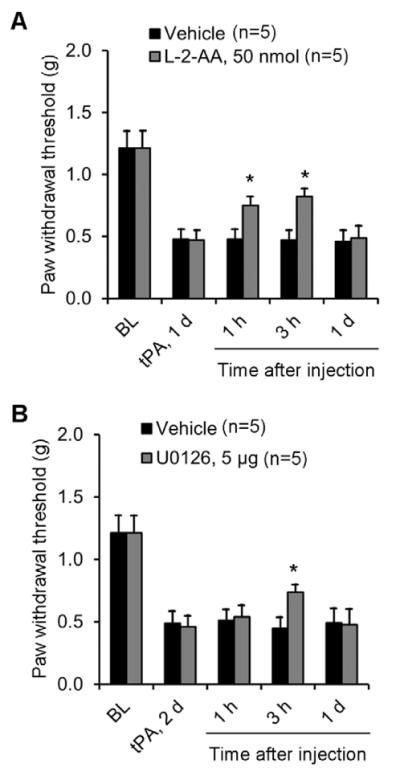
(A, B) Partial reversal of intrathecal tPA (10 μg)-induced mechanical allodynia by intrathecal injection of the astrocyte toxin L-2-AA (A) and the MEK (ERK kinase) inhibitor U0126 (B). *P<0.05, vs. saline control, t-test. n=5 mice.
DISCUSSION
Accumulating evidence suggests a role of tPA in enhancing neuropathic pain (Kozai et al., 2007; Yamanaka et al., 2004; Yamanaka et al., 2005). First, axotomy induced tPA expression in DRG neurons and spinal cord (Yamanaka et al., 2004). Second, axotomy also increased tPA inhibitor expression in DRG neurons (Yamanaka et al., 2005). Third, dorsal root injury induced tPA in DRG neurons and spinal cord astrocytes (Kozai et al., 2007). Fourth, intrathecal injection of a tPA inhibitor attenuated neuropathic pain (Kozai et al., 2007). In this study, we have provided several lines of evidence to support an essential role of tPA in chronic morphine-induced antinociceptive tolerance. First, nociceptive pain and morphine-induced acute analgesia were intact in tPA−/− mice. Second, chronic opioid increased tPA expression in the spinal cord dorsal horn, which was correlated with the development of morphine tolerance. Third, chronic morphine-induced tolerance was largely diminished in tPA−/− mice.
A previous study reported that dorsal root injury induced tPA expression in spinal astrocytes (Kozai et al., 2007). Consistently, we found tPA induction in spinal cord astrocytes after chronic morphine exposure. Since tPA is also expressed in DRG neurons (Yamanaka et al., 2004), some tPA-single-labeled dots in the dorsal horn may be derived from primary afferents (Fig. 2D). Unlike nerve injury, chronic morphine treatment did not up-regulate tPA in DRGs (Yamanaka et al., 2004) (Fig. 2A). Furthermore, chronic morphine induced tPA expression in cultured astrocytes, suggesting a direct modulation of tPA by morphine in astrocytes, although we should not exclude the possibility that morphine may also stimulate neurons or microglia for the secondary activation of astrocytes. Of interest intrathecal tPA only induced astrocytic reaction (GFAP up-regulation) but not microglial reaction (IBA-1 up-regulation) in the spinal cord (Fig. 6A–C). Since astrocyte-derived tPA causes astrocyte activation, tPA should act on astrocytes via autocrine or paracrine signaling, as demonstrated for bFGF (FGF-2) and MMP-2 (Ji et al., 2006; Kawasaki et al., 2008a). Consistently, intrathecal tPA-induced mechanical allodynia was reduced by an astrocyte toxin (Fig. 7A). Although our data supported an importance of astrocyte signaling in the development and maintenance of morphine tolerance, we also recognize a well demonstrated role of spinal microglia in the development of opioid tolerance (Horvath and DeLeo, 2009; Song and Zhao, 2001; Zhou et al., 2010). Chronic opioid was shown to activate p38 MAPK in spinal microglia for eliciting antinociceptive tolerance (Cui et al., 2006; Ji, 2010; Wang et al., 2009; Wen et al., 2011). By contrast, microglia P2X4 signaling was recently shown to modulate chronic opioid-induced hyperalgesia but not tolerance (Ferrini et al., 2013).
