

Abstract
Age-related macular degeneration (AMD) is a complex, degenerative and progressive eye disease that usually does not lead to complete blindness, but can result in severe loss of central vision. Risk factors for AMD include age, genetics, diet, smoking, oxidative stress and many cardiovascular-associated risk factors. Autophagy is a cellular housekeeping process that removes damaged organelles and protein aggregates, whereas heterophagy, in the case of the retinal pigment epithelium (RPE), is the phagocytosis of exogenous photoreceptor outer segments. Numerous studies have demonstrated that both autophagy and heterophagy are highly active in the RPE. To date, there is increasing evidence that constant oxidative stress impairs autophagy and heterophagy, as well as increases protein aggregation and causes inflammasome activation leading to the pathological phenotype of AMD. This review ties together these crucial pathological topics and reflects upon autophagy as a potential therapeutic target in AMD.
Keywords: AMD, autophagy, heterophagy, inflammasome, lysosome, oxidative stress, phagocytosis, proteasome, RPE
Introduction
AMD is a neurodegenerative disease, which is characterized by loss of central vision as a result of cellular dysfunction and cell loss at the macula. The macula is a highly specialized region of the central retina unique to humans and other primates that is normally responsible for achieving high acuity and color vision.1 Macular pathology, as occurs in AMD, results in difficulty in seeing letters on a page, reduced ability to distinguish contrasts, distortion of straight lines, central visual field defects, scotomas and color vision impairment (Fig. 1). AMD is the most common cause of visual impairment in Western countries.2 Worldwide, approximately 50 million elderly people suffer from AMD, and the number of cases is expected to rise 3-fold over the next 20 years, suggesting that AMD is becoming a major public health issue.3,4 Approximately 10 million Americans are affected by AMD, and the costs of treatment are in excess of $340 billion US.5,6 The loss of vision results primarily from the progressive degeneration and death of RPE cells, which secondarily impairs the function of rods and cones. Phenotypically, AMD can be divided into two main forms: dry (atrophic) and wet (exudative) type and further subdivided into early and late stage disease. The early stage of dry AMD is asymptomatic, although pigment mottling, accumulation of intracellular lysosomal lipofuscin, and extracellular drusen deposits can be detected.7,8 The late stage of dry AMD, also known as geographic atrophy, is characterized by discrete areas of RPE loss and impairment of the overlying retinal photoreceptor cells. In wet AMD, aberrant blood vessels sprout from the choroidal capillaries and penetrate through the Bruch’s membrane leading to subretinal membranes, hemorrhage, retinal edema and damage to retinal cells. If left untreated, late stage fibrosis and permanent visual loss may occur. Interestingly, AMD shares several risk factors with other neurodegenerative and aggregation diseases.1 These risk factors include; aging, genetic background, smoking, unhealthy diet, obesity, high blood pressure, hypercholesterolemia and arteriosclerosis (Fig. 2).
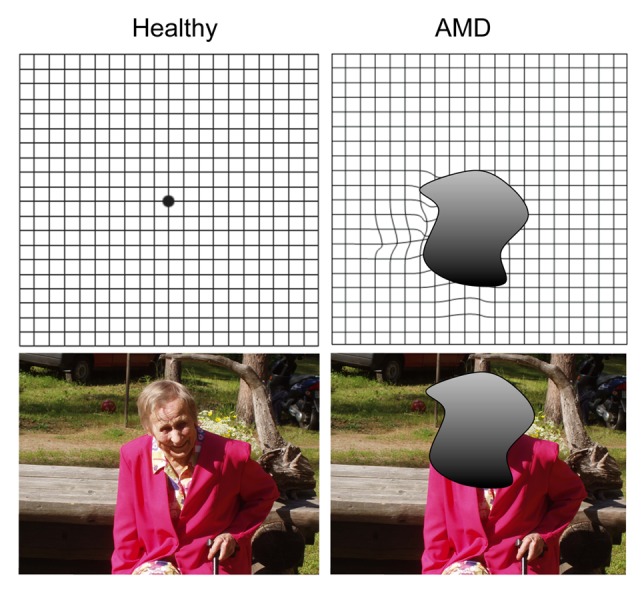
Figure 1. Symptoms of AMD include distortion of straight lines, central visual field defects, central dark spots, contrast sensitivity alterations and color-vision impairment.

Figure 2. AMD risk factors associated with different phenotypes of macular degeneration. Phenotypically, AMD can be divided into early and late stages of dry and wet AMD. (A) Pigment mottling and extracellular drusen deposition can be detected in the the early stage of dry AMD (arrow). (B) The late stage of dry AMD develops a large geographic atrophy lesion in the macula (arrow). (C) Hemorrhages are usually observed in the early neovascularization process in wet AMD (arrow). (D) Fibrotic disciform lesion develops in wet AMD if not treated by anti-VEGF agents.
The degeneration of RPE cells is widely considered to take place first during AMD development. The substantial oxygen consumption, lipid peroxidation products from the ingested photoreceptor outer segments (POS) and almost constant exposure to light, predispose the metabolically active RPE cells to chronic oxidative stress.9 Oxidatively damaged molecules, such as carboxyethylpyrrole, malondialdehyde, 4-hydroxynonenal, and advanced glycation end products, accumulate in the macular area and serve as a source for chronic oxidative stress.10 Age-related increase in oxidative stress is concurrent with increased accumulation of auto-oxidative lipofuscin in the lysosomes of RPE cells, as well as drusen formation in the extracellular space between the RPE and the Bruch’s membrane (Figs. 3 and 4).1,11,12 In addition to oxidative stress and protein aggregation, immunological events are involved in the pathogenesis of AMD. These include the production of several types of inflammation-related molecules, the recruitment of leukocytes, such as macrophages and dendritic cells, as well as activation of the complement pathway, alternate complement pathway, inflammasomes and microglia cells.13-15

Figure 3. Cross-sectional illustration of the eye and retina. Light reflects via the cornea and lens to the retina. RPE cells have a central role in the pathogenesis of AMD. They absorb light, transport metabolites and nutrients between photoreceptors and the choriocapillaris, produce growth factors, control tissue ionic balance, phagocytose shed tips of photoreceptor outer segments, regulate vitamin A metabolism and visual cycle, and create the blood–retinal barrier. Abbreviations used: R, rods; C, cones; RPE, retinal pigment epithelium; POS, photoreceptor outer segments; BM, Bruch’s membrane; L, lipofuscin; D, drusen; ch, choriocapillaris. Red arrow indicates choroidal neovascularization in the wet AMD process.

Figure 4. Optical coherence images from (A) normal macula structure including physiological foveal pit (arrow) and (B) retinal edema (arrow) in wet AMD. After anti-VEGF intravitreal injection treatment for wet AMD the retinal edema usually regresses to near normal retina thickness. Scale bars: 250 μm.
RPE cells in AMD Pathology
The RPE layer consists of hexanocuboidal epithelial cells which have two types of apical microvilli directed toward the interphotoreceptor matrix and the outer segment tips of rods and cones (Fig. 3).16,17 The short microvilli facilitate transepithelial transport. The RPE cells are a central regulator of vision, playing an essential role in maintaining the functionality and survival of photoreceptor cells. The RPE can, for example, metabolize and recycle retinoids, secrete growth factors for photoreceptors and choriocapillaries, and control the transport of nutrients into, and waste products out of, the retina.18,19 Moreover, RPE cells are phagocytically the most active cells in the whole body, whereby up to 10% of the POS length can become phagocytosed by RPE cells on a daily basis in a process called heterophagy.18,19 Since each human RPE cell is responsible for the upkeep of 30–40 photoreceptors, this is an enormous metabolic challenge for the endolysosomal system of the RPE, which is needed for the degradation of the ingested POS. The loss of the normally nondividing RPE cells during aging and AMD increases the metabolic burden on neighboring cells. In the long run, autofluorescent, lipid-protein aggregates called lipofuscin accumulate in the lysosomes of RPE cells.20
The process of photoreceptor disc shedding and renewal and the role of RPE cells in the process were first revealed in the 1960s by Young and colleagues.21-23 The renewal of the POS with the help of RPE is essential for the survival of rods and cones, and the diminished phagocytic capacity of RPE cells has been associated with degenerative diseases of the retina.24,25 Heterophagy of the POS occurs at the apical side of the RPE cells that is intimately associated with the photoreceptor layer. Once the discs have been internalized, the phagosome moves from the apical to the basal surface where the contents of the phagosome become degraded. Although the process is not completely understood, the phagocytosis of POS can be divided into four distinct stages: recognition and attachment of the POS discs, their ingestion, formation of the phagosome and its fusion with a lysosome, and degradation.26
A number of molecules essential for the recognition, binding and internalization of POS have been identified.25,27 The integrin ITGAV-ITGB5 (αVβ5) is, for example, required for the binding of outer segments,28,29 while MERTK (c-mer proto-oncogene tyrosine kinase) is essential for triggering their ingestion.30-32 Protein tyrosine kinase 2 becomes activated through the binding process, being phosphorylated by MERTK, thus linking the signaling between integrin ITGAV-ITGB5 and MERTK.32,33 MFGE8 (milk fat globule-EGF factor 8 protein), a ligand of integrin ITGAV-ITGB5, is needed for the regulation of the circadian rhythm of phagocytosis.34,35 A defect in the initial stage of phagocytosis results in photoreceptor death, as demonstrated in the Royal College of Surgeons rat which carries a mutation in the Mertk gene.30,31 In addition, age-related decrease in the level of integrin ITGAV-ITGB5 leads to the accumulation of lipofuscin within the lysosomes in RPE cells and decreased retinal adhesion, both of which secondarily evoke vision loss.34,36
Although some of the molecular mechanisms involved in the initial stages of phagocytosis or heterophagy are already known (see above), the processes of phagosome maturation and the final degradation of POS still remain obscure. Proteins involved in the maturation of the phagosome have been identified in macrophages—cells in which phagocytosis has been studied much more extensively than other cell types.37 We have also reported that some of the phagosome maturation proteins are expressed in RPE cells of rats, indicating that similar signaling pathways may be involved in the RPE-related phagosome maturation.38 Cytoskeletal elements, particularly actin filaments and microtubule-dependent motor proteins, play a critical role in the internalization of phagosomes by RPE cells.25,39 Abnormal cytoskeletal reorganization that affects POS internalization leads to retina defects as seen in one of the types of Usher’s syndrome, a genetic disorder resulting in a combination of hearing loss and visual impairment.40
Autophagosome and phagosome maturation and fusion with lysosomes happen both in auto- and heterophagy, respectively. When autophagy takes place during cellular remodeling, an autophagosome fuses with a lysosome, which provides hydrolytic enzymes for the degradation of autophagosomal contents (see below).41,42 Our recent findings implicate βA3/A1-crystallin as a novel lysosomal component in the RPE that could regulate both phagocytosis and autophagy.38 Since βA3/A1-crystallin has been found in human drusen material,43 and our recent studies also show that Nuc1 rats with a spontaneous mutation in the Cryba1 gene develop deposits between the basal side of the RPE and Bruch’s membrane during aging (Fig. 5), it is tempting to speculate that perturbation of normal phagocytosis and/or autophagy could lead to some manifestations of AMD. Also, an experimental disruption of lysosomal function has provided supporting evidence for the possible role of lysosomes in the development of AMD.44,45

Figure 5. Transmission electron micrographs of the RPE-Bruch’s membrane/choriocapillaris complex. Typical infoldings of the basal RPE are preserved in wild-type (red arrow, A) and lost in Nuc1 rats (red arrow, B). The cytoplasm of the RPE in Nuc1 rats (red asterisk, B) is more heterogeneous with increased granularity and lipid inclusions as compared with wild type (red asterisk, A). Both thickening and a more heterogeneous composition of Bruch’s membrane (BM) are seen in Nuc1 rats (blue arrowheads, D) as compared with wild type (blue arrowheads, C). A deposit between the basal RPE and BM in Nuc1 rats is indicated in (D) (black arrows). Scale bars: (A and B) 2 μm; (C and D) 500 nm.
