

Abstract
Background
The numerous antigens in the Kell blood group system result from missense nucleotide changes in KEL. Antibodies to antigens in this system can be clinically important. We describe six probands whose plasma contained antibodies to high-prevalence Kell antigens, and discuss their relationship.
Study design and methods
PCR amplification, direct sequencing, RFLP assays, hemagglutination, flow cytometry, and protein modeling were performed by standard methods.
Results
Proband 1 (KUCI), and her serologically-compatible sister, were heterozygous for a nucleotide change in exon 11 (KEL*1271C/T; Ala424Val). Proband 2 (KANT) was heterozygous for KEL*1283G/T (Arg428Leu) and KEL*1216C/T (Arg406Stop) in exon 11. RBCs from Proband 1 and her sister were not agglutinated by plasma from Proband 2; however, RBCs from Proband 2 were agglutinated by plasma from Proband 1. Probands 3, 4, 5, and 6 had the KEL*1391C>T change associated with the previously reported KETI− phenotype. Proband 5 was also homozygous for KEL*905T>C encoding the K11−K17+ phenotype. Hemagglutination studies revealed an association between KUCI, KANT, KETI and K11. Protein modeling indicated that whereas Ala424 and Arg428 are clustered, Val302 and Thr464 are not.
Conclusion
Ala424 in the Kell glycoprotein is associated with the high-prevalence Kell antigen, KUCI (ISBT 006032), which is detected by the antibody of Proband 1. Arg428 is associated with the high-prevalence Kell antigen, KANT (ISBT 006033). The association between KUCI, KANT, KETI, and K11, and the results of protein modeling are discussed.
Introduction
Consisting of over 30 antigens, the Kell blood group system is one of the most polymorphic of the protein blood group systems.1-9). Antibodies to antigens in the Kell blood group system are often clinically-significant and have caused transfusion reactions, and hemolytic disease and anemia of the fetus and newborn.10 Kell antigens are carried on a type II membrane glycoprotein11 that is covalently linked in the RBC membrane to XK, a multipass protein.12 The Kell glycoprotein is a member of the Neprilysin (M13) family of Zinc-endopeptidases and cleaves big endothelins, with a preference for big endothelin-3, to produce bioactive endothelins.13
The molecular bases for the Kell antigens and phenotypes have been described. The antigens are associated with missense nucleotide changes in KEL.5-8,10,14-17 The original report describing cloning of KEL numbered the nucleotides from the first transcribed, not translated, nucleotide (GeneBank accession #M64934).11 Throughout this article, we have used the ISBT recommended system of counting ‘A’ of ‘AUG’ as #1.18 Thus, the numbers for nucleotides will be 120 less than the original numbering. The amino acid numbers remain unchanged.
Weakening of specific Kell antigens is not uncommon and is caused by several different mechanisms. For example, weak expression of K occurs with the Thr to Ser change in amino acid 19319,20 and weak expression of k occurs with a Thr to Val change in amino acid 423.1 All antigens on the same Kell glycoprotein are expressed weakly when amino acid 193 is Arg instead of Thr or Met,21 in the presence of Kpa antigen (Trp281),22,23 or in the absence of Kx protein (McLeod phenotype), and are dramatically weakened on RBCs with the Kmod phenotype.22 Kell antigens, especially K11, are expressed weakly in the absence of glycophorin C/D [Ge:−2,−3,−4 (Leach phenotype)].22 The molecular bases for Kmod and Knull phenotypes include nonsense changes, splice site changes, deletion of nucleotides, and even missense changes 24,25 [see also ISBT Red Cell Immunogenetics and Blood Group Terminology, Web Resources].18
In this report, we describe the serological characteristics and molecular basis of an absence of two new high-prevalence antigens in the Kell blood group system: KUCI (ISBT 006032) and KANT (ISBT 006033). The absence of KUCI or KANT on RBCs is associated with a missense change in exon 11 of KEL, respectively, c.1271C>T and c.1283G>T. While the study was in progress, we tested a third proband with a KEL*1391C>T (Thr464Ile) change but before we applied for an ISBT number for the antigen, the same genotype and phenotype was reported as KETI7 (ISBT 006036). We report here four KETI-negative probands and describe hemagglutination studies that show KUCI− RBCs are also KANT−; that KANT− RBCs are K11+W KUCI+ KETI+W/−; and KETI− RBCs are K11+W/−, and include a discussion of the results of protein modeling based on a published ECE-1 crystal structure.26
Case Histories
Proband 1 (KUCI−) was a 40 year-old Native American (likely Sioux) woman with Anti-Phospholipid Syndrome. She had been pregnant and transfused with two RBC products in 2004 after which she reported back pain, increase in pulse and body temperature, and hematuria. In 2007, her plasma also contained an anti-Fya that reacted (2+) in the indirect antiglobulin test (IAT) and an antibody to a high-prevalence antigen. Plasma from Proband 1 did not react with RBCs from one of her six Fy(a−) siblings (II-6 in Figure 1). The proband’s RBCs reacted very weakly in the direct antiglobulin test (DAT) due to bound IgG and C3. The DAT of the serologically-compatible sister was negative.
Figure 1. Pedigree for family of Proband 1.

The relationship of family members with the interpretation of the sequencing and PCR-RFLP analyses are shown.
Proband 2 (KANT−) was a 38-year-old French woman who had been transfused in 1978 following a peritonitis complication. Four pregnancies (1971, 1974, 1980, and 1985) were without complication. In 1986, during her fifth pregnancy, she was investigated for the identification of an antibody to a high-prevalence antigen that was strongly reactive (3+ in LISS-IAT).
