

Abstract
We hypothesized that rapamycin, through induction of autophagy and promotion of an antiapoptotic phenotype, would permit lentiviral (LV)-based transgene delivery to human T-Rapa cells, which are being tested in phase II clinical trials in the setting of allogeneic hematopoietic cell transplantation. Manufactured T-Rapa cells were exposed to supernatant enriched for a LV vector encoding a fusion protein consisting of truncated CD19 (for cell surface marking) and DTYMK/TMPKΔ, which provides “cell-fate control” due to its ability to phosphorylate (activate) AZT prodrug. LV-transduction in rapamycin-treated T-Rapa cells: (1) resulted in mitochondrial autophagy and a resultant antiapoptotic phenotype, which was reversed by the autophagy inhibitor 3-MA; (2) yielded changes in MAP1LC3B and SQSTM1 expression, which were reversed by 3-MA; and (3) increased T-Rapa cell expression of the CD19-DTYMKΔ fusion protein, despite their reduced proliferative status. Importantly, although the transgene-expressing T-Rapa cells expressed an antiapoptotic phenotype, they were highly susceptible to cell death via AZT exposure both in vitro and in vivo (in a human-into-mouse xenogeneic transplantation model). Therefore, rapamycin induction of T cell autophagy can be used for gene therapy applications, including the CD19-DTYMKΔ cell-fate control axis to improve the safety of T cell immuno-gene therapy.
Keywords: autophagy, DTYMK/TMPK, rapamycin, cell-fate control, suicide gene
Introduction
We have previously shown that rapamycin induces autophagy of primary human CD4+ T cells, resulting in an antiapoptotic T cell phenotype that confers persistent engraftment after adoptive transfer.1 These results, combined with our findings using ex vivo rapamycin in murine allogeneic transplantation models,2,3 indicate that postautophagy “T-Rapa” cells represent a particularly potent cell population for mediation of transplantation responses; indeed, in a phase II clinical trial we have shown that allogeneic donor T-Rapa cells are safely administered in the setting of low-intensity hematopoietic cell transplantation and mediate a potentially favorable balance of pro-engraftment, graft-vs.-tumor, and graft-vs.-host disease (GVHD) effects.4 As such, as we have recently reviewed,5 it is possible to ‘harness’ autophagy for the enhancement of T cell therapy.
An emerging clinical translational discipline consists of T cell immuno-gene therapy whereby ex vivo-manufactured T cells are engineered by viral vectors to express transgenes that can be of utility either for promoting therapeutic efficacy or for increasing T cell safety. In terms of efficacy, T cells expressing T cell receptors or chimeric antigen receptors specific for tumor or viral antigens can enhance anti-cancer or anti-infection effects.6-11 And, as we will focus on in this study, T cells expressing suicide genes, which we prefer to refer to as cell-fate control genes, can be utilized to increase the safety of T cell therapy. In this approach, T cells expressing a cell-fate control gene can be adoptively transferred to mediate a therapeutic effect, with subsequent deletion of the gene-modified T cell population in vivo for prevention or treatment of T cell-mediated adverse effects. T cell toxicity forms the basis for GVHD, which remains the most important complication of allogeneic hematopoietic cell transplantation.12 Cell-fate control of allogeneic T cells has been demonstrated using a TK enzyme/gancyclivor prodrug axis,13 and more recently, by a caspase-9/dimer prodrug axis.14,15 It should be noted that an ability to control the fate of adoptively transferred T cells is important not only for allogeneic transplantation, but also in the autologous transplant setting, where substantial T cell toxicity has also been observed.16-18 Given this emerging need for regulatable T cell-fate control, we have further evaluated a new cell-fate control axis that we previously developed, which includes the use of an optimized (mutated) human DTYMK enzyme that activates (phosphorylates) the prodrug AZT.19,20 This DTYMKΔ-AZT cell fate axis has potential advantages over other previously described systems because: (1) the human DTYMKΔ protein is likely to be nonimmunogenic; and (2) the prodrug AZT is approved by the US Food and Drug Administration (FDA), well-tolerated, and does not abrogate an ability to administer ganciclovir in the event of CMV infection.
To provide both potent therapeutic T cell effects and an enhanced safety profile, it will be necessary to endow T cells of enhanced in vivo efficacy such as the postautophagy, rapamycin-resistant populations, with cell-fate control mechanisms. We initiated the current project to evaluate this possibility, with inclusion of a translational focus through use of primary human CD4+ T cells and an LV manufactured by methods similar to that used for recent clinical trials.8 The specific goals of the current project were to evaluate whether: (1) postautophagy T cells represented an appropriate cellular vehicle for LV-mediated expression of the CD19-DTYMKΔ fusion transgene; and (2) such transgene-expressing T cells might be amenable to deletion by AZT.
Results
Confirmation of postautophagy T cell state at the time of lentiviral transduction
We have previously developed an ex vivo method of human CD4+ T cell manufacturing that involves addition of high-dose rapamycin, which induces BECN1-dependent autophagy as evidenced by alteration of MAP1LC3B expression, reduction in mitochondrial mass and characteristic electron microscopic changes.1,5 Using this ex vivo T cell manufacturing system, we first confirmed here that rapamycin-resistant human T cells had undergone autophagy at the time of exposure to concentrated LV preparations (day 3 of culture). Indeed, at this time point CD4+ T cells demonstrated evidence of autophagy, as defined by conversion of MAP1LC3B-I to the cleaved and lipidated MAP1LC3B-II form21 along with reduction of SQSTM1 protein level22 (Fig. 1A). As anticipated, phosphorylation of RPS6KB1 was severely decreased by rapamycin inhibition of the MTOR pathway (Fig. 1A). We also observed a significant reduction in mitochondrial mass consistent with a mitophagy process (Fig. 1B).23,24 In this setting of reduced mitochondrial mass, T-Rapa cells had a high mitochondrial membrane potential (Fig. 1C).