Impairment of Lysosomal Function in RPE
Lysosomal clearance may be disturbed by various mechanisms during the degeneration of RPE cells and development of AMD.46 Lysosomes possess a multifunctional capacity to cope with different cleaning processes when proteins destined for degradation arrive via endocytosis, phagocytosis (heterophagy) or autophagy. Cathepsins are proteases with a biological task to degrade proteins; so far, CTSA, B, D, E and S have been characterized in RPE cells.47 The main responsibility of CTSD is the degradation of POS and rhodopsin into glycopeptides within the RPE lysosomes;48 CTSD-deficient mice develop retinal degeneration.49 CSTs (cystatins), which are inhibitors of lysosomal cysteine proteases, are highly expressed in RPE cells.50,51 A CST3 (encoding cystatin C) gene variant is associated with an increased risk of developing advanced AMD.52 Due to this cystatin variant, important cellular functions, such as cellular transport and regulation of proteolytic balance in the extracellular space become affected, which are thought to be involved in the RPE degeneration and AMD development.51 This observation is supported by the earlier documentation indicating a relationship between the increased level of serum CST3 and the incidence of AMD.53
Oxidized low-density lipoproteins and lipid peroxidation end products reduce the degradation of phagocytosed POS material, and increase cellular stress in the RPE cell.54-56 The intracellular and lysosomal storage of these metabolites is considered to represent the initial stage of lipofuscinogenesis. The A2-E (N-retinylidene-N-retinylethanol-amine) fluorophore is a harmful component of lipofuscin, the latter being a photosensitizer and auto-oxidant that can increase the mitochondrial stress and irreversibly inhibits lysosomal cathepsin activity upon light exposure, leading to increased damage of the RPE.57-59 It is thought that, once formed, lipofuscin cannot be degraded by proteasomal or lysosomal enzymes or become transported into the extracellular space via exocytosis.60,61 Furthermore, maintenance of an acidic lysosomal pH is critical. Elevated lysosomal pH is observed in RPE cells from ABCA4 knockout mice (a model for Stargardt’s disease) and in cultured human ARPE-19 cells exposed to A2E.62 Thus, POS clearance and lysosomal enzyme activity seem to be related to lysosomal pH.63,64 Clinical findings from AMD patients indicate that mitochondrial damage and excessive lipofuscin accumulation precede the atrophy of outer retinal layers and the subsequent loss of visual function.65,66
Oxidative stress and mitochondrial dysfunction in RPE cells
Oxidative stress can induce electron leakage from the mitochondrial electron transport chain, followed by formation of hydroxyl radicals by Fenton-type reactions, and production of superoxide, hydrogen peroxide and hypochlorite as a consequence of many enzymatic reactions. An imbalance between the generation and the suppression of reactive oxygen species (ROS) can lead to undesirable effects, for example, due to protein unfolding and damage, especially in age-related conditions.67
The central retina is exposed to an exceptionally high burden of oxidative stress, which increases during aging.9,68 Epidemiologic, genetic, and molecular pathology studies support the role of oxidative stress in the pathogenesis of AMD.69 The retina and RPE cells provide an ideal environment for the generation of ROS.68 Oxidative stress is mainly caused by retinal irradiation, lipid peroxidation, photochemical damage of retinal chromophores, and the respiratory burst. These oxidative processes are thought to contribute to the clinical manifestation of pigment dispersion, accumulation of intracellular lysosomal lipofuscin, and extracellular drusen deposits.70
Mitochondrial hydrogen peroxide and lysosomal iron react in the Fenton reaction, producing hydroxyl radicals. Some oxidation products polymerize to form undegradable lipofuscin, which accumulates in the lysosomes.71,72 It has been speculated that lipofuscin itself could be cytotoxic because of its ability to provide a redox-active surface by incorporating oxidatively labile iron.73,74 This could result in the formation of oxygen radicals and cause oxidative modification of proteins, lipids and nucleic acids. For example, many proteins isolated from RPE cell-derived lipofuscin have been modified and decorated by the markers of oxidative stress, such as malondialdehyde, hydroxynonenal, advanced glycation end products and advanced glycosylation end product-specific receptor.71,75 Interestingly, Höhn et al. observed that due to the iron, lipofuscin is able to sustain the production of cytotoxic oxidants independently of the mitochondria.74 Together with an increased accumulation of lipofuscin, impaired lysosomal function, continuous light exposure, and oxidants, this can induce defects in mitochondrial functions, which can further increase the oxidative stress.57,76,77 Due to the high metabolic activity, impaired mitochondrial function is assumed to lead to the accelerated degeneration of RPE cells and secondarily to photoreceptor cell death.78-80 Taken together, the Fenton reaction can occur in mitochondria leading to their damage, but also in lysosomes where it can directly contribute to lipofuscin formation.72-76 However, it must be noted that most of the RPE lipofuscin is derived from retinal POS.20
The concurrent increase in the structural alterations of mitochondria also coincides with the pathology of AMD.65,81 Oxidative stress leads to mitochondrial DNA damage, increases ROS generation and reduces the metabolic capacity. Mitochondrial DNA is more susceptible to oxidative damage and light exposure than nuclear DNA.82,83 Recent findings support the idea that there is increased mitochondrial stress and dysfunction in the RPE cells of AMD patients.84-86 Therefore, selective removal of oxidatively damaged mitochondria by autophagy (called mitophagy) might be essential for cell survival.
Impaired Proteasomal and Autophagosomal Proteolysis in RPE Cells and AMD
Increased oxidative stress evokes misfolding of proteins via oxidation/glutathione conjugation.87,88 Primarily, molecular chaperones (heat shock proteins) repair the misfolding damage, but if this fails, soluble proteins become ubiquitinated and are targeted into proteasomes for degradation (Fig. 6).89,90 The proteasomal activity may also decline during aging, which leads to aggregation of oxidized and ubiquitinated proteins—a process that can occur in RPE cells as well.91-93 The protein aggregates can form juxtanuclear aggresomes after being delivered via the microtubule network from the cellular periphery.94-96 The ubiquitination results in recruitment of adaptor proteins, such as SQSTM1/p62 and microtubule-associated protein 1 light chain 3 α, which link the ubiquitinated proteins to the autophagic complex.97 The mechanisms behind autophagy have been thoroughly reviewed in recent publications.98-101

Figure 6. Crosstalk between heterophagy and autophagy in the regulation of protein aggregation and inflammation in aged RPE cells. Impaired lysosomal POS clearance increases lipofuscin accumulation that increases oxidative stress damage and protein aggregation in the aged RPE cells. Autophagy flux is decreased due to weakened lysosomal function that also increases mitochondrial damage. All these lead to exocytosis of damaged proteins and activation of the NLRP3 inflammasome in association with drusen formation. Abbreviations used: BM, Bruch’s membrane; UB, ubiquitin.
In mammals, three types of autophagy have been described: microautophagy, chaperone-mediated autophagy and macroautophagy.98-101 Microautophagy and chaperone-mediated autophagy are poorly understood in RPE cells, and therefore they are not discussed in this review. Macroautophagy (referred to as autophagy) is the most prevalent form and involves the formation of a double-membrane structure (autophagosome) that engulfs cytoplasmic proteins, lipids and damaged organelles. Thereafter, autophagosomes fuse with primary lysosomes, and their contents become degraded by lysosomal enzymes, such as cathepsins (see above). The autophagy process can be divided into induction, initiation/nucleation, elongation and closure, maturation and fusion, and finally, degradation steps.