Our findings indicated that tPA might produce pronociceptive actions via IL-1β signaling in astrocytes. Although IL-1β antagonist or knockdown via intrathecal route potentiated acute morphine-induced analgesia, acute morphine only increases IL-1β expression in DRG satellite glial cells but not in spinal cord cells (Berta et al., 2012; Hutchinson et al., 2008; Johnston et al., 2004). Deletion or inhibition of IL-1β also inhibited morphine tolerance (DeLeo et al., 2004; Shavit et al., 2005). Furthermore, genetic polymorphism of IL-1R antagonist contributed to the variation in postoperative morphine consumption (Bessler et al., 2006). Inflammation and bone cancer induced IL-1β expression in spinal cord astrocytes (Zhang et al., 2008; Zhang et al., 2005). Our in vivo and in vitro data showed that chronic morphine also increased IL-1β expression in astrocytes. Furthermore, chronic morphine-induced IL-1β expression was abolished in tPA−/− mice (Fig. 3D,F). Upon release from astrocytes, IL-1β may act on surrounding dorsal horn neurons to enhance excitatory synaptic transmission and suppress inhibitory synaptic transmission (Kawasaki et al., 2008b), in part through increasing the phosphorylation of NMDA receptors (Guo et al., 2007; Zhang et al., 2008).
Our findings further showed that tPA enhanced pain via ERK activation in astrocytes. Chronic morphine increased pERK expression in astrocytes, and this increase was abolished in tPA−/− mice (Fig 4B, C). tPA also increased pERK and IL-1β expression in astrocyte cultures (Fig. 4D). In parallel, administration of exogenous tPA via intrathecal route increased ERK activation in spinal astrocytes (Fig. 6E). Notably, tPA-induced mechanical allodynia was inhibited by ERK inhibition (Fig. 7B). pERK induction in spinal cord astrocytes after nerve injury is critical for IL-1β expression. In turn, IL-1β also induces pERK in astrocytes (Kawasaki et al., 2008a). Consistently, chronic morphine also induced pERK in rat spinal cord astrocytes, leading to IL-1β expression (Wang et al., 2009). Further, intrathecal inhibition of ERK was shown to suppress morphine tolerance (Chen and Sommer, 2009; Wang et al., 2009).
While our data support a role of tPA in modulating astrocyte activation and persistent pain (in a few days), we should not exclude an acute role (in a few hours) of tPA in modulating neural plasticity. Indeed, intrathecal injection of tPA induced rapid mechanical allodynia at one hour (Fig. 5A) when astrocyte activation (pERK and GFAP induction) may not occur. tPA has been shown to be rapidly induced during long-term potentiation (Qian et al., 1993) and contribute to LTP and synaptic growth in the hippocampus (Baranes et al., 1998). Further, tPA was shown to convert the precursor proBDNF to the mature BDNF via activating the extracellular protease plasmin, and this conversion is critical for late-phase LTP (Pang et al., 2004). Given an important role of BDNF in inducing spinal cord neural plasticity and pain hypersensitivity (Fukuoka et al., 2001; Mannion et al., 1999), tPA may also module acute pain via BDNF activation.
In conclusion, our findings revealed a novel role of tPA in astrocyte signaling for the development of morphine tolerance and generation of pain hypersensitivity, via ERK activation and IL-1β induction in astrocytes. Targeting tPA signaling may open a new avenue for improving opioid analgesia and alleviating chronic pain.
Highlights
tPA is induced in spinal cord astrocytes after chronic morphine.
Morphine tolerance is abrogated in tPA knockout mice.