Proband 3 was a 62 year-old male Caucasian patient born in Sachsen/Germany. No relationship between his parents was known; however, as they originated from this part of Germany, it is likely they carried the same, rare, allele. In 1986, the patient had an orthopedic operation when he was transfused with RBCs. One year later, when he attended hospital for a hernia operation, his plasma contained anti-K and an antibody to a high-prevalence antigen not reactive with K0 cells. Further surgical procedures, in 1991, 2008, and 2011, were managed with autologous donations. In 2008, his antibody reacted to a titer of 16 with both K+k+ and K+k− RBCs in the column agglutination technique. Papain-treated RBCs were reactive and K+ RBCs reacted more strongly. After complete absorption of the high-prevalence antibody with K−k+ RBCs, a discrete anti-K remained. Absorption with K+k− RBCs removed the anti-K and the antibody to the high-prevalence antigen.
Proband 4 was a 90 year-old Caucasian female with head trauma referred because her plasma contained an antibody to a high-prevalence antigen that initially appeared to be anti-Kpb even though her RBCs typed Kp(a−b+). We were unable to obtain any history of transfusion or pregnancy and the patient was lost to follow-up.
Proband 5 was an 88 year old Caucasian male surgical patient when his sample was investigated in 1996. His RBCs typed K−k+ Kp(a−b+) Js(a−b+) but K:−11,17; however, his serum contained an antibody to a high-prevalence antigen that was not anti-K11. Samples were tested by several reference laboratories but, despite extensive investigations, the antibody remained unidentified. The patient was lost to follow-up.
Proband 6, a 78 year-old Frenchman, was investigated in 1988 prior to surgery when his plasma contained an antibody to a high-prevalence antigen. His plasma was incompatible with all RBC samples lacking high-prevalence Kell antigens known at that time. In 2000, when the patient was hospitalized for a hip replacement, the serology was the same as in 1988. For the current study, we used a sample from our frozen inventory, as the patient was lost to follow-up.
MATERIAL and METHODS
Blood samples
Peripheral blood samples from Proband 1 (KUCI−), her eight siblings, daughter, and maternal uncle, from Proband 3 and Proband 4 were obtained and tested for antibody identification. Plasma and RBC samples from Proband 2 (KANT−), Proband 5, and Proband 6 were from our frozen repository of RBCs or unidentified antibodies to high-prevalence Kell antigens.
DNA Extraction, Polymerase Chain Reaction (PCR) and DNA Sequencing
The procedures and sequence of primers for PCR and for sequencing the amplicons were as described previously.25 The primers were synthesized by Life Technologies, Inc. (Gaithersburg, MD). Briefly, genomic DNA was prepared (QIAamp DNA Blood Mini Kit, QIAGEN, Inc., Valencia, CA) from WBCs from blood, or from buccal epithelial cells. The 19 KEL exons and their flanking intronic regions were amplified by PCR. The PCR products were separated by agarose gel electrophoresis, isolated and sequenced in forward and reverse directions either by the Nucleic Acid Analysis Laboratory of the New York Blood Center on an automated DNA sequencer (model 373XL, version 2.0; Perkin Elmer Life Sciences, Foster City, CA) or by GENEWIZ, Inc. (South Plainfield, NJ). The sequence obtained was compared with the sequence of consensus KEL (GenBank Accession number: M64934 for cDNA and NC000007 for gDNA) using Sequencher v4.9 (GeneCodes, Ann Arbor, MI), or Workbench (SDSC, CA).
Restriction fragment length polymorphism (RFLP) analysis Proband 1 (KUCI−)
Sequence analyses revealed a KEL*1271C>T (previously 1391; Ala424Val) nucleotide change, which ablated an Fnu4HI restriction enzyme site. The region of KEL that included and flanked exon 11 was amplified using the primer pair K11P-F (5′-cctcctagaggccttgctgtcaaattca-3′) and K11R (5′-gtaggaaggggtggagggatgtgg-3′).25 The 422bp PCR products from all family members were digested using Fnu4HI and analyzed on 8% acrylamide gels. The PCR amplicon of consensus KEL yielded two bands of 330 and 92bp, while that of the KUCI− variant remained uncut.
Proband 2 (KANT−)
The KEL*1283G>T (previously 1403; Arg428Leu) change does not alter a restriction enzyme cutting site. The KEL*1216C>T (previously 1336; Arg406Stop) change introduces an NlaIII restriction enzyme site. Exon 11 and its flanking intronic region was amplified as above.25 . Ten μL of the PCR product was digested with 10 units of NlaIII and analyzed on a 4% GelTwin (J.T. Baker, Phillipsburg, NJ). A 375bp PCR amplicon of the consensus KEL was not cut, while that of the KANT− variant yielded two bands of 300 and 75bp.
Probands 3-6 (KETI−)
The KEL*1391C>T (previously 1511; Thr464Ile) change ablates a BsmAI restriction enzyme site. The region of KEL that included and flanked exon 12 was amplified using the primer pair KEL11F-1 (5′-ccaagcccttttccaagggtc-3′) and KELInt13R (5′-gacagagctaagtcacccagg-3′) using PCR conditions as above.25 The 625bp PCR products were digested using BsmAI and analyzed on 8% acrylamide gels. The PCR amplicon of consensus KEL yielded three bands of 264, 195 and 166bp, while that of the KETI− variant resulted in two bands of 430 and 195bp.
RT-PCR analysis
Total RNA from Proband 1 and, as a control, Proband 2 (heterozygous for a nonsense allele and a missense allele) was isolated from 0.2 mL of peripheral blood using the TRIzol® Plus RNA Purification Kit (Invitrogen, Grand Island, NY) and reverse-transcribed using the SuperScript III kit (Invitrogen) using oligo d(T) as a primer. Amplification of the coding sequence of KEL was performed with the primer pair KellX10F (5′-GCACGCAGAAAGCTCAGCCAG-3′) and KellX12R (5′-TGATGAGGGCATCCCGGATCG-3′). Two μL of cDNA were amplified by 5U Taq DNA polymerase (HotStarTaq, QIAGEN Inc.) in a 50 μL reaction mixture containing 2.0mM MgCl2, 1× PCR buffer, 0.2mM dNTPs, and 100ng of each primer. Amplification was achieved over 35 cycles using 64°C as the annealing temperature and a final extension time of 10 minutes.