Oxidative stress, hypoxia, the unfolded protein response or inflammation can activate autophagy, and are present in AMD pathology.1 Autophagy activity is strongly regulated by the signaling of mechanistic target of rapamycin (MTOR), the inhibition of which results in the partial dephosphorylation of ATG13, activation of the ULKs (unc-51-like kinases) and recruitment of RB1CC1 (RB1-inducible coiled-coil 1). The ULK1/2-ATG13-RB1CC1 complex plays a crucial role in the formation of double-membrane autophagic vacuoles, autophagosomes. Other important proteins in autophagosome formation include ATG14, BECN1, PIK3C3, PIK3R4 and UVRAG.98 Microtubule-associated protein 1 light chain 3 α is a ubiquitin-like protein that connects autophagy to the proteasomal clearance system via ubiquitin and SQSTM1 binding sites.97,99
Failure of the RPE cells to use autophagy can result in accumulation of aggregation-prone proteins, cellular degeneration and finally cell death.102 In addition, a decline in the autophagy flux is usually accompanied by the accumulation of SQSTM1 in large perinuclear aggregates or inclusion bodies which are also positive for ubiquitin, as has been reported to happen in numerous neurodegenerative diseases, including Alzheimer, Parkinson and Huntington diseases.103-108
Autophagy decline and inflammasome activation
To date, there is strong evidence showing that decreased autophagy flux is associated with RPE damage and AMD pathology.70,102,109-111 When autophagic capacity declines simultaneously with an increased lipofuscin accumulation, accelerated ROS production and elevated protein aggregation can occur, which may activate an inflammatory response that further provokes long-term, low-grade inflammation in retinal cells, thus, speeding up the aging process (Fig. 6).112 The swelling, destabilization and dysfunction of lysosomes caused by the accumulation of fibrillar amyloid β (Aβ) in the lysosomes of microglia cells results in the activation of NLRP3 (NLR family, pyrin domain containing 3) inflammasomes.113 Additionally, the lysosomal rupture by silica crystals and aluminum salts leads to a similar result.114 Lysosomal destabilization using the lysosmotropic agent L-leucyl-l-leucine methyl ester also activates the NLRP3 inflammasomes in ARPE-19 cells.115 NLRP3 is an intracellular pattern-recognition receptor, which responds to a wide variety of danger signals by inducing the formation of a multiprotein complex called the inflammasome, and thereby initiating an inflammatory response.116 Although the exact mechanism of NLRP3 activation has not been elucidated yet, the contribution of CTSB released from the ruptured lysosomes has been strongly suggested.113,114 Mitochondrial dysfunction and subsequent oxidative stress are also well known activators of the NLRP3 inflammasome.112,117,118 We have recently shown that oxidative stress is capable of inducing activation of the NLRP3 inflammasomes in human ARPE-19 cells.119 Moreover, oxidized mitochondrial DNA leaking out from damaged mitochondria is a direct agonist of NLRP3.120 In normal circumstances, autophagy controls the activation of the NLRP3 inflammasome, for example by degrading the inflammasome components and effector molecules.121,122 Conversely, when autophagy declines, inflammasomes become activated—most probably through the dysregulation of mitochondrial homeostasis.118,123
It is well known that accumulated intracellular lipofuscin and extracellular drusen deposits increase the risk for progression of AMD.66,124,125 Among other effects, drusen may play a role in inflammasome activation. Drusen material isolated from donor AMD eyes can activate the NLRP3 inflammasome pathway in human mononuclear cells.126 Immunohistochemical analyses have revealed that drusen are composed of many intracellular-derived proteins which can regulate proteolytic processes.13,109,127 Drusen formation may be associated with decreased autophagy, and increased transcytosis and exocytosis in RPE cells, which can further be involved in the development of AMD.89,109,128 Some exosome and autophagy markers have been detected in drusen.109 We have observed that SQSTM1 accumulates in the macular RPE cells rather than in peripheral retina, revealing an impaired autophagy flux in AMD.110 In addition, most of the drusen from AMD patients show strong ubiquitin positivity, while the SQSTM1 staining could be observed only intracellularly. This might imply that SQSTM1 is mostly degraded by the autophagic pathway and, unlike ubiquitin, SQSTM1 is not exocytosed to the extracellular space of the RPE cells. Recent reports have argued that ubiquitin and SQSTM1 strongly colocalize in perinuclear aggregates.129 However, we found that only ubiquitin was detectable in the extracellular drusens of AMD samples. As a consequence, we hypothesize that autophagosomes do not fuse with cell membranes and are not involved directly in exocytosis that contributes to AMD pathology.
Complexicity of autophagy as a pharmacological target in AMD therapy
Regulation of the signaling pathways involved in autophagy is certainly a potential therapeutic target for AMD treatment. However, targeting autophagy may be more complex than that, since a) autophagy is a fundamental housekeeping process in all cells, and too little or too much autophagy can result in cellular dysfunction, b) autophagy pathways will differ dependending on the stimulus signal(s), and c) AMD has both degenerative characteristics including protein deposits, and in certain cases proliferative characteristics as occurs in the wet AMD form. To date, there is no consensus as to whether autophagy inhibitors or activators would be beneficial in AMD therapy, and how they should be used in different phenotypes of AMD in prevention or in therapy. The induction of autophagy can be attained through inhibition of the mitogen-activated protein kinase/extracellular signal-regulated kinase, class I phosphoinositide 3-kinase-AKT and MTOR signaling pathways. Autophagy inhibitors can be broadly classified into early or late stage inhibitors depending on the autophagy pathway they act upon (Fig. 7).
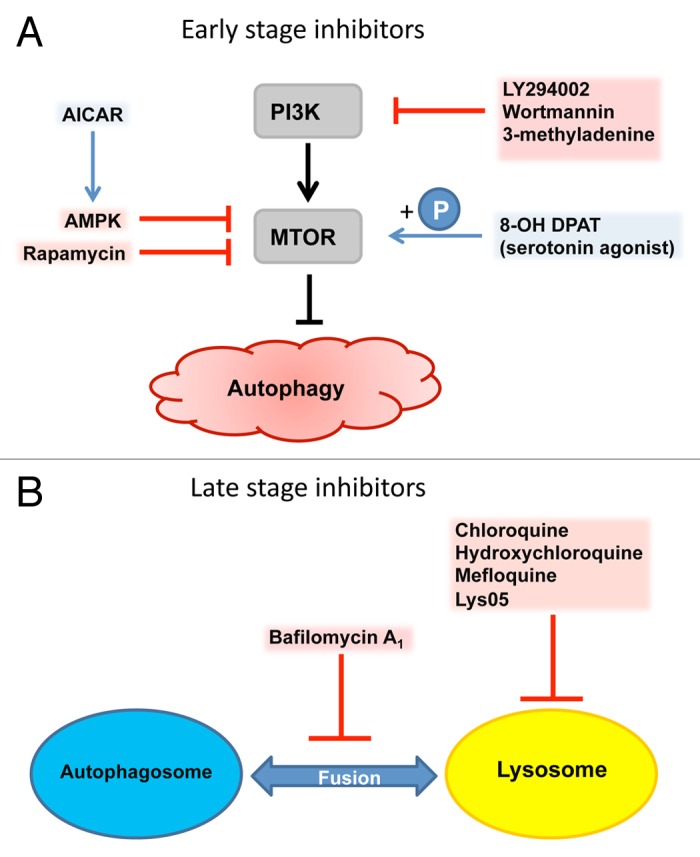
Figure 7. Pharmacological targets of autophagy. (A) Early stage inhibitors acting upon PIK3CA and MTOR, the most important checkpoints of autophagy induction. (B) Late stage inhibitors of autophagy such as chloroquine and its modified derivatives, and bafilomycin A1 act upon the autophagy-lysosomal pathway.
Early stage inhibitors of autophagy, such as wortmannin, 3-methyladenine (3-MA) and LY294002, can act upon an upstream regulator of autophagy, the class III PtdIns3K. In retinal ganglion cells following optic nerve transection, the inhibition of autophagy by wortmannin and 3-MA, as well as the late stage inhibitor bafilomycin A1, can decrease cell viability, suggesting a cell-protective role of autophagy in neurodegenerative diseases.130 Conversely, blocking autophagy with 3-MA in developing retinal neuroepithelium or after optic nerve axotomy results in abnormal retinal tissue formation and function or attenuation of axonal swelling and degeneration, respectively.131,132 Late stage inhibitors of autophagy including antimalarial drugs and broad-spectrum antibiotics such as fluoroquinolones, impose their action on the lysosomal part of the autophago-lysosomal pathway and prevent fusion of autophagosomes with lysosomes. Similarly to early stage inhibitors, the late ones have also been tested in cancer cell lines and cancer animal models. The antimalarial drug chloroquine (CQ) has been used in cancer therapy due to its inhibitory effect on autophagy, besides its additional, autophagy-independent toxic effect on cancer cells.133-136 CQ and other quinines have been associated with cases of retinal toxicity, particularly when provided at higher doses for longer times.137,138 In vitro, CQ can induce lipid accumulation and block phagocytosis in ARPE-19 cells in a dose- and time-dependent manner.44
Neuronal transmitter blockers such as HTR1A (5-hydroxytryptamine (serotonin) receptor 1A, G protein-coupled) agonists offer a therapeutic option for retinal degenerations such as AMD, diabetic retinopathy or retinitis pigmentosa. In both, an in vitro system and an in vivo mice model of atrophic AMD, the HTR1A receptor agonist 8-hydroxy-2-(dipropylamino)tetralin (8-OH DPAT) can protect the retina from degeneration by reducing oxidative damage. This study showed that autophagy- and photo-oxidative stress-derived lipofuscin accumulation can be reduced by 8-OH DPAT in cultured ARPE-19 cells, possibly through stimulation of MTOR phosphorylation, which can lead to decreased autophagy induction (Fig. 7).139
Recent findings have revealed that rapamycin-induced inhibition of MTOR complex 1 (MTORC1), and, therefore, activation of autophagy, can slow the aging and neurodegenerative processes in mice.130,140,141 Interestingly, RPE degeneration is associated with increased sensitivity and enhanced activity of MTORC1 in experimental AMD studies.142,143 Rapamycin prevents the development of harmful AMD-related aging signs in RPE cells. MTOR regulates the detrimental dedifferentiation and hypertrophy of RPE cells exposed to oxidative stress, whereas rapamycin treatment can prevent these effects and preserve photoreceptor functions.142 In addition, rapamycin inhibits choroidal neovascularisation (CNV) by interfering with the function of VEGFA (vascular endothelial growth factor A).143,144 Co-culture assays of RPE and endothelial cells revealed that rapamycin is an effective VEGF inhibitor and it can reduce sprouting of endothelial cells.145 However, even though rapamycin prevents retinal degeneration in animal models, it has a number of off-target effects, which have limited its usefulness in age-related neurological disorders such as Parkinson and Huntington diseases.