Spinal tPA injection induces persistent pain and astrocyte activation.
tPA induces ERK activation and IL-1β expression in astrocytes
ACKNOWLEDGEMENTS
This study is supported by NIH grants DE17794, DE22743, and NS67686 to RRJ and NS82985 to ZZX. TB was supported by a fellowship (PBLAP3-123417 and PA00P3-134165) from Switzerland. YC was supported by a grant from Taiwan National Science Council (97-2918-I-006-012).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- Bahi A, Dreyer JL. Overexpression of plasminogen activators in the nucleus accumbens enhances cocaine-, amphetamine- and morphine-induced reward and behavioral sensitization. Genes Brain Behav. 2008;7:244–256. doi: 10.1111/j.1601-183X.2007.00346.x. [DOI] [PubMed] [Google Scholar]
- Baranes D, Lederfein D, Huang YY, Chen M, Bailey CH, Kandel ER. Tissue plasminogen activator contributes to the late phase of LTP and to synaptic growth in the hippocampal mossy fiber pathway. Neuron. 1998;21:813–825. doi: 10.1016/s0896-6273(00)80597-8. [DOI] [PubMed] [Google Scholar]
- Berta T, Liu T, Liu YC, Xu ZZ, Ji RR. Acute morphine activates satellite glial cells and up-regulates IL-1beta in dorsal root ganglia in mice via matrix metalloprotease-9. Mol Pain. 2012;8:18. doi: 10.1186/1744-8069-8-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bessler H, Shavit Y, Mayburd E, Smirnov G, Beilin B. Postoperative pain, morphine consumption, and genetic polymorphism of IL-1beta and IL-1 receptor antagonist. Neurosci Lett. 2006;404:154–158. doi: 10.1016/j.neulet.2006.05.030. [DOI] [PubMed] [Google Scholar]
- Bukhari N, Torres L, Robinson JK, Tsirka SE. Axonal regrowth after spinal cord injury via chondroitinase and the tissue plasminogen activator (tPA)/plasmin system. J Neurosci. 2011;31:14931–14943. doi: 10.1523/JNEUROSCI.3339-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaplan SR, Bach FW, Pogrel JW, Chung JM, Yaksh TL. Quantitative assessment of tactile allodynia in the rat paw. J Neurosci Methods. 1994;53:55–63. doi: 10.1016/0165-0270(94)90144-9. [DOI] [PubMed] [Google Scholar]
- Chen ML, Cao H, Chu YX, Cheng LZ, Liang LL, Zhang YQ, Zhao ZQ. Role of P2X7 Receptor-Mediated IL-18/IL-18R Signaling in Morphine Tolerance: Multiple Glial-Neuronal Dialogues in the Rat Spinal Cord. J Pain. 2012;13:945–958. doi: 10.1016/j.jpain.2012.06.007. [DOI] [PubMed] [Google Scholar]
- Chen Y, Sommer C. The role of mitogen-activated protein kinase (MAPK) in morphine tolerance and dependence. Mol Neurobiol. 2009;40:101–107. doi: 10.1007/s12035-009-8074-z. [DOI] [PubMed] [Google Scholar]
- Clark AK, Yip PK, Grist J, Gentry C, Staniland AA, Marchand F, Dehvari M, Wotherspoon G, Winter J, Ullah J, Bevan S, Malcangio M. Inhibition of spinal microglial cathepsin S for the reversal of neuropathic pain. Proc Natl Acad Sci U S A. 2007;104:10655–10660. doi: 10.1073/pnas.0610811104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui Y, Chen Y, Zhi JL, Guo RX, Feng JQ, Chen PX. Activation of p38 mitogen-activated protein kinase in spinal microglia mediates morphine antinociceptive tolerance. Brain Res. 2006;1069:235–243. doi: 10.1016/j.brainres.2005.11.066. [DOI] [PubMed] [Google Scholar]
- DeLeo JA, Tanga FY, Tawfik VL. Neuroimmune activation and neuroinflammation in chronic pain and opioid tolerance/hyperalgesia. Neuroscientist. 2004;10:40–52. doi: 10.1177/1073858403259950. [DOI] [PubMed] [Google Scholar]