Serology
Standard hemagglutination tests were performed in tubes or with the column agglutination technique. RBCs were treated with papain, trypsin, α-chymotrypsin, dithiothreitol (DTT), or AET as described.27,28 Eluates were prepared using the Gamma Elu-Kit II™ (Immucor, Norcross, GA). For titration studies, two-fold dilutions of serum or plasma were made in 6% bovine serum albumin (BSA) diluted in phosphate buffered saline at pH 7.2 (PBS). Non-commercial reagents were from our frozen inventories and were from local patients and from numerous colleagues.
Model of the ectodomain of Kell based on the crystal structure of ECE-1
Homology models of the ectodomain of human Kell protein (hKell) were built using the ModWeb29,30 server for comparative protein structure modeling ModBase (see ModBase: Database of Comparative Protein Structure Models, Web Resources). ModWeb server uses comparative modeling by satisfaction of spatial restraints as implemented in Modeller.31 The hKell sequence (P23276; UniProtKB/Swiss-Prot database)was input into the ModWeb server as a Fasta formatted file. The server returned two reliable models (score of 1.0) based on two template proteins from protein data bank (pdb), neprilysin (NEP) with various specific and potent inhibitors (1r1h –chain A; segment 54 - 749)32 and human ECE-1 complexed with phosphoramidon (3dwb – Chain A; segment 101-770).26 However, we selected the model generated from 3dwb target protein due to its higher homology (32% [ECE-1] vs 24% [NEP]) with the hKell sequence (segment 79 -732). The quality of the model was verified using the NIH MBI Laboratory for Structural Genomics and Proteomics Structural Analysis and Verification Server (see Web Resources). The stereochemical properties were verified by Procheck software33 and the overall model was verified by What_Check34 and Verify3D.35
The homology-based model was used to further analyze the effect of variants on the hKell protein conformation using Mutagenesis module of the software Triton 4.0.36 This software uses modeller version 9V3 to model the variant protein. The modeled hKell was analyzed by the PyMol molecular graphics system V 1.50.4 (Schrödinger, LLC).
In order to gain further insight on specific interactions of the amino acids at the variant positions and their possible effect on the antigenicity in the consensus hKell and the variants, we used the Protein Interaction Calculator (PIC) server (see Web Resources, PIC.mbu.iisc.ernet.in) which computes various interactions such as disulphide bonds, hydrogen bonds, ionic interactions (e.g., salt bridge), hydrophobic interactions, aromatic-aromatic interactions, etc. from the three-dimensional coordinates of a protein structure.37
Results
Initial hemagglutination
Plasma from each of the six probands contained an antibody to a high-prevalence antigen in the Kell blood group system. This assumption was based on reactivity with reagent RBCs of common phenotype (untreated or enzyme-treated), the non-reactivity with reagent RBCs treated with DTT (or AET) or with RBCs with the K0 phenotype, and weak reactivity with RBCs with either the Kmod or McLeod phenotype. RBCs (that were negative in the DAT) from the serologically-compatible sister of Proband 1 and from Probands 2 to 6 typed K−k+ Kp(a−b+) Js(a−b+) Ku+ K12+ K13+ K14+ K18+ K19+ K22+ K26+ K27+ K29+ and Kx+. RBCs from Probands 1 and 6 typed K11+, RBCs from Proband 2 typed K11+very weak/−, RBCs from Probands 3 and 4 typed K11+/−, and RBCs from Proband 5 typed K11− K17+. (Table 1)
Table 1.
Summary of serological cross testing results
| RBCs | Anti-KUCI ISBT KEL32 |
Anti-KANT ISBT KEL33 |
Anti-KETI ISBT KEL36 |
Anti-K11 ISBT KEL11 |
Anti-K17 ISBT KEL17 |
|---|---|---|---|---|---|
| Proband 1 KUCI− |
0 | 0 | + | + | 0 |
| Proband 2 KANT− |
+ | 0 | +W/−^ | +W/−^ | 0 |
| Probands 3 and 4 KETI− |
+ | + | 0 | +/−^ | 0 |
| Proband 5 KETI− K11− |
+ | + | 0 | 0 | + |
| Proband 6 KETI− |
+ | + | 0 | + | 0 |
| K11− Control |
+ | + | + | 0 | + |
= positive (weakly) with some examples of the antibody and negative with others
DNA sequence of KEL exons and flanking intron regions and PCR-RFLP analyses
Proband 1 (KUCI−)
The DNA sequences of KEL exons and flanking intronic regions from Proband 1 revealed a heterozygous change at nucleotide KEL*1271 of C>T in exon 11. This change is novel and is predicted to encode Val instead of Ala at amino acid position 424 (Table 2). The serologically-compatible sister had the same heterozygous KEL*1271C/T result. The KEL*1271T allele has been assigned the GenBank Accession number JN020633. Careful visual examination of electropherograms from both sisters did not reveal any other changes in the 19 exons of KEL or in the intronic regions at the exon/intron junctions in either of the alleles.
Table 2.
Summary of DNA analyses
| Proband |
KEL*905T>C (Val302Ala) K11/K17‡ |
KEL*1271C>T (Ala424Val) KUCI+/−‡ |
KEL*1283G>T (Arg428Leu) KANT+/−‡ |
KEL*1391C>T (Thr464Ile) KETI+/−‡ |
|---|---|---|---|---|
| 1. KUCI− | T/T | T/Csilenced† | G/G | C/C |
| 2. KANT− | T/T | C/C | T/Gsilenced^ | C/C |
| 3. KETI− | T/T | C/C | G/G | T/T |
| 4. KETI− | T/T | C/C | G/G | T/T |
| 5. KETI− &K11− |
C/C | C/C | G/G | T/T |
| 6. KETI− | T/T | C/C | G/G | T/Csilenced† |
= despite concerted effort, the reason for silencing of the KEL allele was not determined
= silenced by a nonsense change (KEL*1216C>T; Arg406Stop)
= consensus sequence encodes the antigen listed while the missense nucleotide change encodes the absence of the antigen.
