

Abstract
The class III phosphatidylinositol (PtdIns)-3 kinase, PIK3C3/VPS34, forms multiple complexes and regulates a variety of cellular functions, especially in intracellular vesicle trafficking and autophagy. Even though PtdIns3P, the product of PIK3C3, is thought to be a critical membrane marker for the autophagosome, it is unclear how PIK3C3 is regulated in response to autophagy-inducing stimuli. A complexity of PIK3C3 biology is due in part to the existence of multiple complexes, of which the ATG14- or UVRAG-containing complexes play important roles in autophagy. We recently discovered differential regulation of distinct PIK3C3 complexes in response to energy starvation and showed a mechanism by which AMPK directly phosphorylates PIK3C3 and BECN1 to regulate non- and pro-autophagic PIK3C3 complexes, respectively.
Keywords: VPS34 complexes, AMPK, BECN1, ATG14, autophagy
Extensive studies have established that PIK3C3 produces PtdIns3P on the phagophore to recruit the autophagy machinery. A variety of proteins, such as PIK3R4/VPS15, BECN1, ATG14/Atg14L/Barkor, UVRAG, VMP1, AMBRA1, SH3GLB1/Bif-1, and KIAA0226/Rubicon, bind to PIK3C3 in various combinations to form many different PIK3C3 complexes, indicating a broad function of PIK3C3 in cellular regulation. Indeed, PtdIns3P is a key factor in endocytosis and endosome-to-Golgi retrograde trafficking. Although PtdIns3P is necessary for autophagy, several studies have documented that PIK3C3 activity and PtdIns3P levels are decreased under autophagy-inducing conditions.
In our recent study, we found differential regulation of distinct PIK3C3 complexes in response to energy stress. Glucose starvation decreases the cellular energy level and initiates a wide spectrum of compensatory responses, among which autophagy plays important roles in maintaining cellular homeostasis. To study PIK3C3 regulation, four highly-enriched distinct PIK3C3 complexes, VIC1 (Vps34-containing PtdIns3K-III Complex without BECN1, ATG14, or UVRAG), VIC2 (VIC containing BECN1), VIC3 (VIC containing both BECN1 and ATG14), and VIC4 (VIC containing both BECN1 and UVRAG) were prepared (Fig. 1). Considering that deletion of either ATG14 or UVRAG suppresses autophagy, these four PIK3C3 complexes represent both non- (VIC1 and VIC2) and pro- (VIC3 and VIC4) autophagy complexes. In vitro kinase assays demonstrate that the nonautophagic PIK3C3 complexes are inhibited whereas the pro-autophagic complexes are activated by glucose starvation. Notably, most of the PIK3C3 is not in a complex with ATG14 or UVRAG, and thus exists in nonautophagy complexes. In line with this observation, glucose starvation decreases total cellular PtdIPns3P levels, but the pool of autophagy-related PtdIPns3P is increased. Therefore, different PIK3C3 complexes must be differentially regulated to exert their discrete functions.
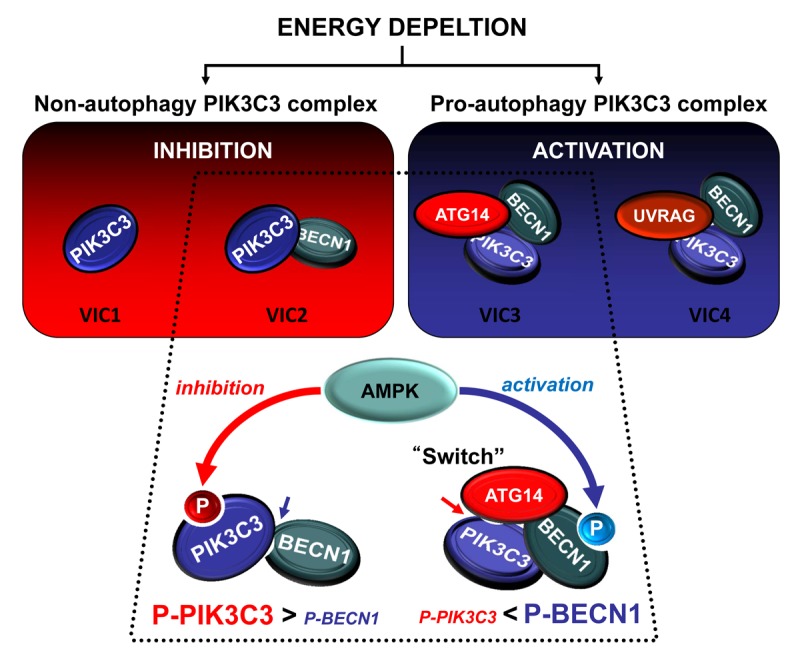
Figure 1. A proposed PIK3C3 regulatory mechanism in response to energy starvation. AMPK phosphorylates PIK3C3 and BECN1 to inhibit the nonautophagy PIK3C3 complex as well as activate the pro-autophagy PIK3C3 complex in response to energy starvation.