AMP-activated protein kinase (AMPK) is classically activated by energy depletion and hypoxia. Moreover, a variety of chemicals including the adenosine analog AICAR (5-aminoimidazole-4-carboxamide ribonucleoside) have been used to investigate the role of AMPK in the regulation of the MTORC1 pathway (Fig. 7). AICAR protects RPE cells from oxidative stress.146 Moreover, AMPK-induced autophagy protects the RPE cells from TNFSF10/TRAIL (tumor necrosis factor (ligand) superfamily, member 10)-induced cell death.147 Our unpublished data reveal that AICAR accelerates cleansing of protesome inhibitor-induced protein aggregation via autophagy and improves cell survival in ARPE-19 cells. Taken together, autophagy is a plausible therapy target in AMD, but may be complex due to a variation in the AMD phenotypes. Dry AMD involves degenerative changes without cellular proliferation, whereas CNV development is based on the choroidal endothelial cell proliferation in wet AMD. The inhibition of autophagy potentiates anti-angiogenic effects and might be used together with anti-VEGF therapy in wet AMD.148,149 However, the autophagy inducer rapamycin functions as a VEGF inhibitor and reduces CNV activity, as discussed above.143,144 Due to these opposite effects, further results and experimental models are required to determine whether autophagy activation or inhibition is a goal in AMD therapy.150 Recent clinical experiences provide the possibility to apply drugs through intravitreal injections. This could elegantly circumvent a number of side effects of putative autophagy-related therapies when applied systemically. Autophagy-targeted gene therapy for treatment of AMD is also an interesting future option.151,152 However, accumulating lysosomal lipofuscin may be a limiting factor in the regulation of autophagy flux. Overall, considering AMD as a degenerative age-related disease it may be tempting to state that functional autophagy may prevent RPE cell degeneration and AMD development, although documentation varies in the different models studied.
Conclusions
During the past several years, our understanding of the mechanisms leading to RPE degeneration and AMD development has greatly increased. Various genetic and environmental risk factors associated with lysosomal damage, including accumulation of lipofuscin and drusen, and induction of chronic inflammation all can lead to decreased autophagy flux in the RPE cells and AMD progression. Therefore, it should be appreciated that autophagy may represent an important therapeutic target in AMD. In particular, the autophagy-regulating kinases AMPK and MTOR can be potential therapeutic targets for preventing RPE cell degeneration and AMD progression either alone or as an adjunct to other treatments.
Acknowledgments
This work was supported by the EVO grants of Kuopio University Hospital (K.K.), the Finnish Cultural Foundation and its North Savo Fund (K.K.), the Finnish Eye Foundation (K.K.), the Finnish Funding Agency for Technology and Innovation (K.K.), Health Research Council of the Academy of Finland (K.K., A.K.), and the Päivikki and Sakari Sohlberg Foundation (A.K.), National Institutes of Health (USA) grants EY019688 and EY021626 (MEB), EY019037-S (D.S.) D.S. is a recipient of the Sybil B. Harrington Special Scholar award for Macular Degeneration from Research to Prevent Blindness, the Hungarian Scientific Research Fund (OTKA PD 101316) and the TÁMOP 4.2.2.A-11/1/KONV-2012-0023 “VÉD-ELEM” project. The project is implemented through the New Hungary Development Plan co-financed by the European Social Fund and the European Regional Development Fund.
Glossary
Abbreviations:
- 3-MA
3-methyladenine
- AMD
age-related macular degeneration
- AMPK
5′ adenosine monophosphate-activated protein kinase
- ATG
autophagy-related
- CNV
choroidal neovascularization
- CQ
chloroquine
- D
drusen
- MERTK
c-mer proto-oncogene tyrosine kinase
- MFGE8
milk fat globule-EGF factor 8 protein
- MTOR
mechanistic target of rapamycin
- MTORC1
MTOR complex 1
- NLR family
pyrin domain containing 3 (NLRP3)
- POS
photoreceptor outer segment
- ROS
reactive oxygen species
- RPE
retinal pigment epithelium
- SQSTM1, sequestosome 1
VEGF, vascular endothelial growth factor
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Footnotes
Previously published online: www.landesbioscience.com/journals/autophagy/article/24546
References
- 1.Kaarniranta K, Salminen A, Haapasalo A, Soininen H, Hiltunen M. Age-related macular degeneration (AMD): Alzheimer’s disease in the eye? J Alzheimers Dis. 2011;24:615–31. doi: 10.3233/JAD-2011-101908. [DOI] [PubMed] [Google Scholar]
- 2.Vision 2020. Right to sight. Blindness and visual impairment: Global facts. Available at: http://vision2020.org/main.cfm?type=FACTS Accessed August 22, 2011.
- 3.Gehrs KM, Anderson DH, Johnson LV, Hageman GS. Age-related macular degeneration--emerging pathogenetic and therapeutic concepts. Ann Med. 2006;38:450–71. doi: 10.1080/07853890600946724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gordois A, Cutler H, Pezzullo L, Gordon K, Cruess A, Winyard S, et al. An estimation of the worldwide economic and health burden of visual impairment. Glob Public Health. 2012;7:465–81. doi: 10.1080/17441692.2011.634815. [DOI] [PubMed] [Google Scholar]
- 5.Friedman DS, O’Colmain BJ, Muñoz B, Tomany SC, McCarty C, de Jong PT, et al. Eye Diseases Prevalence Research Group Prevalence of age-related macular degeneration in the United States. Arch Ophthalmol. 2004;122:564–72. doi: 10.1001/archopht.122.4.564. [DOI] [PubMed] [Google Scholar]
- 6.Weikel KA, Chiu CJ, Taylor A. Nutritional modulation of age-related macular degeneration. Mol Aspects Med. 2012;33:318–75. doi: 10.1016/j.mam.2012.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Algvere PV, Marshall J, Seregard S. Age-related maculopathy and the impact of blue light hazard. Acta Ophthalmol Scand. 2006;84:4–15. doi: 10.1111/j.1600-0420.2005.00627.x. [DOI] [PubMed] [Google Scholar]
- 8.Kinnunen K, Petrovski G, Moe MC, Berta A, Kaarniranta K. Molecular mechanisms of retinal pigment epithelium damage and development of age-related macular degeneration. Acta Ophthalmol. 2012;90:299–309. doi: 10.1111/j.1755-3768.2011.02179.x. [DOI] [PubMed] [Google Scholar]
- 9.Winkler BS, Boulton ME, Gottsch JD, Sternberg P. Oxidative damage and age-related macular degeneration. Mol Vis. 1999;5:32. [PMC free article] [PubMed] [Google Scholar]
- 10.Handa JT. How does the macula protect itself from oxidative stress? Mol Aspects Med. 2012;33:418–35. doi: 10.1016/j.mam.2012.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jarrett SG, Boulton ME. Consequences of oxidative stress in age-related macular degeneration. Mol Aspects Med. 2012;33:399–417. doi: 10.1016/j.mam.2012.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Plafker SM, O’Mealey GB, Szweda LI. Mechanisms for countering oxidative stress and damage in retinal pigment epithelium. Int Rev Cell Mol Biol. 2012;298:135–77. doi: 10.1016/B978-0-12-394309-5.00004-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hageman GS, Luthert PJ, Victor Chong NH, Johnson LV, Anderson DH, Mullins RF. An integrated hypothesis that considers drusen as biomarkers of immune-mediated processes at the RPE-Bruch’s membrane interface in aging and age-related macular degeneration. Prog Retin Eye Res. 2001;20:705–32. doi: 10.1016/S1350-9462(01)00010-6. [DOI] [PubMed] [Google Scholar]
- 14.Tuo J, Grob S, Zhang K, Chan CC. Genetics of immunological and inflammatory components in age-related macular degeneration. Ocul Immunol Inflamm. 2012;20:27–36. doi: 10.3109/09273948.2011.628432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ambati J, Fowler BJ. Mechanisms of age-related macular degeneration. Neuron. 2012;75:26–39. doi: 10.1016/j.neuron.2012.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Garron LK. The ultrastructure of the retinal pigment epithelium with observations on the choriocapillaris and Bruch's membrane. Trans Am Ophthalmol Soc. 1963;61:545–88. [PMC free article] [PubMed] [Google Scholar]
- 17.Marmorstein AD, Finnemann SC, Bonilha VL, Rodriguez-Boulan E. Morphogenesis of the retinal pigment epithelium: toward understanding retinal degenerative diseases. Ann N Y Acad Sci. 1998;857:1–12. doi: 10.1111/j.1749-6632.1998.tb10102.x. [DOI] [PubMed] [Google Scholar]
- 18.Bok D. The retinal pigment epithelium: a versatile partner in vision. J Cell Sci Suppl. 1993;17:189–95. doi: 10.1242/jcs.1993.supplement_17.27. [DOI] [PubMed] [Google Scholar]