PCR-RFLP analyses using Fnu4HI confirmed that Proband 1 and her serologically-compatible sister were heterozygous KEL*1271C/T. . Presumably KEL*1271C is in the in trans allele, which is silenced by an undetermined mechanism. The PCR-RFLP assay showed that of the proband’s serologically-incompatible siblings, three were also heterozygous KEL*1271C/T. The interpretations of PCR-RFLP of testing family members from Proband 1 are given in Figure 1.
RT-PCR showed expected quality and quantity of cDNA. The 205bp PCR products obtained from the RT-PCR analysis of RNA from Proband 1 and, as a control, Proband 2, gave the expected banding pattern when digested using Fnu4HI. This finding indicates that unstable mRNA was not the reason for the silencing of the in trans KEL allele in Proband 1. It also provided additional verification that Proband 1 was heterozygous for KEL*1271C/T.
Proband 2 (KANT−)
Proband 2 had a KEL*1283G>T change in exon 11 in one allele, which is novel and predicted to encode Leu instead of Arg at amino acid position 428, and a KEL*1216C>T (Arg406Stop) change in the other allele, which is known to silence KEL24 (Table 2). The GenBank accession number for the KEL*1283T allele is JN038573.
PCR-RFLP analysis of the 375bp amplicon produced using primers, In10F and Kllp-R, from Proband 2 confirmed heterozygosity for the predicted nucleotide change, KEL*1216C/T, showing 75 and 300bp and uncut 375bp bands.
Probands 3-6 (KETI−)
Sequence analyses revealed that Probands 3 and 4 had a homozygous KEL*1391C>T change, which is predicted to encode Ile at position 464 in place of Thr.7 This was confirmed by PCR-RFLP using BsmAI. Sequence analyses and PCR-RFLP (BsmAI for KEL*1391 and HaeIII for KEL*905; −/+ WT/variant38) showed that Proband 5 was homozygous not only for KEL*1391T but also for KEL*905T>C (previously 1025), which is associated with Ala in place of Val at position 302 and explains his K11−,17+ phenotype. Proband 6 was heterozygous for KEL*1391C/T (Table 2); the cause of silencing of the apparent consensus allele was not determined.
Further analyses by hemagglutination
To provide evidence that the in trans allele was silenced in Proband 1, titration studies were performed in an attempt to detect any weakening of k antigen. RBCs from Proband 1 and her compatible sibling reacted with anti-k to a titer of 32, with scores of, respectively, 47 and 45; RBCs from Proband 2 and a K−k+ control reacted to a titer of 16, with scores of, respectively, 32 and 39. Additional titration studies using three other anti-k and RBC from Proband 1 and, her compatible sister did not reveal weakening of k on KUCI− RBCs; controls used included RBCs from two incompatible siblings with KEL*1271C/T (where the allele with the consensus sequence was expressed), Proband 2 (KANT variant and KEL silenced by a nucleotide change), and those with the K−k+, K+k+, or K+k− phenotype. Similarly, numerous experiments by flow cytometry using monoclonal anti-k (MIMA16), anti-Kpb/c (MIMA9), and anti-K14 (6-22/20W3) against RBCs from all available family members of Proband 1 and numerous controls failed to show weakening of these Kell antigens.
Titration studies were also performed with a polyclonal anti-Kx (RM) to determine if the strength of expression of Kx was increased, as might be expected of RBCs from a person heterozygous for a K0 alelle. The difference in titers and scores between the K+k+ and K0 RBC controls was not great (titer of 64, with scores of, respectively, 50, and 57). In addition, the testing with Proband 1 and her compatible sibling, two incompatible heterozyous siblings (without a silenced KEL), and Proband 2 (variant in trans to K0) were not informative.
Testing the RBCs from Proband 1 with a battery of sera from our collection of antibodies to unidentified Kell system antigens, revealed Proband 2 because the plasma (containing anti-KANT) weakly agglutinated (same strength as the DAT) RBCs from Proband 1 and was non-reactive with the DAT-negative RBCs from her serologically-compatible sister. In contrast, an eluate containing the Kell antibody from Proband 1 (anti-KUCI) agglutinated RBCs from Proband 2, showing that anti-KUCI and anti-KANT were not mutually compatible.
We also cross-tested plasma and RBCs from all six probands.. RBCs from Proband 1 were KUCI− KANT− KETI+ and K11+, while RBCs from Proband 2 were KUCI+ KANT-KETI+W/− and K11+W/−. RBCS from Probands 3, 4, 5, and 6 were KUCI+ KANT+ KETI−. RBCs from Probands 3, 4 and 6 were K11+W/− and from Proband 5 were K11−. Tests of RBCs and plasma from Probands 3, 4, 5 and 6 showed mutual compatibility. Tests showed that RBCs with the K11−17+ phenotype are KUCI+, KANT+, and KETI+. (Table 1)
Modeling of the ectodomain of Kell based on the crystal structure of ECE-1
Based on the 3D homology model of the human Kell ectodomain, we modeled the four variants (Val302Ala, Ala424Val, Arg428Leu and Thr464Ile) of hKell representing, respectively, an absence of K11, KUCI, KANT, and KETI. The predicted locations of amino acids associated with these antigens are shown in Figure 2. In addition, to gain more insights on the critical interactions in the antigenic sites we used the Protein Interaction Calculation (PIC) server.37 The calculation discerned several key interactions of residues at and near the mutation sites. For example, in the consensus hKell, Val302 interacted with Leu307, Leu335 and Val337. In the Val302Ala variant, similar interactions were preserved; however, due to the much smaller size of alanine residue the effect of hydrophobic interaction will be reduced. Similarly, the Ala424Val variant gained two additional interactions with Leu425 and Ala485 in addition to the interaction with Val427 that was also observed in the hKell. As expected, Thr464 did not have hydrophobic interactions but the variant Thr464Ile showed hydrophobic interactions with Met467 and Leu604. Concerning the hydrogen bond interactions of the residues at the variant positions, only Thr464 forms a hydrogen bond with Asn461 in the consensus type. The hydrogen bond was preserved when it was mutated to 464Ile. The PIC analysis also indicated a major salt bridge formation between Arg428 and Glu145 residues. This critical interaction is lost in the variant Leu428.