The regulation of PIK3C3 complex activity by glucose starvation is abolished in PRKAA/AMPKα1/α2 double knockout MEFs. In vitro AMPK treatment results in inhibition and activation of nonautophagic and pro-autophagic PIK3C3 complexes, respectively. Furthermore, this in vitro regulation is reversed by lambda phosphatase treatment. These results demonstrate that AMPK regulates PIK3C3 complexes by direct phosphorylation. PIK3C3 T163/S165 and BECN1 S91/S94 were identified to be direct AMPK phosphorylation sites. Functional analyses indicate that phosphorylation of PIK3C3 T163/S165 inhibits nonautophagic PIK3C3 complexes, whereas phosphorylation of BECN1 S91/S94 activates pro-autophagic PIK3C3 complexes. MEFs expressing BECN1 S91/S94A mutant were defective in autophagy in response to glucose starvation. However, MEFs expressing PIK3C3 T163/S165A were normal in autophagy but failed to decrease total PtdIPns3P upon glucose starvation. These data support the idea that PIK3C3 and BECN1 phosphorylation are required for different cellular functions; inhibition of nonautophagy PIK3C3 complexes and activation of pro-autophagy complexes, respectively.
Similar to glucose starvation, amino acid starvation also differentially regulates PIK3C3 complexes inhibiting nonautophagic PIK3C3 complexes and activating pro-autophagic complexes. However, neither AMPK nor BECN1 S91/S94 phosphorylation is required for pro-autophagy PIK3C3 activation by amino acid starvation, suggesting that an additional layer of mechanism is involved in PIK3C3 regulation.
It is intriguing how AMPK exerts opposite effects on VIC2 and VIC3 although both contain PIK3C3 and BECN1. We observed that the preference of AMPK-mediated phosphorylation was different between VIC2 and VIC3. BECN1 S91/S94 phosphorylation was higher in VIC3 than VIC2, and the opposite was observed for PIK3C3 T163/S165 phosphorylation. Also, the low PIK3C3 phosphorylation in VIC3 was hardly affected by glucose starvation, whereas phosphorylation of PIK3C3 in VIC2 was high and robustly induced. These data explain why nonautophagic VIC2 is inactivated by AMPK-dependent phosphorylation of PIK3C3, whereas pro-autophagic VIC3 is activated by phosphorylation of BECN1. It is ATG14 that makes VIC3 different from VIC2. The autophagy-specific subunit ATG14 functions to target the PIK3C3 complex to the phagophore. Our study reveals another critical function for ATG14 as a switch converting the inhibitory effect of AMPK to activation of the PIK3C3 complex. ATG14 functions not only to prevent the inhibitory phosphorylation in PIK3C3, but also to promote the activating phosphorylation in BECN1 by AMPK. ATG14 binding may induce conformational changes in the PIK3C3 complex to mask the availability of T163/S165 in PIK3C3 and promote the availability of S91/S94 in BECN1 for phosphorylation by AMPK.
Our recent study also shows that inhibition of nonautophagy PIK3C3 complexes by AMPK is important for stress response. Although autophagy remains normal, cells expressing the PIK3C3 T163/165A mutant are sensitive to starvation, as indicated by elevated cell death. We speculate that a decrease of nonautophagy PtdIns3P reduces cellular activities associated with growth/proliferation to maintain viability upon energy depletion. In fact, accumulating reports have suggested a function of PIK3C3 in cell cycle, growth and cytokinesis. Therefore, inhibition of nonautophagy PIK3C3 represents an important, although little understood, physiological stress response.
Given the existence of multiple complexes and their broad functions in cellular regulation, PIK3C3 regulation is likely to be complex and additional signaling molecules, such as TOR and ULK, may be involved in response to various stresses.
Acknowledgments
This work was supported by NIH grant (CA108941 and GM51586 to K.L.G.) and National Research Foundation of Korea (NRF) grant (MEST, 2012R1A5A2051387 to J.K.).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Footnotes
Previously published online: www.landesbioscience.com/journals/autophagy/article/24877