- 19.Strauss O. The retinal pigment epithelium in visual function. Physiol Rev. 2005;85:845–81. doi: 10.1152/physrev.00021.2004. [DOI] [PubMed] [Google Scholar]
- 20.Feeney-Burns L, Eldred GE. The fate of the phagosome: conversion to ‘age pigment’ and impact in human retinal pigment epithelium. Trans Ophthalmol Soc U K. 1983;103:416–21. [PubMed] [Google Scholar]
- 21.Young RW. The renewal of photoreceptor cell outer segments. J Cell Biol. 1967;33:61–72. doi: 10.1083/jcb.33.1.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Young RW, Droz B. The renewal of protein in retinal rods and cones. J Cell Biol. 1968;39:169–84. doi: 10.1083/jcb.39.1.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Young RW, Bok D. Participation of the retinal pigment epithelium in the rod outer segment renewal process. J Cell Biol. 1969;42:392–403. doi: 10.1083/jcb.42.2.392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Travis GH, Golczak M, Moise AR, Palczewski K. Diseases caused by defects in the visual cycle: retinoids as potential therapeutic agents. Annu Rev Pharmacol Toxicol. 2007;47:469–512. doi: 10.1146/annurev.pharmtox.47.120505.105225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kevany BM, Palczewski K. Phagocytosis of retinal rod and cone photoreceptors. Physiology (Bethesda) 2010;25:8–15. doi: 10.1152/physiol.00038.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bosch E, Horwitz J, Bok D. Phagocytosis of outer segments by retinal pigment epithelium: phagosome-lysosome interaction. J Histochem Cytochem. 1993;41:253–63. doi: 10.1177/41.2.8419462. [DOI] [PubMed] [Google Scholar]
- 27.Caberoy NB, Zhou Y, Li W. Tubby and tubby-like protein 1 are new MerTK ligands for phagocytosis. EMBO J. 2010;29:3898–910. doi: 10.1038/emboj.2010.265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Finnemann SC, Bonilha VL, Marmorstein AD, Rodriguez-Boulan E. Phagocytosis of rod outer segments by retinal pigment epithelial cells requires α(v)β5 integrin for binding but not for internalization. Proc Natl Acad Sci U S A. 1997;94:12932–7. doi: 10.1073/pnas.94.24.12932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nandrot EF, Kim Y, Brodie SE, Huang X, Sheppard D, Finnemann SC. Loss of synchronized retinal phagocytosis and age-related blindness in mice lacking alphavbeta5 integrin. J Exp Med. 2004;200:1539–45. doi: 10.1084/jem.20041447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.D’Cruz PM, Yasumura D, Weir J, Matthes MT, Abderrahim H, LaVail MM, et al. Mutation of the receptor tyrosine kinase gene Mertk in the retinal dystrophic RCS rat. Hum Mol Genet. 2000;9:645–51. doi: 10.1093/hmg/9.4.645. [DOI] [PubMed] [Google Scholar]
- 31.Gal A, Li Y, Thompson DA, Weir J, Orth U, Jacobson SG, et al. Mutations in MERTK, the human orthologue of the RCS rat retinal dystrophy gene, cause retinitis pigmentosa. Nat Genet. 2000;26:270–1. doi: 10.1038/81555. [DOI] [PubMed] [Google Scholar]
- 32.Feng W, Yasumura D, Matthes MT, LaVail MM, Vollrath D. Mertk triggers uptake of photoreceptor outer segments during phagocytosis by cultured retinal pigment epithelial cells. J Biol Chem. 2002;277:17016–22. doi: 10.1074/jbc.M107876200. [DOI] [PubMed] [Google Scholar]
- 33.Finnemann SC. Focal adhesion kinase signaling promotes phagocytosis of integrin-bound photoreceptors. EMBO J. 2003;22:4143–54. doi: 10.1093/emboj/cdg416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nandrot EF, Anand M, Almeida D, Atabai K, Sheppard D, Finnemann SC. Essential role for MFG-E8 as ligand for alphavbeta5 integrin in diurnal retinal phagocytosis. Proc Natl Acad Sci U S A. 2007;104:12005–10. doi: 10.1073/pnas.0704756104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nandrot EF, Finnemann SC. Lack of alphavbeta5 integrin receptor or its ligand MFG-E8: distinct effects on retinal function. Ophthalmic Res. 2008;40:120–3. doi: 10.1159/000119861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mallavarapu M, Finnemann SC. Neural retina and MerTK-independent apical polarity of alphavbeta5 integrin receptors in the retinal pigment epithelium. Adv Exp Med Biol. 2010;664:123–31. doi: 10.1007/978-1-4419-1399-9_15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kinchen JM, Ravichandran KS. Phagosome maturation: going through the acid test. Nat Rev Mol Cell Biol. 2008;9:781–95. doi: 10.1038/nrm2515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zigler JS, Jr., Zhang C, Grebe R, Sehrawat G, Hackler L, Jr., Adhya S, et al. Mutation in the βA3/A1-crystallin gene impairs phagosome degradation in the retinal pigmented epithelium of the rat. J Cell Sci. 2011;124:523–31. doi: 10.1242/jcs.078790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Marmor MF, Wolfensberger TJ, eds. The Retinal Pigment Epithelium: Function and Disease. In The retinal pigment epithelium cytoskeleton. Burnside B. and Bost-Usinger L., eds. New York: Oxford University Press; 1998. [Google Scholar]
- 40.Gibbs D, Kitamoto J, Williams DS. Abnormal phagocytosis by retinal pigmented epithelium that lacks myosin VIIa, the Usher syndrome 1B protein. Proc Natl Acad Sci U S A. 2003;100:6481–6. doi: 10.1073/pnas.1130432100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Glick D, Barth S, Macleod KF. Autophagy: cellular and molecular mechanisms. J Pathol. 2010;221:3–12. doi: 10.1002/path.2697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tong J, Yan X, Yu L. The late stage of autophagy: cellular events and molecular regulation. Protein Cell. 2010;1:907–15. doi: 10.1007/s13238-010-0121-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Crabb JW, Miyagi M, Gu X, Shadrach K, West KA, Sakaguchi H, et al. Drusen proteome analysis: an approach to the etiology of age-related macular degeneration. Proc Natl Acad Sci U S A. 2002;99:14682–7. doi: 10.1073/pnas.222551899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chen PM, Gombart ZJ, Chen JW. Chloroquine treatment of ARPE-19 cells leads to lysosome dilation and intracellular lipid accumulation: possible implications of lysosomal dysfunction in macular degeneration. Cell Biosci. 2011;1:10. doi: 10.1186/2045-3701-1-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sundelin S, Wihlmark U, Nilsson SE, Brunk UT. Lipofuscin accumulation in cultured retinal pigment epithelial cells reduces their phagocytic capacity. Curr Eye Res. 1998;17:851–7. doi: 10.1080/02713689808951268. [DOI] [PubMed] [Google Scholar]
- 46.Kaarniranta K, Kauppinen A, Blasiak J, Salminen A. Autophagy regulating kinases as potential therapeutic targets for age-related macular degeneration. Future Med Chem. 2012;4:2153–61. doi: 10.4155/fmc.12.169. [DOI] [PubMed] [Google Scholar]
- 47.Im E, Kazlauskas A. The role of cathepsins in ocular physiology and pathology. Exp Eye Res. 2007;84:383–8. doi: 10.1016/j.exer.2006.05.017. [DOI] [PubMed] [Google Scholar]
- 48.el-Hifnawi E. Localization of cathepsin D in rat ocular tissues. An immunohistochemical study. Ann Anat. 1995;177:11–7. doi: 10.1016/S0940-9602(11)80123-8. [DOI] [PubMed] [Google Scholar]
- 49.Koike M, Shibata M, Ohsawa Y, Nakanishi H, Koga T, Kametaka S, et al. Involvement of two different cell death pathways in retinal atrophy of cathepsin D-deficient mice. Mol Cell Neurosci. 2003;22:146–61. doi: 10.1016/S1044-7431(03)00035-6. [DOI] [PubMed] [Google Scholar]
- 50.Paraoan L, Grierson I, Maden BE. Analysis of expressed sequence tags of retinal pigment epithelium: cystatin C is an abundant transcript. Int J Biochem Cell Biol. 2000;32:417–26. doi: 10.1016/S1357-2725(99)00143-0. [DOI] [PubMed] [Google Scholar]
- 51.Paraoan L, Hiscott P, Gosden C, Grierson I. Cystatin C in macular and neuronal degenerations: implications for mechanism(s) of age-related macular degeneration. Vision Res. 2010;50:737–42. doi: 10.1016/j.visres.2009.10.022. [DOI] [PubMed] [Google Scholar]