Figure 2. A ribbon diagram of the homology model of hKell based on the x-ray structure of ECE-1 (3dwb, chain A).
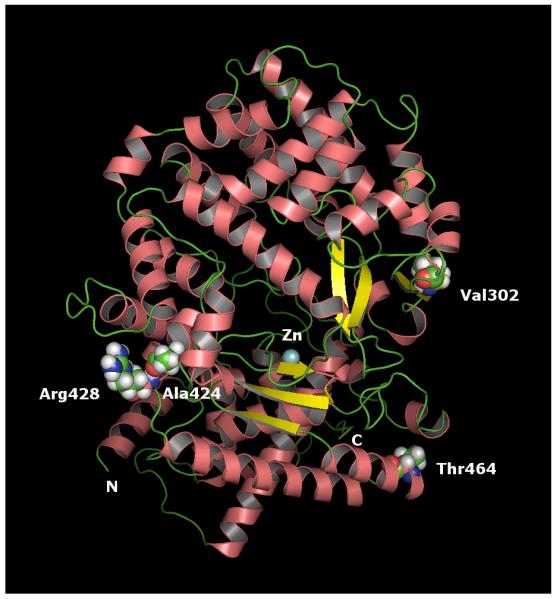
The model indicates that the structure is highly helical in nature. Helices are indicated in pink salmon, the β-sheets are in yellow and the Zinc (Zn) in cyan. The variant residues are indicated in space-filling model as spheres. The model indicates that Ala424 and Arg428 are clustered whereas Val302 and Thr464 residues are not.
Discussion
We report several novel findings: two new high-prevalence antigens in the Kell blood group system that show one-way compatibility, the first silenced KEL allele in a Native American,39 and one allele encoding an absence of two high-prevalence antigens from a variant Kell glycoprotein. We also show that K11 is expressed weakly on RBCs with either the KANT-negative or the KETI-negative phenotype while K11-negative RBCs have an apparently normal expression of both KANT and KETI antigens. Furthermore, KANT-negative RBCs have a weak expression of KETI.
Interestingly, while protein modeling did not show that an absence of KUCI, KANT, KETI, or K11 caused a noticeable change in conformation of the Kell glycoprotein, it did reveal a key interaction: the disruption of the predicted salt bridge interaction between Arg428 and Glu145, which could induce subtle changes in conformation but are not evident on the 3D model. These changes could conceivably cause the reduced expression of any Kell antigen that is in the spatial vicinity of these interactions.
The two new high-prevalence Kell antigens are named KUCI and KANT and have been assigned the ISBT numbers, respectively, KEL32 (006032) and KEL33 (006033). Both are named “K” for Kell followed by part of the probands’ names, with their permission. Our molecular analyses revealed that the two antigens are only four amino acids apart and, thereby provide an explanation for the one-way serological compatibility between these two people. Neither antigen was sensitive to protease treatment of antigen-positive RBCs.
Proband 1 and Proband 6 were heterozygous for a missense nucleotide change (respectively, KEL*1271C>T and KEL*1391C>T), which are both predicted to ablate expression of respectively, KUCI and KETI. As both probands made an alloantibody, compatible with RBCs from individuals with the same nucleotide change (respectively, anti-KUCI and anti-KETI), it is reasonable to presume that the in trans KEL allele was silenced. Despite extensive and multifaceted analyses, the reason for the silencing was not determined. However, the serologically-incompatible siblings of Proband 1 who were heterozygous KEL*1271C/T, show that their in trans allele encoded KUCI. This shows that the KEL*1271T allele does not, in some unexplained manner, silence the KEL*1271C consensus allele. We have previously studied (unpublished data) a serologically-defined K0 case (GH) in whom one allele had a nonsense nucleotide change, KEL*1719C/T (previously1839) in exon 16, but an in trans apparently normal KEL that, despite extensive analyses that included the promoter region, did not reveal the cause of the silencing. The KEL*1719C/T nonsense change has been previously published in a person with a concomitant KEL*2107G>A (previously 2227) change in exon 19 (Gly703Arg) with the Kmod phenotype.16,24,40 It is possible that nucleotide changes in the promoter or other regulatory regions could silence KEL; this possibility was not ruled. In addition, any nucleotide change in the region targeted by a primer could result in lack of amplification of specific exons, thereby leading to allelic drop out. A similar case with an undefined silenced KEL allele was reported by Moulds, et al., in 2011.41 Thus, there are a growing number of individuals who have a silenced KEL that is unexplained by current analyses at the DNA and RNA levels.
In Proband 2, the KEL*1283T allele encoding Leu in place of Arg at residue 428, confers the KANT-negative phenotype: the in trans allele (KEL*1216T) encodes a truncated Kell glycoprotein (Arg406Stop). This is consistent with the presence of anti-KANT in plasma from Proband 2. To date, no homozygous KEL*1283T person has been found and no family members from Proband 2 were available for testing. The predicted amino acid change associated with the KUCI− phenotype (Ala424Val) is only four residues from that associated with the KANT− phenotype (Arg428Leu). It is interesting that while KUCI− RBCs typed as KANT− (insufficient anti-KANT was available to perform absorption and elution studies to detect a low expression level), KANT− RBCs are KUCI+. Clearly, the 424Val change (in the KUCI− phenotype) disrupts not only KUCI but also KANT, at least as recognized by the only example of anti-KANT known. However, this change seemingly does not affect the reactivity of anti-KETI or anti-K11 with their respective epitopes. The 428Leu change in KANT− RBCs disrupts KANT but not KUCI and weakens expression of KETI and K11. Kell antigens are more likely to be conformational (discontinuous epitope) than linear (continuous epitope). Of note, when compared to alanine, valine has two additional methyl groups on its side-chain, making it physically larger and having the potential to disrupt an epitope close by, i.e., involving Arg428. In respect to the KANT epitope, the hydrophobic nature of leucine, compared to arginine, could orient it farther away from the surface of the Kell glycoprotein but not to the extent that it affects expression of KUCI. The inability of anti-KANT (KANT is at amino acid 428) to detect the KUCI antigen (at amino acid 424) is similar to that described in the Colton blood group system where a Gln47Arg amino acid change alters the folding of the Aquaporin-1 around Ala45Val (Coa/Cob), inhibiting expression of Coa or Cob as well as Co4.42
As amino acids 424 (KUCI) and 428 (KANT) are predicted to be spatially distant from amino acid 302 (K11) (Figure 2), it is hard to envision why a change at only one of these locations affects expression of K11. The valine (nonpolar) at 424 (KUCI−), instead of alanine (also nonpolar) in the consensus (KUCI+), does not seemingly affect expression of K11; however, leucine (nonpolar) at 428 (KANT−), instead of arginine (polar, positively charged) in the consensus (KANT+) does weaken expression of K11.