Figure 1. Autophagic phenotype of human T cells after treatment with rapamycin. CD4+ T cells were cultured in the presence of 1 μM rapamycin during 3 d and evaluated for autophagic markers. (A) The cells were lysed and subjected to western blotting using antibodies that detect both MAP1LC3B-I and MAP1LC3B-II (18 KDa and 16 KDa), as well as antibodies against SQSTM1 (62 KDa), phospho-RPS6KB1 (70 KDa) and ACTB (45 KDa). (B) Cells were also stained with MitoTracker Green FM (100 nM) and the mitochondrial mass examined by flow cytometry. A representative histogram is shown (left panel; Rapa treatment, black line; control, gray line; negative control, light gray line) and mean (± SEM) of the mean fluorescence intensity (MFI) of staining was determined by pooling results from three independent experiments (right panel; *p < 0.0374). (C) Mitochondrial membrane potential was evaluated by labeling cells with JC1 reagent and analyzing by flow cytometry at FL1 and FL2 channel. The ratio FL2 to FL1 (aggregate/monomer) was calculated and expressed as mean (± SEM) of four independent experiments (**p < 0.0014).
Rapamycin-treated T cells stably express high levels of CD19-DTYMKΔ transgene.
Due to the fusion protein design of our LV, the CD19 cell-surface marker and the DTYMKΔ enzyme are necessarily coexpressed in a 1:1 ratio.20 After LV transduction, T-Rapa cells and control T cells not manufactured in rapamycin had similar frequencies of transgene-expressing populations at multiplicity of infection (MOI) values that ranged from 1 to 110 (Fig. 2A). At the higher MOI values, > 80% of cells expressed CD19-DTYMKΔ, indicating that the cell-fate gene was effectively expressed in the great majority of the input T cell population even without a cell selection or enrichment step. At each condition of transduction, relative to control T cells, T-Rapa cells had higher transgene expression at each of the six MOI conditions evaluated (Fig. 2B). Furthermore, long-term transgene expression was observed in T-Rapa cells maintained in culture for approximately one month (Fig. 2C). In a second experiment, transgene expression was again relatively preserved after prolonged culture of T-Rapa cells but was greatly diminished in control T cells (Fig. 2D).

Figure 2. T-Rapa cells have higher CD19-DTYMKΔ transgene expression. CD4+ cells cultured in the presence of recombinant human IL4 were treated (rapamycin) or not (control) with rapamycin at 1 μM final concentration during three days. After that cells were washed and resuspended in fresh media containing the CD19-DTYMKΔ virus suspension at multiplicity of infection (MOI) of 1, 6, 12, 24, 61 or 110. On day 7 cells were stained with anti-human CD19 and anti-human CD4 antibodies and (A) the frequency of human CD19+ cells analyzed by flow cytometry. The data represent the mean (± SEM) of two independent experiments. (B) A representative histogram (T-Rapa cells [black line], control [gray line], nontransduced [light gray line]) of CD19 expression is shown. (C) A representative histogram of CD19 expression in T-Rapa cells on day 7 and 30 of culture, and (D) in T-Rapa cells (+R, black line) and control T cells (-R, gray line) on day 6 and 24 (left panel, experiment 1) is shown. The MFI is represented as mean (± SEM) of three independent experiments (top right of each panel); day 21: T-Rapa vs. control cells **p < 0.0064.
Inhibition of autophagy by 3-MA reverses T-Rapa cell antiapoptotic phenotype
To examine the effect of autophagy on LV infection, T-Rapa cells were treated with 3-methyladenine (3-MA), a widely used and specific inhibitor of autophagy in mammalian cells.25 Addition of 3-MA to the CD4+ T cell culture abrogated the autophagy induced by rapamycin, as evidenced by lack of MAP1LC3B-II conversion and stabilization of SQSTM1 levels (Fig. 3A).

Figure 3. Inhibition of autophagy reduces the frequency of LV infected cells and viability of T-Rapa cells. Human CD4+ T cells treated during three days with rapamycin and/or 3-MA were transduced with GFP-LV at an MOI of 15. (A) Protein extraction was obtained immediately before LV infection and analyzed by western blotting using anti-MAP1LC3B-I and MAP1LC3B-II (18 KDa and 16 KDa), anti-SQSTM1 (62 KDa) and anti-ACTB (45 KDa) antibodies. (B) The transgene expression was evaluated by flow cytometry on day 7 (left panel) and day 10 (right panel; **p < 0.0048; *p < 0.0163) post-LV infection. (C) Cells subjected to GFP-LV infection were also stained with annexin V on day 7 and analyzed by flow cytometry (***p < 0.0003; *p < 0.0120). The results are shown as mean (± SEM) of three independent experiments.
Three days post-treatment with rapamycin in the presence or absence of 3-MA, CD4+ T cells were infected with GFP-expressing LV. Initial analysis of transgene expression showed a similar frequency of transduction among cells treated with or without rapamycin (Fig. 3B). However, by day 10 of cell culture, there was increased transgene expression in T-Rapa cells relative to T cells cultured without rapamycin; addition of 3-MA to the T-Rapa cell culture abrogated the increase in transgene expression (Fig. 3B). Moreover, the transgene-expressing T-Rapa cells also had reduced apoptosis; the reduction in apoptosis was also autophagy-dependent, as the effect was abrogated by inclusion of 3-MA (Fig. 3C).
Rapamycin inhibits proliferation of CD4+ T cells without impairing LV transduction
Antagonists of MTOR such as rapamycin are known to inhibit proliferation of T cells.26,27 As such, cell yield in T-Rapa cell cultures can be influenced by both reduction in apoptosis and reduction in proliferation; to evaluate the relative contribution of these effects, we used dye labeling to track cell proliferation. As anticipated, both nontransduced (NT) and GFP-transduced T-Rapa cells had reduced proliferation relative to control T cells from day 6 to day 8 of culture (Fig. 4A and B). And, T-Rapa cells had reduced apoptosis relative to control T cells (Fig. 4C). This combination of reduced proliferation and reduced apoptosis in the T-Rapa cells resulted in an overall cell growth curve that was comparable to that of control T cells through day 27 of culture (Fig. 4D).