- Ferrini F, Trang T, Mattioli TA, Laffray S, Del'guidice T, Lorenzo LE, Castonguay A, Doyon N, Zhang W, Godin AG, Mohr D, Beggs S, Vandal K, Beaulieu JM, Cahill CM, Salter MW, De KY. Morphine hyperalgesia gated through microglia-mediated disruption of neuronal Cl(-) homeostasis. Nat Neurosci. 2013 doi: 10.1038/nn.3295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuoka T, Kondo E, Dai Y, Hashimoto N, Noguchi K. Brain-derived neurotrophic factor increases in the uninjured dorsal root ganglion neurons in selective spinal nerve ligation model. J Neurosci. 2001;21:4891–4900. doi: 10.1523/JNEUROSCI.21-13-04891.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao YJ, Ji RR. Light touch induces ERK activation in superficial dorsal horn neurons after inflammation: involvement of spinal astrocytes and JNK signaling in touch-evoked central sensitization and mechanical allodynia. J Neurochem. 2010a;115:505–514. doi: 10.1111/j.1471-4159.2010.06946.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao YJ, Ji RR. Targeting astrocyte signaling for chronic pain. Neurotherapeutics. 2010b;7:482–493. doi: 10.1016/j.nurt.2010.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao YJ, Xu ZZ, Liu YC, Wen YR, Decosterd I, Ji RR. The c-Jun N-terminal kinase 1 (JNK1) in spinal astrocytes is required for the maintenance of bilateral mechanical allodynia under a persistent inflammatory pain condition. Pain. 2010a;148:309–319. doi: 10.1016/j.pain.2009.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao YJ, Zhang L, Ji RR. Spinal injection of TNF-alpha-activated astrocytes produces persistent pain symptom mechanical allodynia by releasing monocyte chemoattractant protein-1. Glia. 2010b;58:1871–1880. doi: 10.1002/glia.21056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao YJ, Zhang L, Samad OA, Suter MR, Yasuhiko K, Xu ZZ, Park JY, Lind AL, Ma Q, Ji RR. JNK-induced MCP-1 production in spinal cord astrocytes contributes to central sensitization and neuropathic pain. J Neurosci. 2009;29:4096–4108. doi: 10.1523/JNEUROSCI.3623-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrison CJ, Dougherty PM, Carlton SM. GFAP expression in lumbar spinal cord of naive and neuropathic rats treated with MK-801. Exp Neurol. 1994;129:237–243. doi: 10.1006/exnr.1994.1165. [DOI] [PubMed] [Google Scholar]
- Garrison CJ, Dougherty PM, Kajander KC, Carlton SM. Staining of glial fibrillary acidic protein (GFAP) in lumbar spinal cord increases following a sciatic nerve constriction injury. Brain Res. 1991;565:1–7. doi: 10.1016/0006-8993(91)91729-k. [DOI] [PubMed] [Google Scholar]
- Guo W, Wang H, Watanabe M, Shimizu K, Zou S, LaGraize SC, Wei F, Dubner R, Ren K. Glialcytokine-neuronal interactions underlying the mechanisms of persistent pain. J Neurosci. 2007;27:6006–6018. doi: 10.1523/JNEUROSCI.0176-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gwak YS, Kang J, Unabia GC, Hulsebosch CE. Spatial and temporal activation of spinal glial cells: role of gliopathy in central neuropathic pain following spinal cord injury in rats. Exp Neurol. 2012;234:362–372. doi: 10.1016/j.expneurol.2011.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hargreaves K, Dubner R, Brown F, Flores C, Joris J. A new and sensitive method for measuring thermal nociception in cutaneous hyperalgesia. Pain. 1988;32:77–88. doi: 10.1016/0304-3959(88)90026-7. [DOI] [PubMed] [Google Scholar]