While investigating Probands 1 and 2, we were investigating the molecular basis of Probands 3, 4, 5, and 6 Probands 3 and 4 were homozygous for KEL*1391T. Proband 5 was extremely unusual in that he was homozygous both for KEL*1391T (KETI−) and for the missense change (KEL*905T>C) that is associated with the KEL11−11 phenotype. Proband 6 had a KEL*1391T allele and an in trans presumed silenced KEL*1391C allele. As we were investigating these four probands, the same genotype was reported with the name KETI7 (ISBT 00036). We found that KETI− RBCs had a weak expression of K11, and as such gave a connection with the KANT− phenotype. In contrast, K11− RBCs have an apparently normal expression of KUCI, KANT, and KETI antigens.
Finally, KANT− RBCs had a weak expression of KETI, but KETI− RBCs had an apparently normal expression of KANT. Although protein modeling did not reveal an explanation, the association between KUCI and KANT, between KANT and KETI, and that of K11 with KANT and KETI, are likely a consequence of conformation of the Kell glycoprotein, which is known to be highly folded.14 Proband 5 provides the first example of an allele that encodes for absence of two high-prevalence antigens on a Kell glycoprotein: the RBCs are K11−− and KETI−. This novel finding gives strength to the interpretation of Yazdanbakhsh and colleagues, who showed by transfection experiments that not finding two low-prevalence Kell antigens on the same glycoprotein is due to statistical probability and not to any constraints imposed by the structure of the glycoprotein.43
Acknowledgments
We thank Susanne Heck for performing the flow cytometry experiments, Xu Wu, Carolyn Beatty and Laima Sausais for technical assistance, Gregory Halverson for monoclonal antibodies, the staff of the Nucleic Acid Analysis Laboratory of the New York Blood Center for DNA sequencing, and Robert Ratner for assistance in preparing the manuscript and figures. We thank Rebecca Thomas from Memorial Blood Centers, St Paul, MN for technical assistance, and Jane Schafer and Dennis Reinke from Medcenter One, Bismarck, ND for clinical consultation. We also thank numerous colleagues for reagent RBCs and plasma.
Statement of Disclaimer for WF: The views expressed do not necessarily represent the view of the National Institutes of Health, the Department of Health and Human Services, or the U.S. Federal Government.
The authors certify that they have no affiliation with or financial involvement in any organization or entity with a direct financial interest in the subject matter or materials discussed in this manuscript.
This study was funded in part by NIH grant R01 HL075716 (SL)
Footnotes
Web Resources
International Society of Blood Transfusion (ISBT) Working Party on Red Cell Immunogenetics and Blood Group Terminology; ISBT on a global scale [website]; www.isbtweb.org > Working Parties > Red Cell Immunogenetics and Blood Group Terminology.
ModBase: Database of Comparative Protein Structure Models, maintained by Department of Bioengineering and Therapeutic Sciences and California Institute for Quantitative Biomedical Research, Mission Bay Campus, Byers Hall, University of California San Francisco, San Francisco, California; http://modbase.compbio.ucsf.edu
Protein Interactions Calculator (PIC), Molecular Biophysics Unit, Indians Institute of Science, Bangalore, India; http://pic.mbu.iisc.ernet.in/
Proteomics NMLoSGa. Structural Analysis and Verification Server, Los Angeles, CA: University of California, Los Angeles; 2012. http://nihserver.mbi.ucla.edu/SAVES_3/
References
- 1.Lee S. Molecular basis of Kell blood group phenotypes. Vox Sang. 1997;73:1–11. doi: 10.1046/j.1423-0410.1997.7310001.x. [DOI] [PubMed] [Google Scholar]
- 2.Lee S, Russo D, Redman CM. The Kell blood group system: Kell and XK membrane proteins. Seminars in Hematology. 2000;37:113–21. doi: 10.1016/s0037-1963(00)90036-2. [DOI] [PubMed] [Google Scholar]
- 3.Lee S, Debnath AK, Wu X, Scofield T, George T, Kakaiya RM, Yogore MG, III, Sausais L, Yacob M, Lomas-Francis C, Reid ME. Molecular basis of two novel high prevalence antigens in the Kell blood group system, KALT and KTIM. Transfusion. 2006;46:1323–7. doi: 10.1111/j.1537-2995.2006.00899.x. [DOI] [PubMed] [Google Scholar]
- 4.Uchikawa M, Onodera T, Ogasawara K, Tsuneyama H, Toyoda C, Yabe R, Enomoto T, Satake M, Nakajima K. Molecular basis for a novel low-frequency antigen in the Kell blood group system, KYO (Abstract) Vox Sanguinis. 2006;91(suppl 3):136. [Google Scholar]
- 5.Karamatic Crew V, Poole J, Watson T, Bullock T, Burton N, Daniels G. KASH (KEL34): A novel high incidence antigen in the Kell blood group system (Abstract) Vox Sanguinis. 2010;99(Suppl 1):357. [Google Scholar]