Figure 4. Tracking cell proliferation of CD4+ T cells post-treatment with rapamycin. T-Rapa and control cells were transduced with GFP-LV on day 3 of cell culture. (A) Cells were labeled with cell proliferation dye e670 (CPe670) on day 4, day after LV infection, and evaluated by flow cytometry on day 6, 7 and 8 of cell culture. Flow cytometry data represents the CPe670 dilution (black line; modeled populations of proliferating cells in red line) in GFP-LV transduced and NT T-Rapa and control cells on day 6. The number of cell division is shown in the top of each panel. (B) Percentage of cells divided was calculated using FlowJo software. (*p < 0.0174; **p < 0.0017). (C) At day 8 of cell proliferation analysis, the cells were also stained with annexin V. (GFP: *p < 0.0122; NT: *p < 0.0401). (D) At day 6, cells were harvested, counted and replated at 0.5 × 106 cells/ml. NT-CD4+ T cells were also evaluated. Cells were maintained under CD3 and CD28 costimulation in media containing cytokines and enumerated every 3 d. The data represent the mean (± SEM) of three independent experiments.
T-Rapa cells are susceptible to apoptosis through the DTYMKΔ-AZT axis
Previous research, which was performed primarily on tumor cell lines, demonstrated that AZT caused the death of DTYMKΔ-transduced cells by apoptosis induction.19,20 However, we found here that postautophagy primary human CD4+ T cells were apoptosis-resistant (Figs. 3C and 4C), and as such, we first evaluated whether such T cells would be susceptible to apoptosis through the DTYMKΔ-AZT cell-fate control axis. For these experiments, postautophagy T cells and control T cells were infected at an MOI of 110 in order to: (a) eliminate the need for an enrichment method for positive cells; and (b) evaluate AZT sensitivity of a relatively heterogenous T-Rapa cell population that contained low, medium, or high levels of the CD19-DTYMKΔ fusion protein. Relative to nontransduced T cells, DTYMKΔ-expressing T-Rapa cells experienced high levels of apoptosis when exposed to 50 μM AZT (Fig. 5A) with a resultant reduction in the absolute number of live cells within 72 h of prodrug exposure (Fig. 5B); similar results were obtained with 100 μM AZT treatment (Fig. 5A and B). The frequency of apoptotic DTYMKΔ-engineered T-Rapa cells increased when a higher dose of AZT was used (200 μM; Fig. 5A), which resulted in a more rapid reduction in the absolute number of live cells (Fig. 5B). In general, the sensitivity to AZT was similar between DTYMKΔ-transduced T-Rapa cells and control T cells (Fig. 5A and B).

Figure 5. AZT induces apoptosis of CD19-DTYMKΔ-expressing T-Rapa cells. Non-enriched CD19-DTYMKΔ-transduced T-Rapa and control cells were treated with AZT at final concentrations of 50 μM, 100 μM and 200 μM from day 0 to day 3. NT T cells were used as negative control. (A) Cells were stained with annexin V and analyzed by flow cytometry at 24, 48 and 72 h of AZT treatment. (B) The absolute number of viable cells was determined by Trypan Blue exclusion method. The results are shown as mean (± SEM) of three or four independent experiments. Statistical analyses were performed by comparison of results between CD19-DTYMKΔ-transduced and NT T cells. p values were smaller than *p < 0.0337; **p < 0.0082; ***p < 0.0007.
AZT selectively kills DTYMKΔ-expressing T-Rapa cells.
Having shown that DTYMKΔ-expressing T-Rapa cells were highly sensitive to apoptosis after AZT exposure, we next characterized the kinetics of the subsequent T cell death. We found that the viability of CD19-DTYMKΔ-expressing T-Rapa cells progressively declined after treatment with AZT (Fig. 6). In marked contrast, addition of stavudine (d4T), which is a thymidine prodrug molecule that is not predicted to be activated by DTYMKΔ,28 did not mediate selective killing of CD19-DTYMKΔ-expressing T-Rapa cells (Fig. 6A; stavudine concentrations between 20 to 200 μM were tested). The highest dose of AZT tested (200 μM) killed approximately 50% of transduced-cells within 24 h, whereas only 15% of cells were killed at AZT concentrations of 50 and 100 μM (Fig. 6B–D). Importantly, although these data indicate that the killing rate was dependent upon AZT dose, exhaustive depletion of all T cells (> 99.9%) was observed at each AZT concentration by days 7, 9, or 10 after initiation of AZT exposure (200, 100 and 50 μM AZT conditions, respectively). There was no significant difference in cell death induced by AZT between cells treated or not with rapamycin.

Figure 6. AZT kills CD19-DTYMKΔ-expressing T-Rapa cells. Growth curve kinetics of NT and CD19-DTYMKΔ-expressing T-Rapa and control cells, (A) nontreated or treated with 150 μM stavudine were evaluated side-by-side with cells treated with AZT at final concentrations of (B) 50 μM, (C) 100 μM and (D) 200 μM from day 0 to day 3. Cell viability was evaluated by Trypan Blue exclusion test. The results are shown as mean (± SEM) of three independent experiments [50 μM (*p < 0.0110; **p < 0.0056); 100 μM (**p < 0.0071; **p < 0.0090) and 200 μM (*p < 0.0125; *p < 0.0139)].