- 52.Zurdel J, Finckh U, Menzer G, Nitsch RM, Richard G. CST3 genotype associated with exudative age related macular degeneration. Br J Ophthalmol. 2002;86:214–9. doi: 10.1136/bjo.86.2.214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Klein R, Knudtson MD, Lee KE, Klein BE. Serum cystatin C level, kidney disease markers, and incidence of age-related macular degeneration: the Beaver Dam Eye Study. Arch Ophthalmol. 2009;127:193–9. doi: 10.1001/archophthalmol.2008.551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Finnemann SC, Leung LW, Rodriguez-Boulan E. The lipofuscin component A2E selectively inhibits phagolysosomal degradation of photoreceptor phospholipid by the retinal pigment epithelium. Proc Natl Acad Sci U S A. 2002;99:3842–7. doi: 10.1073/pnas.052025899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kaarniranta K, Ryhänen T, Karjalainen HM, Lammi MJ, Suuronen T, Huhtala A, et al. Geldanamycin increases 4-hydroxynonenal (HNE)-induced cell death in human retinal pigment epithelial cells. Neurosci Lett. 2005;382:185–90. doi: 10.1016/j.neulet.2005.03.009. [DOI] [PubMed] [Google Scholar]
- 56.Kaemmerer E, Schutt F, Krohne TU, Holz FG, Kopitz J. Effects of lipid peroxidation-related protein modifications on RPE lysosomal functions and POS phagocytosis. Invest Ophthalmol Vis Sci. 2007;48:1342–7. doi: 10.1167/iovs.06-0549. [DOI] [PubMed] [Google Scholar]
- 57.Bergmann M, Schütt F, Holz FG, Kopitz J. Inhibition of the ATP-driven proton pump in RPE lysosomes by the major lipofuscin fluorophore A2-E may contribute to the pathogenesis of age-related macular degeneration. FASEB J. 2004;18:562–4. doi: 10.1096/fj.03-0289fje. [DOI] [PubMed] [Google Scholar]
- 58.Hammer M, Richter S, Guehrs KH, Schweitzer D. Retinal pigment epithelium cell damage by A2-E and its photo-derivatives. Mol Vis. 2006;12:1348–54. [PubMed] [Google Scholar]
- 59.Vives-Bauza C, Anand M, Shirazi AK, Magrane J, Gao J, Vollmer-Snarr HR, et al. The age lipid A2E and mitochondrial dysfunction synergistically impair phagocytosis by retinal pigment epithelial cells. J Biol Chem. 2008;283:24770–80. doi: 10.1074/jbc.M800706200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Jung T, Bader N, Grune T. Lipofuscin: formation, distribution, and metabolic consequences. Ann N Y Acad Sci. 2007;1119:97–111. doi: 10.1196/annals.1404.008. [DOI] [PubMed] [Google Scholar]
- 61.Wang AL, Lukas TJ, Yuan M, Du N, Tso MO, Neufeld AH. Autophagy and exosomes in the aged retinal pigment epithelium: possible relevance to drusen formation and age-related macular degeneration. PLoS One. 2009;4:e4160. doi: 10.1371/journal.pone.0004160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Liu J, Lu W, Reigada D, Nguyen J, Laties AM, Mitchell CH. Restoration of lysosomal pH in RPE cells from cultured human and ABCA4(-/-) mice: pharmacologic approaches and functional recovery. Invest Ophthalmol Vis Sci. 2008;49:772–80. doi: 10.1167/iovs.07-0675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Baltazar GC, Guha S, Lu W, Lim J, Boesze-Battaglia K, Laties AM, et al. Acidic nanoparticles are trafficked to lysosomes and restore an acidic lysosomal pH and degradative function to compromised ARPE-19 cells. PLoS One. 2012;7:e49635. doi: 10.1371/journal.pone.0049635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kurz T, Karlsson M, Brunk UT, Nilsson SE, Frennesson C. ARPE-19 retinal pigment epithelial cells are highly resistant to oxidative stress and exercise strict control over their lysosomal redox-active iron. Autophagy. 2009;5:494–501. doi: 10.4161/auto.5.4.7961. [DOI] [PubMed] [Google Scholar]
- 65.Feher J, Kovacs I, Artico M, Cavallotti C, Papale A, Balacco Gabrieli C. Mitochondrial alterations of retinal pigment epithelium in age-related macular degeneration. Neurobiol Aging. 2006;27:983–93. doi: 10.1016/j.neurobiolaging.2005.05.012. [DOI] [PubMed] [Google Scholar]
- 66.Holz FG, Bindewald-Wittich A, Fleckenstein M, Dreyhaupt J, Scholl HP, Schmitz-Valckenberg S, FAM-Study Group Progression of geographic atrophy and impact of fundus autofluorescence patterns in age-related macular degeneration. Am J Ophthalmol. 2007;143:463–72. doi: 10.1016/j.ajo.2006.11.041. [DOI] [PubMed] [Google Scholar]
- 67.Alexeyev MF. Is there more to aging than mitochondrial DNA and reactive oxygen species? FEBS J. 2009;276:5768–87. doi: 10.1111/j.1742-4658.2009.07269.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Beatty S, Koh H, Phil M, Henson D, Boulton M. The role of oxidative stress in the pathogenesis of age-related macular degeneration. Surv Ophthalmol. 2000;45:115–34. doi: 10.1016/S0039-6257(00)00140-5. [DOI] [PubMed] [Google Scholar]
- 69.Katta S, Kaur I, Chakrabarti S. The molecular genetic basis of age-related macular degeneration: an overview. J Genet. 2009;88:425–49. doi: 10.1007/s12041-009-0064-4. [DOI] [PubMed] [Google Scholar]
- 70.Kaarniranta K, Hyttinen J, Ryhanen T, Viiri J, Paimela T, Toropainen E, et al. Mechanisms of protein aggregation in the retinal pigment epithelial cells. Front Biosci (Elite Ed) 2010;2:1374–84. doi: 10.2741/E198. [DOI] [PubMed] [Google Scholar]
- 71.Schutt F, Ueberle B, Schnölzer M, Holz FG, Kopitz J. Proteome analysis of lipofuscin in human retinal pigment epithelial cells. FEBS Lett. 2002;528:217–21. doi: 10.1016/S0014-5793(02)03312-4. [DOI] [PubMed] [Google Scholar]
- 72.Terman A, Kurz T, Navratil M, Arriaga EA, Brunk UT. Mitochondrial turnover and aging of long-lived postmitotic cells: the mitochondrial-lysosomal axis theory of aging. Antioxid Redox Signal. 2010;12:503–35. doi: 10.1089/ars.2009.2598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kurz T, Terman A, Gustafsson B, Brunk UT. Lysosomes and oxidative stress in aging and apoptosis. Biochim Biophys Acta. 2008;1780:1291–303. doi: 10.1016/j.bbagen.2008.01.009. [DOI] [PubMed] [Google Scholar]
- 74.Höhn A, Jung T, Grimm S, Grune T. Lipofuscin-bound iron is a major intracellular source of oxidants: role in senescent cells. Free Radic Biol Med. 2010;48:1100–8. doi: 10.1016/j.freeradbiomed.2010.01.030. [DOI] [PubMed] [Google Scholar]
- 75.Schutt F, Bergmann M, Holz FG, Kopitz J. Proteins modified by malondialdehyde, 4-hydroxynonenal, or advanced glycation end products in lipofuscin of human retinal pigment epithelium. Invest Ophthalmol Vis Sci. 2003;44:3663–8. doi: 10.1167/iovs.03-0172. [DOI] [PubMed] [Google Scholar]
- 76.Brunk UT, Wihlmark U, Wrigstad A, Roberg K, Nilsson SE. Accumulation of lipofuscin within retinal pigment epithelial cells results in enhanced sensitivity to photo-oxidation. Gerontology. 1995;41(Suppl 2):201–12. doi: 10.1159/000213743. [DOI] [PubMed] [Google Scholar]
- 77.Jia L, Liu Z, Sun L, Miller SS, Ames BN, Cotman CW, et al. Acrolein, a toxicant in cigarette smoke, causes oxidative damage and mitochondrial dysfunction in RPE cells: protection by (R)-alpha-lipoic acid. Invest Ophthalmol Vis Sci. 2007;48:339–48. doi: 10.1167/iovs.06-0248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Barron MJ, Johnson MA, Andrews RM, Clarke MP, Griffiths PG, Bristow E, et al. Mitochondrial abnormalities in ageing macular photoreceptors. Invest Ophthalmol Vis Sci. 2001;42:3016–22. [PubMed] [Google Scholar]
- 79.Barreau E, Brossas JY, Courtois Y, Tréton JA. Accumulation of mitochondrial DNA deletions in human retina during aging. Invest Ophthalmol Vis Sci. 1996;37:384–91. [PubMed] [Google Scholar]
- 80.Wallace DC. A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: a dawn for evolutionary medicine. Annu Rev Genet. 2005;39:359–407. doi: 10.1146/annurev.genet.39.110304.095751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Navarro A, Boveris A. The mitochondrial energy transduction system and the aging process. Am J Physiol Cell Physiol. 2007;292:C670–86. doi: 10.1152/ajpcell.00213.2006. [DOI] [PubMed] [Google Scholar]
- 82.King A, Gottlieb E, Brooks DG, Murphy MP, Dunaief JL. Mitochondria-derived reactive oxygen species mediate blue light-induced death of retinal pigment epithelial cells. Photochem Photobiol. 2004;79:470–5. doi: 10.1562/LE-03-17.1. [DOI] [PubMed] [Google Scholar]
- 83.Godley BF, Shamsi FA, Liang FQ, Jarrett SG, Davies S, Boulton M. Blue light induces mitochondrial DNA damage and free radical production in epithelial cells. J Biol Chem. 2005;280:21061–6. doi: 10.1074/jbc.M502194200. [DOI] [PubMed] [Google Scholar]