- 6.Karamatic Crew V, Poole J, Bullock T, Malde R, Burton N, Daniels G. KELP (KEL35): A new high incidence antigen in the Kell blood group defined by two homozygous missense mutations in KEL (Abstract) Transfusion Medicine. 2010;20(Suppl 1):30. [Google Scholar]
- 7.Karamatic Crew V, Poole J, Bullock T, Regan P, Burton N, Daniels G. KETI, a novel high incidence antigen in the Kell blood group system: A serological and molecular study (Abstract) Vox Sanguinis. 2011;101(Suppl. 1):19. [Google Scholar]
- 8.Vege S, Lomas-Francis C, Velliquette RW, Rodberg K, Bailey D, Horn T, Westhoff CM. A new high prevalence antigen (KHUL) in the Kell blood group system (Abstract) Transfusion. 2011;51(Suppl):25A–6A. [Google Scholar]
- 9.Lomas-Francis C, Fuuchiswa A, Uchikawa M, Tani Y, Vege S, Westhoff CW. A new high prevalence Kell antigen KYOR, antithetical to the low-prevalence antigen KYO, is the second trypsin-senstive Kell antigen. Transfusion. 2012;52(Suppl):158A–9A. [Google Scholar]
- 10.Reid ME, Lomas-Francis C, Olsson ML. The Blood Group Antigen FactsBook. 3rd ed Elsevier; 2012. [Google Scholar]
- 11.Lee S, Zambas ED, Marsh WL, Redman CM. Molecular cloning and primary structure of Kell blood group protein. Proc.Natl.Acad.Sci.USA. 1991;88:6353–7. doi: 10.1073/pnas.88.14.6353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ho M, Chelly J, Carter N, Danek A, Crocker P, Monaco AP. Isolation of the gene for McLeod syndrome that encodes a novel membrane transport protein. Cell. 1994;77:869–80. doi: 10.1016/0092-8674(94)90136-8. [DOI] [PubMed] [Google Scholar]
- 13.Lee S, Lin M, Mele A, Cao Y, Farmar J, Russo D, Redman C. Proteolytic processing of big endothelin-3 by the Kell blood group protein. Blood. 1999;94:1440–50. [PubMed] [Google Scholar]
- 14.Lee S, Russo D, Redman C. Functional and structural aspects of the Kell blood group system. Transfusion Medicine Reviews. 2000;14:93–103. doi: 10.1016/s0887-7963(00)80001-2. [DOI] [PubMed] [Google Scholar]
- 15.Lee S, Reid ME, Redman CM. Point mutations in KEL exon 8 determine a high incidence (RAZ) and a low incidence (KEL25, VLAN) antigen of the Kell blood group system. Vox Sang. 2001;81:259–63. doi: 10.1046/j.1423-0410.2001.00119.x. [DOI] [PubMed] [Google Scholar]
- 16.Lee S. The value of DNA analysis for antigens of the Kell and Kx blood group systems. Transfusion. 2007;47(Suppl):32S–9S. doi: 10.1111/j.1537-2995.2007.01308.x. [DOI] [PubMed] [Google Scholar]
- 17.Daniels G, Castilho L, Flegel WA, Fletcher A, Garratty G, Levene C, Lomas-Francis C, Moulds JM, Moulds JJ, Olsson ML, Overbeeke M, Poole J, Reid ME, Rouger P, van der Schoot E, Scott M, Sistonen P, Smart E, Storry JR, Tani Y, Yu L-C, Wendel S, Westhoff C, Yahalom V, Zelinski T. International Society of Blood Transfusion Committee on Terminology for Red Cell Surface Antigens: Macao report. Vox Sanguinis. 2009;96:153–6. doi: 10.1111/j.1423-0410.2008.01133.x. [DOI] [PubMed] [Google Scholar]
- 18.Storry JR, Castilho L, Daniels G, Flegel WA, Garratty G, Francis CL, Moulds JM, Moulds JJ, Olsson ML, Poole J, Reid ME, Rouger P, van der Schoot E, Scott M, Smart E, Tani Y, Yu LC, Wendel S, Westhoff C, Yahalom V, Zelinski T. International Society of Blood Transfusion Working Party on red cell immunogenetics and blood group terminology: Berlin report. Vox Sanguinis. 2011;101:77–82. doi: 10.1111/j.1423-0410.2010.01462.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Poole J, Warke N, Hustinx H, Taleghani BM, Martin P, Finning K, Crew VK, Green C, Bromilow I, Daniels G. A KEL gene encoding serine at position 193 of the Kell glycoprotein results in expression of KEL1 antigen. Transfusion. 2006;46:1879–85. doi: 10.1111/j.1537-2995.2006.00993.x. [DOI] [PubMed] [Google Scholar]
- 20.Lee-Stroka H, Slezak SL, Adams S, Martin J, Robbins FM, Caruccio L, Byrne KM, Stroncek DF. Another example of a KEL1 variant red cell phenotype due to a threonine to serine change at position 193 of Kell glycoprotein. Transfusion. 2008;48:925–9. doi: 10.1111/j.1537-2995.2007.01623.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Uchikawa M, Onodera T, Tsuneyama H, Enomoto T, Ishijima A, Yuasa S, Murata S, Tadokoro K, Nakajima K, Juji T. Molecular basis of unusual Kmod phenotype with K+wk− (Abstract) Vox Sang. 2000;78(Suppl 1):0011. [Google Scholar]
- 22.Øyen R, Halverson GR, Reid ME. Review: Conditions causing weak expression of Kell system antigens. Immunohematology. 1997;13:75–9. [PubMed] [Google Scholar]
- 23.Kormoczi GF, Scharberg EA, Gassner C. A novel KEL*1,3 allele with weak Kell antigen expression confirming the cis-modifier effect of KEL3. Transfusion. 2009;49:733–9. doi: 10.1111/j.1537-2995.2008.02031.x. [DOI] [PubMed] [Google Scholar]