AZT therapy controls the in vivo fate of DTYMK-expressing T-Rapa cells
Finally, using a human-into-mouse xenogeneic transplantation model, we evaluated whether CD19-DTYMKΔ-expressing T-Rapa cells were sensitive to in vivo AZT prodrug therapy; for these experiments, we used an AZT dose previously shown to be nontoxic to unmodified human T cells.29 Murine recipients of human CD19-DTYMKΔ-expressing T-Rapa cells that received control saline injections post-transplant had an easily detectable population of human transgene-expressing T cells present in the spleen at day 30 post-transplant (Fig. 7). Relative to this control cohort, murine recipients of human CD19-DTYMKΔ-expressing T-Rapa cells that received AZT prodrug injections post-transplant had an approximate 30-fold reduction in the absolute number of human CD19-DTYMKΔ-expressing T-Rapa cells (Fig. 7).
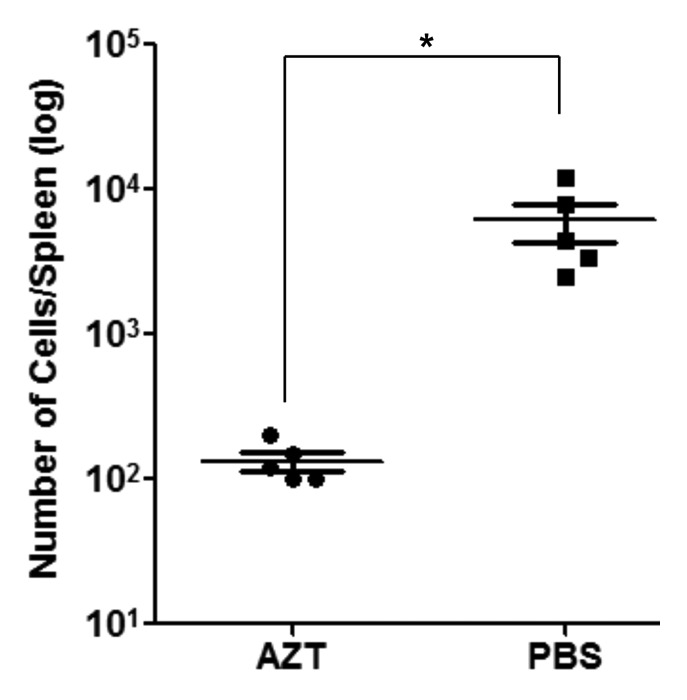
Figure 7. In vivo depletion of CD19-DTYMKΔ-modified T-Rapa cells by AZT therapy. T-Rapa cells were transduced with CD19-DTYMKΔ LV (MOI 110) and injected intravenously into NSG mice (1 million cells/ mouse). For the first 7 d after adoptive transfer, recipients were treated with either AZT (25 mg/kg/2×/day) or PBS. Thirty days later, human T cell engraftment was analyzed in the spleen. Splenocytes were counted and stained with anti-human CD19 and anti-human CD45 antibodies. The frequencies of human CD45+CD19+ cells were examined by flow cytometry. Results shown are absolute number of cells per spleen and mean (± SEM) of 5 mice per group (*p < 0.0288).
Discussion
Effective adoptive T cell therapy is facilitated by the infusion of antiapoptotic T cells such as the postautophagy T-Rapa cell population that persists and mediates increased effector function in vivo; however, the success of such effective T cell therapy is in part limited by immune-mediated toxicity against normal host tissue. Immuno-gene therapy that incorporates a cell-fate control switch can provide a safety mechanism to selectively eliminate deleterious T cell responses. The two cell-fate control approaches that have been translated to the clinic have shown promise but also may have some limitations. First, the HSV-TK enzyme used in initial clinical efforts is highly immunogenic, thereby likely limiting the in vivo persistence and immune therapeutic efficacy of transgene-expressing T cells.13,30 In a second approach, T cell expression of iCasp9 allowed dimerization of proapoptotic molecules thus resulting in rapid elimination of transgene-expressing T cells and subsequent reduction in GVHD;15 however, the dimerizing agent prodrug is not FDA approved and further clinical trials will be necessary to assure safe application of this cell-fate control axis. As an alternative to these systems, we have developed the CD19-DTYMKΔ transgene, which is a variant of human DTYMK that is unlikely to be immunogenic and has activity on dividing and non-dividing cells.19 Furthermore, the chemical compound of choice for DTYMKΔ is AZT, which is a widely utilized and FDA-approved drug; AZT is converted from a monophosphate to a diphosphate form by the catalytic activity of DTYMKΔ and then to a final triphosphate form by other enzymes.31 In this study, we have shown that the DTYMKΔ-AZT prodrug axis is suitable for cell fate control of a clinically relevant T cell population, the postautophagy primary human T-Rapa cell subset.
First, we demonstrated effective and stable LV transduction of T-Rapa cells. Rapamycin is an immunosuppressant that inhibits MTOR, thereby interfering with multiple signaling pathways that control cell proliferation, protein translation and protein phosphorylation; importantly, rapamycin also induces autophagy of mammalian cells, including T cells.1,32-34 Because rapamycin inhibits cytokine signaling and the cell cycle machinery required for T cell proliferation35,36 one might hypothesize that T cells treated in vitro with rapamycin would be refractory to LV transduction. That is, LV is known for transducing many types of nondividing cells and also for promoting stable gene expression, but one limitation is its relative inability to infect resting T cells;37 consistent with this point, in previous studies, T cell transduction in the setting of increased IL2 signaling was permissive for LV infection.38 In our system, we determined that LV transduction of T cells treated with rapamycin occurred in the relatively nonproliferative and postautophagy state, as evidenced by the reduction of MAP1LC3B-I to MAP1LC3B-II, decrease in SQSTM1 levels and by major reduction in the T cell mitochondrial mass;21 to our knowledge, there exist no reports in the literature with respect to the susceptibility of postautophagy cell populations for viral transduction. Therefore, our study shows that postautophagy T cells, the T-Rapa cell population, are actually increased in their ability to incorporate and functionally express transgenes via LV-mediated transduction. The mechanism(s) whereby the postautophagy state is permissive for viral transduction are not known and will require further investigation. Potentially, postautophagy T cells are simply able to tolerate greater viral loads on the basis of their antiapoptotic phenotype; alternatively, the membrane changes that occur during autophagy may allow for more efficient viral trafficking and integration. Nonetheless, increased transgene expression is a crucial element for the effectiveness of the cell-fate control gene therapy, and as such, we have shown that autophagy represents a novel cellular pathway to potentially improve this form of immuno-gene therapy.