- 84.Nordgaard CL, Karunadharma PP, Feng X, Olsen TW, Ferrington DA. Mitochondrial proteomics of the retinal pigment epithelium at progressive stages of age-related macular degeneration. Invest Ophthalmol Vis Sci. 2008;49:2848–55. doi: 10.1167/iovs.07-1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Karunadharma PP, Nordgaard CL, Olsen TW, Ferrington DA. Mitochondrial DNA damage as a potential mechanism for age-related macular degeneration. Invest Ophthalmol Vis Sci. 2010;51:5470–9. doi: 10.1167/iovs.10-5429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Lin H, Xu H, Liang FQ, Liang H, Gupta P, Havey AN, et al. Mitochondrial DNA damage and repair in RPE associated with aging and age-related macular degeneration. Invest Ophthalmol Vis Sci. 2011;52:3521–9. doi: 10.1167/iovs.10-6163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Nakamura T, Lipton SA. Molecular mechanisms of nitrosative stress-mediated protein misfolding in neurodegenerative diseases. Cell Mol Life Sci. 2007;64:1609–20. doi: 10.1007/s00018-007-6525-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Grune T, Jung T, Merker K, Davies KJ. Decreased proteolysis caused by protein aggregates, inclusion bodies, plaques, lipofuscin, ceroid, and ‘aggresomes’ during oxidative stress, aging, and disease. Int J Biochem Cell Biol. 2004;36:2519–30. doi: 10.1016/j.biocel.2004.04.020. [DOI] [PubMed] [Google Scholar]
- 89.Kaarniranta K, Salminen A, Eskelinen E-L, Kopitz J. Heat shock proteins as gatekeepers of proteolytic pathways-Implications for age-related macular degeneration (AMD) Ageing Res Rev. 2009;8:128–39. doi: 10.1016/j.arr.2009.01.001. [DOI] [PubMed] [Google Scholar]
- 90.Kannan R, Sreekumar PG, Hinton DR. Novel roles for α-crystallins in retinal function and disease. Prog Retin Eye Res. 2012;31:576–604. doi: 10.1016/j.preteyeres.2012.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Fernandes AF, Zhou J, Zhang X, Bian Q, Sparrow J, Taylor A, et al. Oxidative inactivation of the proteasome in retinal pigment epithelial cells. A potential link between oxidative stress and up-regulation of interleukin-8. J Biol Chem. 2008;283:20745–53. doi: 10.1074/jbc.M800268200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Li Y, Wang YS, Shen XF, Hui YN, Han J, Zhao W, et al. Alterations of activity and intracellular distribution of the 20S proteasome in ageing retinal pigment epithelial cells. Exp Gerontol. 2008;43:1114–22. doi: 10.1016/j.exger.2008.08.052. [DOI] [PubMed] [Google Scholar]
- 93.Jung T, Catalgol B, Grune T. The proteasomal system. Mol Aspects Med. 2009;30:191–296. doi: 10.1016/j.mam.2009.04.001. [DOI] [PubMed] [Google Scholar]
- 94.Kawaguchi Y, Kovacs JJ, McLaurin A, Vance JM, Ito A, Yao TP. The deacetylase HDAC6 regulates aggresome formation and cell viability in response to misfolded protein stress. Cell. 2003;115:727–38. doi: 10.1016/S0092-8674(03)00939-5. [DOI] [PubMed] [Google Scholar]
- 95.Boyault C, Zhang Y, Fritah S, Caron C, Gilquin B, Kwon SH, et al. HDAC6 controls major cell response pathways to cytotoxic accumulation of protein aggregates. Genes Dev. 2007;21:2172–81. doi: 10.1101/gad.436407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Ryhänen T, Viiri J, Hyttinen JM, Uusitalo H, Salminen A, Kaarniranta K. Influence of Hsp90 and HDAC inhibition and tubulin acetylation on perinuclear protein aggregation in human retinal pigment epithelial cells. J Biomed Biotechnol. 2011;2011:798052. doi: 10.1155/2011/798052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, Outzen H, et al. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem. 2007;282:24131–45. doi: 10.1074/jbc.M702824200. [DOI] [PubMed] [Google Scholar]
- 98.Yang Z, Klionsky DJ. Mammalian autophagy: core molecular machinery and signaling regulation. Curr Opin Cell Biol. 2010;22:124–31. doi: 10.1016/j.ceb.2009.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell. 2011;147:728–41. doi: 10.1016/j.cell.2011.10.026. [DOI] [PubMed] [Google Scholar]
- 100.Reggiori F, Komatsu M, Finley K, Simonsen A. Selective types of autophagy. Int J Cell Biol. 2012;2012:156272. doi: 10.1155/2012/156272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Russo R, Berliocchi L, Adornetto A, Amantea D, Nucci C, Tassorelli C, et al. In search of new targets for retinal neuroprotection: is there a role for autophagy? Curr Opin Pharmacol. 2013;13:72–7. doi: 10.1016/j.coph.2012.09.004. [DOI] [PubMed] [Google Scholar]
- 102.Ryhänen T, Hyttinen JM, Kopitz J, Rilla K, Kuusisto E, Mannermaa E, et al. Crosstalk between Hsp70 molecular chaperone, lysosomes and proteasomes in autophagy-mediated proteolysis in human retinal pigment epithelial cells. J Cell Mol Med. 2009;13(9B):3616–31. doi: 10.1111/j.1582-4934.2008.00577.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Kuusisto E, Salminen A, Alafuzoff I. Ubiquitin-binding protein p62 is present in neuronal and glial inclusions in human tauopathies and synucleinopathies. Neuroreport. 2001;12:2085–90. doi: 10.1097/00001756-200107200-00009. [DOI] [PubMed] [Google Scholar]
- 104.Kuusisto E, Salminen A, Alafuzoff I. Early accumulation of p62 in neurofibrillary tangles in Alzheimer’s disease: possible role in tangle formation. Neuropathol Appl Neurobiol. 2002;28:228–37. doi: 10.1046/j.1365-2990.2002.00394.x. [DOI] [PubMed] [Google Scholar]
- 105.Zatloukal K, Stumptner C, Fuchsbichler A, Heid H, Schnoelzer M, Kenner L, et al. p62 Is a common component of cytoplasmic inclusions in protein aggregation diseases. Am J Pathol. 2002;160:255–63. doi: 10.1016/S0002-9440(10)64369-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Braak H, Thal DR, Del Tredici K. Nerve cells immunoreactive for p62 in select hypothalamic and brainstem nuclei of controls and Parkinson’s disease cases. J Neural Transm. 2011;118:809–19. doi: 10.1007/s00702-010-0508-2. [DOI] [PubMed] [Google Scholar]
- 107.Salminen A, Kaarniranta K, Haapasalo A, Hiltunen M, Soininen H, Alafuzoff I. Emerging role of p62/sequestosome-1 in the pathogenesis of Alzheimer’s disease. Prog Neurobiol. 2012;96:87–95. doi: 10.1016/j.pneurobio.2011.11.005. [DOI] [PubMed] [Google Scholar]
- 108.Geetha T, Vishwaprakash N, Sycheva M, Babu JR. Sequestosome 1/p62: across diseases. Biomarkers. 2012;17:99–103. doi: 10.3109/1354750X.2011.653986. [DOI] [PubMed] [Google Scholar]
- 109.Wang AL, Lukas TJ, Yuan M, Du N, Tso MO, Neufeld AH. Autophagy, exosomes and drusen formation in age-related macular degeneration. Autophagy. 2009;5:563–4. doi: 10.4161/auto.5.4.8163. [DOI] [PubMed] [Google Scholar]
- 110.Viiri J, Hyttinen JM, Ryhänen T, Rilla K, Paimela T, Kuusisto E, et al. p62/sequestosome 1 as a regulator of proteasome inhibitor-induced autophagy in human retinal pigment epithelial cells. Mol Vis. 2010;16:1399–414. [PMC free article] [PubMed] [Google Scholar]
- 111.Mitter SK, Rao HV, Qi X, Cai J, Sugrue A, Dunn WA, Jr., et al. Autophagy in the retina: a potential role in age-related macular degeneration. Adv Exp Med Biol. 2012;723:83–90. doi: 10.1007/978-1-4614-0631-0_12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Salminen A, Kaarniranta K, Kauppinen A. Inflammaging: disturbed interplay between autophagy and inflammasomes. Aging (Albany NY) 2012;4:166–75. doi: 10.18632/aging.100444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Halle A, Hornung V, Petzold GC, Stewart CR, Monks BG, Reinheckel T, et al. The NALP3 inflammasome is involved in the innate immune response to amyloid-beta. Nat Immunol. 2008;9:857–65. doi: 10.1038/ni.1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Hornung V, Bauernfeind F, Halle A, Samstad EO, Kono H, Rock KL, et al. Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat Immunol. 2008;9:847–56. doi: 10.1038/ni.1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Tseng WA, Thein T, Kinnunen K, Lashkari K, Gregory MS, D’Amore PA, et al. NLRP3 inflammasome activation in retinal pigment epithelial cells by lysosomal destabilization: implications for age-related macular degeneration. Invest Ophthalmol Vis Sci. 2013;54:110–20. doi: 10.1167/iovs.12-10655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Schroder K, Tschopp J. The inflammasomes. Cell. 2010;140:821–32. doi: 10.1016/j.cell.2010.01.040. [DOI] [PubMed] [Google Scholar]
- 117.Tschopp J, Schroder K. NLRP3 inflammasome activation: The convergence of multiple signalling pathways on ROS production? Nat Rev Immunol. 2010;10:210–5. doi: 10.1038/nri2725. [DOI] [PubMed] [Google Scholar]
- 118.Zhou R, Yazdi AS, Menu P, Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature. 2011;469:221–5. doi: 10.1038/nature09663. [DOI] [PubMed] [Google Scholar]
- 119.Kauppinen A, Niskanen H, Suuronen T, Kinnunen K, Salminen A, Kaarniranta K. Oxidative stress activates NLRP3 inflammasomes in ARPE-19 cells--implications for age-related macular degeneration (AMD) Immunol Lett. 2012;147:29–33. doi: 10.1016/j.imlet.2012.05.005. [DOI] [PubMed] [Google Scholar]