- Horvath RJ, DeLeo JA. Morphine enhances microglial migration through modulation of P2X4 receptor signaling. J Neurosci. 2009;29:998–1005. doi: 10.1523/JNEUROSCI.4595-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hulsebosch CE, Hains BC, Crown ED, Carlton SM. Mechanisms of chronic central neuropathic pain after spinal cord injury. Brain Res Rev. 2009;60:202–213. doi: 10.1016/j.brainresrev.2008.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutchinson MR, Coats BD, Lewis SS, Zhang Y, Sprunger DB, Rezvani N, Baker EM, Jekich BM, Wieseler JL, Somogyi AA, Martin D, Poole S, Judd CM, Maier SF, Watkins LR. Proinflammatory cytokines oppose opioid-induced acute and chronic analgesia. Brain Behav Immun. 2008;22:1178–1189. doi: 10.1016/j.bbi.2008.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hylden JL, Wilcox GL. Intrathecal morphine in mice: a new technique. Eur J Pharmacol. 1980;67:313–316. doi: 10.1016/0014-2999(80)90515-4. [DOI] [PubMed] [Google Scholar]
- Ji RR. Targeting microglial purinergic signaling to improve morphine analgesia. Pain. 2010;150:377–378. doi: 10.1016/j.pain.2010.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji RR, Kawasaki Y, Zhuang ZY, Wen YR, Decosterd I. Possible role of spinal astrocytes in maintaining chronic pain sensitization: review of current evidence with focus on bFGF/JNK pathway. Neuron Glia Biol. 2006;2:259–269. doi: 10.1017/S1740925X07000403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji RR, Xu ZZ, Wang X, Lo EH. Matrix metallopr otease regulation of neuropathic pain. Trends Pharmacol Sci. 2009;30:336–340. doi: 10.1016/j.tips.2009.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston IN, Milligan ED, Wieseler-Frank J, Frank MG, Zapata V, Campisi J, Langer S, Martin D, Green P, Fleshner M, Leinwand L, Maier SF, Watkins LR. A role for proinflammatory cytokines and fractalkine in analgesia, tolerance, and subsequent pain facilitation induced by chronic intrathecal morphine. J Neurosci. 2004;24:7353–7365. doi: 10.1523/JNEUROSCI.1850-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawasaki Y, Xu ZZ, Wang X, Park JY, Zhuang ZY, Tan PH, Gao YJ, Roy K, Corfas G, Lo EH, Ji RR. Distinct roles of matrix metalloproteases in the early- and late-phase development of neuropathic pain. Nat Med. 2008a;14:331–336. doi: 10.1038/nm1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawasaki Y, Zhang L, Cheng JK, Ji RR. Cytokine mechanisms of central sensitization: distinct and overlapping role of interleukin-1beta, interleukin-6, and tumor necrosis factor-alpha in regulating synaptic and neuronal activity in the superficial spinal cord. J Neurosci. 2008b;28:5189–5194. doi: 10.1523/JNEUROSCI.3338-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozai T, Yamanaka H, Dai Y, Obata K, Kobayashi K, Mashimo T, Noguchi K. Tissue type plasminogen activator induced in rat dorsal horn astrocytes contributes to mechanical hypersensitivity following dorsal root injury. Glia. 2007;55:595–603. doi: 10.1002/glia.20483. [DOI] [PubMed] [Google Scholar]
- Liu T, Berta T, Xu ZZ, Park CK, Zhang L, Lu N, Liu Q, Liu Y, Gao YJ, Liu YC, Ma Q, Dong X, Ji RR. TLR3 deficiency impairs spinal cord synaptic transmission, central sensitization, and pruritus in mice. J Clin Invest. 2012;122:2195–2207. doi: 10.1172/JCI45414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mannion RJ, Costigan M, Decosterd I, Amaya F, Ma QP, Holstege JC, Ji RR, Acheson A, Lindsay RM, Wilkinson GA, Woolf CJ. Neurotrophins: peripherally and centrally acting modulators of tactile stimulus-induced inflammatory pain hypersensitivity. Proc Natl Acad Sci U S A. 1999;96:9385–9390. doi: 10.1073/pnas.96.16.9385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milligan ED, Watkins LR. Pathological and protective roles of glia in chronic pain. Nat Rev Neurosci. 