Second, in this study we showed that AZT addition drives CD19-DTYMKΔ expressing postautophagy T cells to a “suicide” pathway. We and others have showed that ex vivo T cells cultured in the presence of rapamycin are more robust T cell phenotype due in part to their high expression of antiapoptotic molecules such as BCL2L1.1,3,39 Of note, the DTYMKΔ-AZT axis executes an apoptotic cascade through disruption of the mitochondrial inner membrane and activation of caspase 3, thereby resulting in apoptosis.19 Because of this potential mechanistic antagonism (favorable mitochondrial function of T-Rapa cells; mitochondrial action of DTYMKΔ-AZT axis), we reasoned that the T-Rapa cells would represent a stringent population for testing the capability of the DTYMKΔ-AZT cell-fate control axis to eliminate potentially resistant T cell populations. Indeed, after prolonged AZT exposure, even at lower doses, viable transgene positive cells could not be detected. Apoptosis of DTYMKΔ-expressing T-Rapa cells was seen across a wide range of AZT concentrations, thus confirming the functionality of DTYMKΔ-AZT axis even in an apoptosis-resistant T cell population.
Finally, we also demonstrated T-Rapa cell fate control in vivo, as a significant reduction in the absolute number of engrafted T-Rapa cells was achieved using a clinically relevant therapeutic dose of AZT. Our experiments using the clinically relevant primary human T-Rapa cell population therefore extend prior observations, where the DTYMKΔ-AZT axis was found to control the fate of tumor cell lines in vivo.19,20 Because the T-Rapa cell population has been evaluated in phase II clinical trials performed in the setting of HLA-matched sibling allogeneic transplantation and has shown evidence of therapeutic efficacy,40 our current results project that this type of cell therapy may be further optimized. Specifically, an ability to control T-Rapa cell-fate control may facilitate allogeneic T cell therapy in settings associated with increased toxicity such as haplo-identical transplantation. In sum, these collective results should pave the way for additional research to explore the potential role of autophagy induction in the improved application of gene therapy.
Materials and Methods
Manufactured T-Rapa cells and CD4+ T cells culture
Normal donor human CD4+ cells were obtained according to an IRB-approved protocol. Cells were cultured in the presence of recombinant human (rh) IL4 (1000 IU/ml) (PeproTech, 200-04), rhIL2 (20 IU/ml) (Peprotech, 200-02) and anti-CD3/CD28 microbeads (3 beads per 1 cell; antibody-coated beads kindly provided by Dr. Bruce Levine, University of Pennsylvania) in X-VIVO 20 media supplemented with 5% human AB serum (Gem Cell, 100-318). T-Rapa cells were obtained by the addition of rapamycin (Rapamune, Wyeth Pharmaceuticals Company), at the final concentration of 1 μM on day 0. T-Rapa cells were cultured during 3 d in the presence of rapamycin before harvesting. In some conditions, 3-methyladenine (3-MA) (Sigma, M9281) was also added at the final concentration of 100 μM on day 0.
Lentivector construction and preparation
The vector construction was described previously.20 The CD19-DTYMKΔF105Y fusion cassette, containing six amino-acid linker peptide between both fusion domains, was subcloned into the self-inactivating (SIN) HIV-based lentiviral backbone (pDY’cPPT-EF1α.woodchuck hepatitis virus post-transcriptional regulatory element (WPRE)).20 Lentivirus was prepared following good manufacturing practices at Lentigen Corp.
The enhanced GFP-LV was prepared by cotransfection of three plasmids, pCMV-R8.91∆, pMD.G and the GFP transfer vector into HEK-293T cells.41 The DNA was mixed into a PEI solution and then added dropwise to the cells. After 48 and 72 h of incubation at 37°C, the viral supernatants were collected and concentrated by ultracentrifugation. Viral titer was determined by serially diluted transductions of HEK-293T cells and analyzed in a FACSCalibur (Becton Dickinson) flow cytometry.
Lentivirus transduction
T-Rapa cells obtained on day 3 after treatment with rapamycin were washed and resuspended at 1.5 × 106 cells/ml with the LV supernatant. Protamine sulfate (Sigma, P4020) was added at 8 μg/ml. Half volume of fresh media was added to the cell culture at 6 h after infection. The next day, cells were centrifuged and resuspended in fresh media containing cytokines. Four days later a small aliquot of cell culture was stained with anti-human CD4 (BD PharMingen, 555346) and anti-human CD19 (BD PharMingen, 555415) antibodies for analysis of functional transduction efficiency by flow cytometry.
Western blot analysis
Human CD4+ T cells cultured during three days in supplemented media containing 1 μM rapamycin and/or 100 μM 3-MA (Sigma, M9281) were harvested and lysed in RIPA lysis buffer containing protease inhibitor cocktail (Thermo Scientific, 78429). Cell lysates were then subjected to 10 to 20% SDS-PAGE and analyzed by immunobloting. Membranes were probed using anti-MAP1LC3B (Cell Signaling, 2775), anti-SQSTM1/p62 (Cell Signaling, 5114), anti-phospho RPS6KB1 (Thr421/Ser424; Cell Signaling, 9208) and anti-ACTB (Cell Signaling, 4967) antibodies, followed by anti-rabbit immunoglobulin. The results were compared with nonrapamycin treated cells.
Mitochondrial mass analysis
MitoTracker Green FM (Molecular Probes, M7514) stock solution was prepared in DMSO according to the manufacturer's instructions. For the experiments the MitoTracker Green stock solution was further diluted in cell culture media (without serum). T-Rapa cells and control cells were stained with 100 nM MitoTracker Green FM for 30 min at 37°C. Cells were washed and the staining analyzed by flow cytometry.