- 24.Kormoczi GF, Wagner T, Jungbauer C, Vadon M, Ahrens N, Moll W, Muhlbacher A, Ozgul-Gulce S, Kleinrath T, Kilga-Nogler S, Schonitzer D, Gassner C. Genetic diversity of KELnull and KELel: a nationwide Austrian survey. Transfusion. 2007;47:703–14. doi: 10.1111/j.1537-2995.2007.01174.x. [DOI] [PubMed] [Google Scholar]
- 25.Lee S, Russo DCW, Reiner AP, Lee JH, Sy MY, Telen MJ, Judd WJ, Simon P, Rodrigues MJ, Chabert T, Poole J, Jovanovic-Srzentic S, Levene C, Yahalom V, Redman CM. Molecular defects underlying the Kell null phenotype. Journal of Biological Chemistry. 2001;276:27281–9. doi: 10.1074/jbc.M103433200. [DOI] [PubMed] [Google Scholar]
- 26.Schulz H, Dale GE, Karimi-Nejad Y, Oefner C. Structure of human endothelin-converting enzyme I complexed with phosphoramidon. Journal of molecular biology. 2009;385:178–87. doi: 10.1016/j.jmb.2008.10.052. [DOI] [PubMed] [Google Scholar]
- 27.Judd WJ, Johnson ST, Storry JR. Judd’s Methods in Immunohematology. 3rd ed. AABB Press; Bethesda, MD: 2008. [Google Scholar]
- 28.Mallory D. Immunohematology Methods and Procedures. American Red Cross; Rockville, MD: 1993. [Google Scholar]
- 29.Pieper U, Webb BM, Barkan DT, Schneidman-Duhovny D, Schlessinger A, Braberg H, Yang Z, Meng EC, Pettersen EF, Huang CC, Datta RS, Sampathkumar P, Madhusudhan MS, Sjolander K, Ferrin TE, Burley SK, Sali A. ModBase, a database of annotated comparative protein structure models, and associated resources. Nucleic acids research. 2011;39:D465–74. doi: 10.1093/nar/gkq1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Eswar N, John B, Mirkovic N, Fiser A, Ilyin VA, Pieper U, Stuart AC, Marti-Renom MA, Madhusudhan MS, Yerkovich B, Sali A. Tools for comparative protein structure modeling and analysis. Nucleic acids research. 2003;31:3375–80. doi: 10.1093/nar/gkg543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sali A, Blundell TL. Comparative protein modelling by satisfaction of spatial restraints. Journal of molecular biology. 1993;234:779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- 32.Oefner C, Roques BP, Fournie-Zaluski MC, Dale GE. Structural analysis of neprilysin with various specific and potent inhibitors. Acta crystallographica. Section D, Biological crystallography. 2004;60:392–6. doi: 10.1107/S0907444903027410. [DOI] [PubMed] [Google Scholar]
- 33.Laskowski RA, MacArthur MW, Moss DS, Thornton JM. PROCHECK - a program to check the stereochemical quality of protein structures. Journal of Applied Crystallography. 1993;26:283–91. [Google Scholar]
- 34.Hooft RW, Vriend G, Sander C, Abola EE. Errors in protein structures. Nature. 1996;381:272. doi: 10.1038/381272a0. [DOI] [PubMed] [Google Scholar]
- 35.Eisenberg D, Luthy R, Bowie JU. VERIFY3D: assessment of protein models with three-dimensional profiles. Methods in enzymology. 1997;277:396–404. doi: 10.1016/s0076-6879(97)77022-8. [DOI] [PubMed] [Google Scholar]
- 36.Prokop M, Adam J, Kriz Z, Wimmerova M, Koca J. TRITON: a graphical tool for ligand-binding protein engineering. Bioinformatics. 2008;24:1955–6. doi: 10.1093/bioinformatics/btn344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tina KG, Bhadra R, Srinivasan N. PIC: Protein Interactions Calculator. Nucleic acids research. 2007;35:W473–6. doi: 10.1093/nar/gkm423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lee S, Wu X, Son S, Naime D, Reid M, Okudo Y, Sistonen P, Redman C. Point mutations characterize KEL10, the KEL3, KEL4, and KEL21 alleles, and the KEL17 and KEL11 alleles. Transfusion. 1996;36:490–4. doi: 10.1046/j.1537-2995.1996.36696269505.x. [DOI] [PubMed] [Google Scholar]
- 39.Velliquette RW, Sausais L, Lomas-Francis C, Wu X, Lee S, Reid ME, Gillen B, Beatty CE, Thomas R, Schierts JD, Gentzkow K, Schafer J, Reinke D. Two novel and related high-prevalence antigens in the Kell blood group system (Abstract) Transfusion. 2007;47(Suppl):164A–5A. [Google Scholar]
- 40.Lee S, Russo DCW, Reid ME, Redman CM. Mutations that diminish expression of Kell surface protein and lead to the Kellmod red cell phenotype. Transfusion. 2003;43:1121–5. doi: 10.1046/j.1537-2995.2003.00472.x. [DOI] [PubMed] [Google Scholar]
- 41.Moulds JM, Persa R, Hue-Roye K, Reid ME. Characterization of the first K0 with anti-Ku found in an Apache Indian (Abstract) Transfusion. 2011;51(Suppl):135A. [Google Scholar]
- 42.Arnaud L, Helias V, Menanteau C, Peyrard T, Lucien N, Ripoche P, Lapegue R, Pham BN, Le Pennec PY, Moulds JJ, Cartron JP. A functional AQP1 allele producing a Co(a−b−) phenotype revises and extends the Colton blood group system. Transfusion. 2010;50:2106–16. doi: 10.1111/j.1537-2995.2010.02687.x. [DOI] [PubMed] [Google Scholar]
- 43.Yazdanbakhsh K, Lee S, Yu Q, Reid ME. Identification of a defect in the intracellular trafficking of a Kell blood group variant. Blood. 1999;94:310–8. [PubMed] [Google Scholar]