2009;10:23–36. doi: 10.1038/nrn2533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyoshi K, Obata K, Kondo T, Okamura H, Noguchi K. Interleukin-18-mediated microglia/astrocyte interaction in the spinal cord enhances neuropathic pain processing after nerve injury. J Neurosci. 2008;28:12775–12787. doi: 10.1523/JNEUROSCI.3512-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagai T, Kamei H, Ito M, Hashimoto K, Takuma K, Nabeshima T, Yamada K. Modification by the tissue plasminogen activator-plasmin system of morphine-induced dopamine release and hyperlocomotion, but not anti-nociceptive effect in mice. J Neurochem. 2005;93:1272–1279. doi: 10.1111/j.1471-4159.2005.03117.x. [DOI] [PubMed] [Google Scholar]
- Nagai T, Yamada K, Yoshimura M, Ishikawa K, Miyamoto Y, Hashimoto K, Noda Y, Nitta A, Nabeshima T. The tissue plasminogen activator-plasmin system participates in the rewarding effect of morphine by regulating dopamine release. Proc Natl Acad Sci U S A. 2004;101:3650–3655. doi: 10.1073/pnas.0306587101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang PT, Teng HK, Zaitsev E, Woo NT, Sakata K, Zhen S, Teng KK, Yung WH, Hempstead BL, Lu B. Cleavage of proBDNF by tPA/plasmin is essential for long-term hippocampal plasticity. Science. 2004;306:487–491. doi: 10.1126/science.1100135. [DOI] [PubMed] [Google Scholar]
- Qian Z, Gilbert ME, Colicos MA, Kandel ER, Kuhl D. Tissue-plasminogen activator is induced as an immediate-early gene during seizure, kindling and long-term potentiation. Nature. 1993;361:453–457. doi: 10.1038/361453a0. [DOI] [PubMed] [Google Scholar]
- Raghavendra V, Rutkowski MD, DeLeo JA. The role of spinal neuroimmune activation in morphine tolerance/hyperalgesia in neuropathic and sham-operated rats. J Neurosci. 2002;22:9980–9989. doi: 10.1523/JNEUROSCI.22-22-09980.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raghavendra V, Tanga FY, DeLeo JA. Complete Freunds adjuvant-induced peripheral inflammation evokes glial activation and proinflammatory cytokine expression in the CNS. Eur J Neurosci. 2004;20:467–473. doi: 10.1111/j.1460-9568.2004.03514.x. [DOI] [PubMed] [Google Scholar]
- Ren K, Dubner R. Interactions between the immune and nervous systems in pain. Nat Med. 2010;16:1267–1276. doi: 10.1038/nm.2234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shavit Y, Wolf G, Goshen I, Livshits D, Yirmiya R. Interleukin-1 antagonizes morphine analgesia and underlies morphine tolerance. Pain. 2005;115:50–59. doi: 10.1016/j.pain.2005.02.003. [DOI] [PubMed] [Google Scholar]
- Shi Y, Gelman BB, Lisinicchia JG, Tang SJ. Chronic-pain-associated astrocytic reaction in the spinal cord dorsal horn of human immunodeficiency virus-infected patients. J Neurosci. 2012;32:10833–10840. doi: 10.1523/JNEUROSCI.5628-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song P, Zhao ZQ. The involvement of glial cells in the development of morphine tolerance. Neurosci Res. 2001;39:281–286. doi: 10.1016/s0168-0102(00)00226-1. [DOI] [PubMed] [Google Scholar]
- Stone LS, MacMillan LB, Kitto KF, Limbird LE, Wilcox GL. The alpha2a adrenergic receptor subtype mediates spinal analgesia evoked by alpha2 agonists and is necessary for spinal adrenergic-opioid synergy. J Neurosci. 1997;17:7157–7165. doi: 10.1523/JNEUROSCI.17-18-07157.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svensson CI, Brodin E. Spinal astrocytes in pain processing: non-neuronal cells as therapeutic targets. Mol Interv. 2010;10:25–38. doi: 10.1124/mi.10.1.6. [DOI] [PubMed] [Google Scholar]