Mitochondrial membrane potential assay
Human CD4+ T cells were stained with JC1 (Molecular Probes, M34152) on day 3 of cell culture following the manufacturer’s instructions. The JC1 monomer and aggregate fluorescence signal were detected on the FL1 and FL2 channels, respectively. The JC1 reagent shows potential-dependent accumulation in mitochondria that is observed by a fluorescence shift from green to orange.
Cell proliferation analysis
T-Rapa and control cells were harvested day after post-LV infection, on day 4 of cell culture. Cells were washed in PBS (phosphate-buffered saline), then stained with 10 μM Cell Proliferation Dye eFluor 670 (CPe670; eBioscience, 65-0840) during 10 min at 37°C in the dark, according to the instructions provided by the manufacturer. After that, the cells were washed and grown in supplemented cell culture media in the presence of CD3 and CD28 costimulation, and cytokines. Cell proliferation was evaluated by the dye dilution at day 6, 7 and 8 of cell culture (i.e., 48, 72 and 96 h after cell labeling). The percentage of divided (precursor) cells was calculated using the cell proliferation platform on FlowJo software (version 9.3.2). At day 8, cells were also stained with annexin V for evaluation of apoptosis.
Apoptosis analysis
T-Rapa cells and control T cells expressing the CD19-DTYMKΔ transgene or nontransduced cells were treated with AZT (Retrovir, Glaxosmithkline) at 50 μM, 100 μM and 200 μM from day 0 to day 3. Cells were harvested at 24, 48 and 72 h and processed for staining of apoptotic using annexin V PE (BD PharMingen, 559763) and 7-AAD. Analyses were performed by flow cytometry. The frequency of apoptotic cells was calculated by subtracting the amount of spontaneous apoptosis in media alone. In some conditions, human CD4+ T cells cultured in the presence of rapamycin and/or 3-MA were stained with annexin V on day 7 post-infection with GFP-LV.
Cell viability in vitro
CD19-DTYMKΔ-expressing T-Rapa cells and nontransduced cells were plated at 2 x 105 cells/ml and treated with 50 μM, 100 μM and 200 μM of AZT. AZT was added every day from day 0 to day 3. Nontransduced cells were used as control to examine background prodrug toxicity. The cells were stained with Trypan Blue and the number of live cells counted with a hemocytometer. In some experiments modified T-Rapa cells were treated with different concentrations of stavudine (Zerit, Bristol-Myers Squibb) from 20 to 200 μM.
Xenogeneic transplantation model
T-Rapa cells were transduced with the CD19-DTYMKΔ LV at an MOI of 110. A total of 1 × 106 cells were injected intravenously via retro-orbital injection into NOD/SCID/IL2Rγnull (NSG, Jackson) mice (6–8 weeks old). Mice also received 1 × 106 human monocytes to cross-present xenoantigen. The mice were treated from day 0 to day 7 with intraperitoneal injections of AZT at 25 mg/kg twice per day. A group of mice did not receive AZT and were injected with PBS. Thirty days after adoptive transfer, spleens were harvested and single cells stained with anti-human CD45 (BD PharMingen, 555483) and anti-human CD19. All experiments were performed in accordance with the guidelines of NCI Animal Care and Use Committee.
Statistical analysis
Statistical determinations of the difference between means of experimental groups were performed using Student’s 2-tailed t tests (GraphPad Prism version 5.0 software). Differences were considered statistically significant when p < 0.05.
Acknowledgments
This work was funded in part through a cooperative research agreement between NCI and Lentigen. J.A.M. and D.H.F. are inventors on a patent relating to the TMPK-AZT cell fate system. This work was supported by the Center for Cancer Research, National Cancer Institute, Intramural Research Program and by a cooperative research agreement between the NCI and Lentigen. The authors would like to thank Drs. Bruce Levine and Carl June at the Abramson Family Cancer Research Institute at the University of Pennsylvania Cancer Center for the provision of reagents and for review of the manuscript.
Glossary
Abbreviations:
- 3-MA
3-methyladenine
- ACTB
actin, beta
- AZT
azidothymidine
- BCL2L1
BCL2-like 1
- BECN1
Beclin 1
- CMV
cytomegalovirus
- DTYMK/TMPK
deoxythymidylate kinase (thymidylate kinase)
- FDA
Food and Drug Administration
- GFP
green fluorescent protein
- GVHD
graft-versus-host-disease
- HLA
human leukocyte antigen
- iCasp9
inducible caspase 9
- LV
lentiviral vector
- MAP1LC3B
microtubule-associated protein 1 light chain 3 beta
- MAP1LC3B-II
microtubule-associated protein 1 light chain 3 beta-II (the 16 kDa, cleaved and lipidated form of the 18 kDa MAP1LC3B/MAP1LC3B-I)
- MFI
mean intensity fluorescence
- MOI
multiplicity of infection
- MTOR
mechanistic target of rapamycin
- NT
nontransduced
- Rapa
rapamycin
- Rh
recombinant human
- RPS6KB1
ribosomal protein S6 kinase, 70 kDa, polypeptide 1
- SQSTM1
sequestosome 1
- TK
thymidine kinase
Disclosure of Potential Conflicts of Interest
The authors declare no conflict of interest.