- 120.Shimada K, Crother TR, Karlin J, Dagvadorj J, Chiba N, Chen S, et al. Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity. 2012;36:401–14. doi: 10.1016/j.immuni.2012.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Shi CS, Shenderov K, Huang NN, Kabat J, Abu-Asab M, Fitzgerald KA, et al. Activation of autophagy by inflammatory signals limits IL-1β production by targeting ubiquitinated inflammasomes for destruction. Nat Immunol. 2012;13:255–63. doi: 10.1038/ni.2215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Harris J, Hartman M, Roche C, Zeng SG, O’Shea A, Sharp FA, et al. Autophagy controls IL-1beta secretion by targeting pro-IL-1beta for degradation. J Biol Chem. 2011;286:9587–97. doi: 10.1074/jbc.M110.202911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Nakahira K, Haspel JA, Rathinam VA, Lee SJ, Dolinay T, Lam HC, et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol. 2011;12:222–30. doi: 10.1038/ni.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Pauleikhoff D, Barondes MJ, Minassian D, Chisholm IH, Bird AC. Drusen as risk factors in age-related macular disease. Am J Ophthalmol. 1990;109:38–43. doi: 10.1016/s0002-9394(14)75576-x. [DOI] [PubMed] [Google Scholar]
- 125.Holz FG, Schütt F, Kopitz J, Eldred GE, Kruse FE, Völcker HE, et al. Inhibition of lysosomal degradative functions in RPE cells by a retinoid component of lipofuscin. Invest Ophthalmol Vis Sci. 1999;40:737–43. [PubMed] [Google Scholar]
- 126.Doyle SL, Campbell M, Ozaki E, Salomon RG, Mori A, Kenna PF, et al. NLRP3 has a protective role in age-related macular degeneration through the induction of IL-18 by drusen components. Nat Med. 2012;18:791–8. doi: 10.1038/nm.2717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Mullins RF, Russell SR, Anderson DH, Hageman GS. Drusen associated with aging and age-related macular degeneration contain proteins common to extracellular deposits associated with atherosclerosis, elastosis, amyloidosis, and dense deposit disease. FASEB J. 2000;14:835–46. [PubMed] [Google Scholar]
- 128.Krohne TU, Holz FG, Kopitz J. Apical-to-basolateral transcytosis of photoreceptor outer segments induced by lipid peroxidation products in human retinal pigment epithelial cells. Invest Ophthalmol Vis Sci. 2010;51:553–60. doi: 10.1167/iovs.09-3755. [DOI] [PubMed] [Google Scholar]
- 129.Korolchuk VI, Mansilla A, Menzies FM, Rubinsztein DC. Autophagy inhibition compromises degradation of ubiquitin-proteasome pathway substrates. Mol Cell. 2009;33:517–27. doi: 10.1016/j.molcel.2009.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Kim SH, Munemasa Y, Kwong JM, Ahn JH, Mareninov S, Gordon LK, et al. Activation of autophagy in retinal ganglion cells. J Neurosci Res. 2008;86:2943–51. doi: 10.1002/jnr.21738. [DOI] [PubMed] [Google Scholar]
- 131.Knöferle J, Koch JC, Ostendorf T, Michel U, Planchamp V, Vutova P, et al. Mechanisms of acute axonal degeneration in the optic nerve in vivo. Proc Natl Acad Sci U S A. 2010;107:6064–9. doi: 10.1073/pnas.0909794107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Mellén MA, de la Rosa EJ, Boya P. The autophagic machinery is necessary for removal of cell corpses from the developing retinal neuroepithelium. Cell Death Differ. 2008;15:1279–90. doi: 10.1038/cdd.2008.40. [DOI] [PubMed] [Google Scholar]
- 133.Yang ZJ, Chee CE, Huang S, Sinicrope FA. The role of autophagy in cancer: therapeutic implications. Mol Cancer Ther. 2011;10:1533–41. doi: 10.1158/1535-7163.MCT-11-0047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Wang Y, Peng RQ, Li DD, Ding Y, Wu XQ, Zeng YX, et al. Chloroquine enhances the cytotoxicity of topotecan by inhibiting autophagy in lung cancer cells. Chin J Cancer. 2011;30:690–700. doi: 10.5732/cjc.011.10056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Choi JH, Yoon JS, Won YW, Park BB, Lee YY. Chloroquine enhances the chemotherapeutic activity of 5-fluorouracil in a colon cancer cell line via cell cycle alteration. APMIS. 2012;120:597–604. doi: 10.1111/j.1600-0463.2012.02876.x. [DOI] [PubMed] [Google Scholar]
- 136.Maycotte P, Aryal S, Cummings CT, Thorburn J, Morgan MJ, Thorburn A. Chloroquine sensitizes breast cancer cells to chemotherapy independent of autophagy. Autophagy. 2012;8:200–12. doi: 10.4161/auto.8.2.18554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Michaelides M, Stover NB, Francis PJ, Weleber RG. Retinal toxicity associated with hydroxychloroquine and chloroquine: risk factors, screening, and progression despite cessation of therapy. Arch Ophthalmol. 2011;129:30–9. doi: 10.1001/archophthalmol.2010.321. [DOI] [PubMed] [Google Scholar]
- 138.Hickley NM, Al-Maskari A, McKibbin M. Chloroquine and hydroxychloroquine toxicity. Arch Ophthalmol. 2011;129:1506–7. doi: 10.1001/archophthalmol.2011.321. [DOI] [PubMed] [Google Scholar]
- 139.Thampi P, Rao HV, Mitter SK, Cai J, Mao H, Li H, et al. The 5HT1a receptor agonist 8-Oh DPAT induces protection from lipofuscin accumulation and oxidative stress in the retinal pigment epithelium. PLoS One. 2012;7:e34468. doi: 10.1371/journal.pone.0034468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Wilkinson JE, Burmeister L, Brooks SV, Chan CC, Friedline S, Harrison DE, et al. Rapamycin slows aging in mice. Aging Cell. 2012;11:675–82. doi: 10.1111/j.1474-9726.2012.00832.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Rodríguez-Muela N, Germain F, Mariño G, Fitze PS, Boya P. Autophagy promotes survival of retinal ganglion cells after optic nerve axotomy in mice. Cell Death Differ. 2012;19:162–9. doi: 10.1038/cdd.2011.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Zhao C, Yasumura D, Li X, Matthes M, Lloyd M, Nielsen G, et al. mTOR-mediated dedifferentiation of the retinal pigment epithelium initiates photoreceptor degeneration in mice. J Clin Invest. 2011;121:369–83. doi: 10.1172/JCI44303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Li Y, Huang D, Xia X, Wang Z, Luo L, Wen R. CCR3 and choroidal neovascularization. PLoS One. 2011;6:e17106. doi: 10.1371/journal.pone.0017106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Dejneka NS, Kuroki AM, Fosnot J, Tang W, Tolentino MJ, Bennett J. Systemic rapamycin inhibits retinal and choroidal neovascularization in mice. Mol Vis. 2004;10:964–72. [PubMed] [Google Scholar]
- 145.Stahl A, Paschek L, Martin G, Gross NJ, Feltgen N, Hansen LL, et al. Rapamycin reduces VEGF expression in retinal pigment epithelium (RPE) and inhibits RPE-induced sprouting angiogenesis in vitro. FEBS Lett. 2008;582:3097–102. doi: 10.1016/j.febslet.2008.08.005. [DOI] [PubMed] [Google Scholar]
- 146.Qin S, De Vries GW. α2 But not α1 AMP-activated protein kinase mediates oxidative stress-induced inhibition of retinal pigment epithelium cell phagocytosis of photoreceptor outer segments. J Biol Chem. 2008;283:6744–51. doi: 10.1074/jbc.M708848200. [DOI] [PubMed] [Google Scholar]
- 147.Herrero-Martín G, Høyer-Hansen M, García-García C, Fumarola C, Farkas T, López-Rivas A, et al. TAK1 activates AMPK-dependent cytoprotective autophagy in TRAIL-treated epithelial cells. EMBO J. 2009;28:677–85. doi: 10.1038/emboj.2009.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Nishikawa T, Tsuno NH, Okaji Y, Sunami E, Shuno Y, Sasaki K, et al. The inhibition of autophagy potentiates anti-angiogenic effects of sulforaphane by inducing apoptosis. Angiogenesis. 2010;13:227–38. doi: 10.1007/s10456-010-9180-2. [DOI] [PubMed] [Google Scholar]
- 149.Chekhonin VP, Shein SA, Korchagina AA, Gurina OI. Vegf in tumor progression and targeted therapy. Curr Cancer Drug Targets. 2012 doi: 10.2174/15680096113139990074. [DOI] [PubMed] [Google Scholar]
- 150.Juuti-Uusitalo K, Vaajasaari H, Ryhänen T, Narkilahti S, Suuronen R, Mannermaa E, et al. Efflux protein expression in human stem cell-derived retinal pigment epithelial cells. PLoS One. 2012;7:e30089. doi: 10.1371/journal.pone.0030089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Rolling F, Le Meur G, Stieger K, Smith AJ, Weber M, Deschamps JY, et al. Gene therapeutic prospects in early onset of severe retinal dystrophy: restoration of vision in RPE65 Briard dogs using an AAV serotype 4 vector that specifically targets the retinal pigmented epithelium. Bull Mem Acad R Med Belg. 2006;161:497–508, discussion 508-9. [PubMed] [Google Scholar]
- 152.Pastore N, Blomenkamp K, Annunziata F, Piccolo P, Mithbaokar P, Maria Sepe R, et al. Gene transfer of master autophagy regulator TFEB results in clearance of toxic protein and correction of hepatic disease in alpha-1-anti-trypsin deficiency. EMBO Mol Med. 2013;5:397–412. doi: 10.1002/emmm.201202046. [DOI] [PMC free article] [PubMed] [Google Scholar]