- Wang X, Lee SR, Arai K, Lee SR, Tsuji K, Rebeck GW, Lo EH. Lipoprotein receptor-mediated induction of matrix metalloproteinase by tissue plasminogen activator. Nat Med. 2003;9:1313–1317. doi: 10.1038/nm926. [DOI] [PubMed] [Google Scholar]
- Wang Z, Ma W, Chabot JG, Quirion R. Cell-type specific activation of p38 and ERK mediates calcitonin gene-related peptide involvement in tolerance to morphine-induced analgesia. FASEB J. 2009;23:2576–2586. doi: 10.1096/fj.08-128348. [DOI] [PubMed] [Google Scholar]
- Watkins LR, Hutchinson MR, Johnston IN, Maier SF. Glia: novel counter-regulators of opioid analgesia. Trends Neurosci. 2005;28:661–669. doi: 10.1016/j.tins.2005.10.001. [DOI] [PubMed] [Google Scholar]
- Wen YR, Tan PH, Cheng JK, Liu YC, Ji RR. Microglia: a promising target for treating neuropathic and postoperative pain, and morphine tolerance. J Formos Med Assoc. 2011;110:487–494. doi: 10.1016/S0929-6646(11)60074-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamanaka H, Obata K, Fukuoka T, Dai Y, Kobayashi K, Tokunaga A, Noguchi K. Tissue plasminogen activator in primary afferents induces dorsal horn excitability and pain response after peripheral nerve injury. Eur J Neurosci. 2004;19:93–102. doi: 10.1046/j.1460-9568.2003.03080.x. [DOI] [PubMed] [Google Scholar]
- Yamanaka H, Obata K, Fukuoka T, Dai Y, Kobayashi K, Tokunaga A, Noguchi K. Induction of plasminogen activator inhibitor-1 and -2 in dorsal root ganglion neurons after peripheral nerve injury. Neuroscience. 2005;132:183–191. doi: 10.1016/j.neuroscience.2004.12.003. [DOI] [PubMed] [Google Scholar]
- Yan Y, Yamada K, Mizoguchi H, Noda Y, Nagai T, Nitta A, Nabeshima T. Reinforcing effects of morphine are reduced in tissue plasminogen activator-knockout mice. Neuroscience. 2007;146:50–59. doi: 10.1016/j.neuroscience.2007.01.011. [DOI] [PubMed] [Google Scholar]
- Zhang J, De Koninck Y. Spatial and temporal relationship between monocyte chemoattractant protein-1 expression and spinal glial activation following peripheral nerve injury. J Neurochem. 2006;97:772–783. doi: 10.1111/j.1471-4159.2006.03746.x. [DOI] [PubMed] [Google Scholar]
- Zhang RX, Li A, Liu B, Wang L, Ren K, Zhang H, Berman BM, Lao L. IL-1ra alleviates inflammatory hyperalgesia through preventing phosphorylation of NMDA receptor NR-1 subunit in rats. Pain. 2008;135:232–239. doi: 10.1016/j.pain.2007.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang RX, Liu B, Wang L, Ren K, Qiao JT, Berman BM, Lao L. Spinal glial activation in a new rat model of bone cancer pain produced by prostate cancer cell inoculation of the tibia. Pain. 2005;118:125–136. doi: 10.1016/j.pain.2005.08.001. [DOI] [PubMed] [Google Scholar]
- Zhou D, Chen ML, Zhang YQ, Zhao ZQ. Involvement of spinal microglial P2X7 receptor in generation of tolerance to morphine analgesia in rats. J Neurosci. 2010;30:8042–8047. doi: 10.1523/JNEUROSCI.5377-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuang ZY, Gerner P, Woolf CJ, Ji RR. ERK is sequentially activated in neurons, microglia, and astrocytes by spinal nerve ligation and contributes to mechanical allodynia in this neuropathic pain model. Pain. 2005;114:149–159. doi: 10.1016/j.pain.2004.12.022. [DOI] [PubMed] [Google Scholar]
- Zhuang ZY, Wen YR, Zhang DR, Borsello T, Bonny C, Strichartz GR, Decosterd I, Ji RR. A peptide c-Jun N-terminal kinase (JNK) inhibitor blocks mechanical allodynia after spinal nerve ligation: respective roles of JNK activation in primary sensory neurons and spinal astrocytes for neuropathic pain development and maintenance. J Neurosci. 2006;26:3551–3560. doi: 10.1523/JNEUROSCI.5290-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]