Footnotes
Previously published online: www.landesbioscience.com/journals/autophagy/article/24639
References
- 1.Amarnath S, Flomerfelt FA, Costanzo CM, Foley JE, Mariotti J, Konecki DM, et al. Rapamycin generates anti-apoptotic human Th1/Tc1 cells via autophagy for induction of xenogeneic GVHD. Autophagy. 2010;6:523–41. doi: 10.4161/auto.6.4.11811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Foley JE, Jung U, Miera A, Borenstein T, Mariotti J, Eckhaus M, et al. Ex vivo rapamycin generates donor Th2 cells that potently inhibit graft-versus-host disease and graft-versus-tumor effects via an IL-4-dependent mechanism. J Immunol. 2005;175:5732–43. doi: 10.4049/jimmunol.175.9.5732. [DOI] [PubMed] [Google Scholar]
- 3.Mariotti J, Foley J, Jung U, Borenstein T, Kantardzic N, Han S, et al. Ex vivo rapamycin generates apoptosis-resistant donor Th2 cells that persist in vivo and prevent hemopoietic stem cell graft rejection. J Immunol. 2008;180:89–105. doi: 10.4049/jimmunol.180.1.89. [DOI] [PubMed] [Google Scholar]
- 4.Mossoba MEMJ, Yan X-Y, Gangopadhyay A, Winterton M, Sabatino M, Khuu H, et al. Fowler DH. T-Rapa Cell Clinical Products Contain a Balance of Minimally Differentiated Th2/Th1 Effector Cells Depleted of Treg Cells. ASH Annual Meeting, 2010:352. [Google Scholar]
- 5.Amarnath S, Fowler DH. Harnessing autophagy for adoptive T-cell therapy. Immunotherapy. 2012;4:1–4. doi: 10.2217/imt.11.144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Morgan RA, Dudley ME, Wunderlich JR, Hughes MS, Yang JC, Sherry RM, et al. Cancer regression in patients after transfer of genetically engineered lymphocytes. Science. 2006;314:126–9. doi: 10.1126/science.1129003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cohen CJ, Li YF, El-Gamil M, Robbins PF, Rosenberg SA, Morgan RA. Enhanced antitumor activity of T cells engineered to express T-cell receptors with a second disulfide bond. Cancer Res. 2007;67:3898–903. doi: 10.1158/0008-5472.CAN-06-3986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Milone MC, Fish JD, Carpenito C, Carroll RG, Binder GK, Teachey D, et al. Chimeric receptors containing CD137 signal transduction domains mediate enhanced survival of T cells and increased antileukemic efficacy in vivo. Mol Ther. 2009;17:1453–64. doi: 10.1038/mt.2009.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Carpenito C, Milone MC, Hassan R, Simonet JC, Lakhal M, Suhoski MM, et al. Control of large, established tumor xenografts with genetically retargeted human T cells containing CD28 and CD137 domains. Proc Natl Acad Sci U S A. 2009;106:3360–5. doi: 10.1073/pnas.0813101106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Porter DL, Levine BL, Kalos M, Bagg A, June CH. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N Engl J Med. 2011;365:725–33. doi: 10.1056/NEJMoa1103849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shirakura Y, Mizuno Y, Wang L, Imai N, Amaike C, Sato E, et al. T-cell receptor gene therapy targeting melanoma-associated antigen-A4 inhibits human tumor growth in non-obese diabetic/SCID/γcnull mice. Cancer Sci. 2012;103:17–25. doi: 10.1111/j.1349-7006.2011.02111.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gooley TA, Chien JW, Pergam SA, Hingorani S, Sorror ML, Boeckh M, et al. Reduced mortality after allogeneic hematopoietic-cell transplantation. N Engl J Med. 2010;363:2091–101. doi: 10.1056/NEJMoa1004383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bonini C, Ferrari G, Verzeletti S, Servida P, Zappone E, Ruggieri L, et al. HSV-TK gene transfer into donor lymphocytes for control of allogeneic graft-versus-leukemia. Science. 1997;276:1719–24. doi: 10.1126/science.276.5319.1719. [DOI] [PubMed] [Google Scholar]
- 14.Tey SK, Dotti G, Rooney CM, Heslop HE, Brenner MK. Inducible caspase 9 suicide gene to improve the safety of allodepleted T cells after haploidentical stem cell transplantation. Biol Blood Marrow Transplant. 2007;13:913–24. doi: 10.1016/j.bbmt.2007.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Di Stasi A, Tey SK, Dotti G, Fujita Y, Kennedy-Nasser A, Martinez C, et al. Inducible apoptosis as a safety switch for adoptive cell therapy. N Engl J Med. 2011;365:1673–83. doi: 10.1056/NEJMoa1106152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bolaños-Meade J, Garrett-Mayer E, Luznik L, Anders V, Webb J, Fuchs EJ, et al. Induction of autologous graft-versus-host disease: results of a randomized prospective clinical trial in patients with poor risk lymphoma. Biol Blood Marrow Transplant. 2007;13:1185–91. doi: 10.1016/j.bbmt.2007.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kline J, Subbiah S, Lazarus HM, van Besien K. Autologous graft-versus-host disease: harnessing anti-tumor immunity through impaired self-tolerance. Bone Marrow Transplant. 2008;41:505–13. doi: 10.1038/sj.bmt.1705931. [DOI] [PubMed] [Google Scholar]
- 18.Cogbill CH, Drobyski WR, Komorowski RA. Gastrointestinal pathology of autologous graft-versus-host disease following hematopoietic stem cell transplantation: a clinicopathological study of 17 cases. Mod Pathol. 2011;24:117–25. doi: 10.1038/modpathol.2010.163. [DOI] [PubMed] [Google Scholar]
- 19.Sato T, Neschadim A, Konrad M, Fowler DH, Lavie A, Medin JA. Engineered human tmpk/AZT as a novel enzyme/prodrug axis for suicide gene therapy. Mol Ther. 2007;15:962–70. doi: 10.1038/mt.sj.6300122. [DOI] [PubMed] [Google Scholar]
- 20.Scaife M, Pacienza N, Au BC, Wang JC, Devine S, Scheid E, et al. Engineered human Tmpk fused with truncated cell-surface markers: versatile cell-fate control safety cassettes. Gene Ther. 2013;20:24–34. doi: 10.1038/gt.2011.210. [DOI] [PubMed] [Google Scholar]
- 21.Mizushima N, Yoshimori T. How to interpret LC3 immunoblotting. Autophagy. 2007;3:542–5. doi: 10.4161/auto.4600. [DOI] [PubMed] [Google Scholar]
- 22.Bjørkøy G, Lamark T, Pankiv S, Øvervatn A, Brech A, Johansen T. Monitoring autophagic degradation of p62/SQSTM1. Methods Enzymol. 2009;452:181–97. doi: 10.1016/S0076-6879(08)03612-4. [DOI] [PubMed] [Google Scholar]
- 23.Rodriguez-Enriquez S, Kim I, Currin RT, Lemasters JJ. Tracker dyes to probe mitochondrial autophagy (mitophagy) in rat hepatocytes. Autophagy. 2006;2:39–46. doi: 10.4161/auto.2229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kim I, Rodriguez-Enriquez S, Lemasters JJ. Selective degradation of mitochondria by mitophagy. Arch Biochem Biophys. 2007;462:245–53. doi: 10.1016/j.abb.2007.03.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Seglen PO, Gordon PB. 3-Methyladenine: specific inhibitor of autophagic/lysosomal protein degradation in isolated rat hepatocytes. Proc Natl Acad Sci U S A. 1982;79:1889–92. doi: 10.1073/pnas.79.6.1889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mourtada-Maarabouni M, Hasan AM, Farzaneh F, Williams GT. Inhibition of human T-cell proliferation by mammalian target of rapamycin (mTOR) antagonists requires noncoding RNA growth-arrest-specific transcript 5 (GAS5) Mol Pharmacol. 2010;78:19–28. doi: 10.1124/mol.110.064055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lin JT, Lineberry NB, Kattah MG, Su LL, Utz PJ, Fathman CG, et al. Naive CD4 t cell proliferation is controlled by mammalian target of rapamycin regulation of GRAIL expression. J Immunol. 2009;182:5919–28. doi: 10.4049/jimmunol.0803986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Khan M. Immunopharmacology. Springer Science+Business Media, LLC, 2008 [Google Scholar]
- 29.Amarnath S, Mangus CW, Wang JC, Wei F, He A, Kapoor V, et al. The PDL1-PD1 axis converts human TH1 cells into regulatory T cells. Sci Transl Med. 2011;3:ra120. doi: 10.1126/scitranslmed.3003130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Berger C, Flowers ME, Warren EH, Riddell SR. Analysis of transgene-specific immune responses that limit the in vivo persistence of adoptively transferred HSV-TK-modified donor T cells after allogeneic hematopoietic cell transplantation. Blood. 2006;107:2294–302. doi: 10.1182/blood-2005-08-3503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Furman PA, Fyfe JA, St Clair MH, Weinhold K, Rideout JL, Freeman GA, et al. Phosphorylation of 3′-azido-3′-deoxythymidine and selective interaction of the 5′-triphosphate with human immunodeficiency virus reverse transcriptase. Proc Natl Acad Sci U S A. 1986;83:8333–7. doi: 10.1073/pnas.83.21.8333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Noda T, Ohsumi Y. Tor, a phosphatidylinositol kinase homologue, controls autophagy in yeast. J Biol Chem. 1998;273:3963–6. doi: 10.1074/jbc.273.7.3963. [DOI] [PubMed] [Google Scholar]
- 33.Wullschleger S, Loewith R, Hall MN. TOR signaling in growth and metabolism. Cell. 2006;124:471–84. doi: 10.1016/j.cell.2006.01.016. [DOI] [PubMed] [Google Scholar]
- 34.Díaz-Troya S, Pérez-Pérez ME, Florencio FJ, Crespo JL. The role of TOR in autophagy regulation from yeast to plants and mammals. Autophagy. 2008;4:851–65. doi: 10.4161/auto.6555. [DOI] [PubMed] [Google Scholar]
- 35.Kuo CJ, Chung J, Fiorentino DF, Flanagan WM, Blenis J, Crabtree GR. Rapamycin selectively inhibits interleukin-2 activation of p70 S6 kinase. Nature. 1992;358:70–3. doi: 10.1038/358070a0. [DOI] [PubMed] [Google Scholar]
- 36.Brennan P, Babbage JW, Thomas G, Cantrell D. p70(s6k) integrates phosphatidylinositol 3-kinase and rapamycin-regulated signals for E2F regulation in T lymphocytes. Mol Cell Biol. 1999;19:4729–38. doi: 10.1128/mcb.19.7.4729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Korin YD, Zack JA. Progression to the G1b phase of the cell cycle is required for completion of human immunodeficiency virus type 1 reverse transcription in T cells. J Virol. 1998;72:3161–8. doi: 10.1128/jvi.72.4.3161-3168.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Unutmaz D, KewalRamani VN, Marmon S, Littman DR. Cytokine signals are sufficient for HIV-1 infection of resting human T lymphocytes. J Exp Med. 1999;189:1735–46. doi: 10.1084/jem.189.11.1735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Slavik JM, Lim DG, Burakoff SJ, Hafler DA. Rapamycin-resistant proliferation of CD8+ T cells correlates with p27kip1 down-regulation and bcl-xL induction, and is prevented by an inhibitor of phosphoinositide 3-kinase activity. J Biol Chem. 2004;279:910–9. doi: 10.1074/jbc.M209733200. [DOI] [PubMed] [Google Scholar]
- 40.Fowler DHMM, Schuver BB, Layton P, Hakin FT, Kurlander R, et al. Adoptive transfer of Treg-depleted donor Th1 and Th2 cells safely accelerates alloengraftment after low-intensity chemotherapy. Blood (ASH Annual Meeting Abstracts) 2010:521. [Google Scholar]
- 41.Felizardo TC, Wang JC, McGray RA, Evelegh C, Spaner DE, Fowler DH, et al. Differential immune responses mediated by adenovirus- and lentivirus-transduced DCs in a HER-2/neu overexpressing tumor model. Gene Ther. 2011;18:986–95. doi: 10.1038/gt.2011.53. [DOI] [PMC free article] [PubMed] [Google Scholar]