

Abstract
Listeria monocytogenes is a bacterial pathogen that can escape the phagosome and replicate in the cytosol of host cells during infection. We previously observed that a population (up to 35%) of L. monocytogenes strain 10403S colocalize with the macroautophagy marker LC3 at 1 h postinfection. This is thought to give rise to spacious Listeria-containing phagosomes (SLAPs), a membrane-bound compartment harboring slow-growing bacteria that is associated with persistent infection. Here, we examined the host and bacterial factors that mediate LC3 recruitment to bacteria at 1 h postinfection. At this early time point, LC3+ bacteria were present within single-membrane phagosomes that are LAMP1+. Protein ubiquitination is known to play a role in targeting cytosolic L. monocytogenes to macroautophagy. However, we found that neither protein ubiquitination nor the ubiquitin-binding adaptor SQSTM1/p62 are associated with LC3+ bacteria at 1 h postinfection. Reactive oxygen species (ROS) production by the CYBB/NOX2 NADPH oxidase was also required for LC3 recruitment to bacteria at 1 h postinfection and for subsequent SLAP formation. Diacylglycerol is an upstream activator of the CYBB/NOX2 NADPH oxidase, and its production by both bacterial and host phospholipases was required for LC3 recruitment to bacteria. Our data suggest that the LC3-associated phagocytosis (LAP) pathway, which is distinct from macroautophagy, targets L. monocytogenes during the early stage of infection within host macrophages and allows establishment of an intracellular niche (SLAPs) associated with persistent infection.
Keywords: autophagy, diacylglycerol, innate immunity, LC3, LC3-associated phagocytosis, Listeria monocytogenes, reactive oxygen species, ubiquitin
Introduction
Macroautophagy (hereafter referred to as autophagy) is a key component of host innate immune defense against many pathogenic microorganisms,1 limiting bacterial escape from the phagosome2-4 as well as targeting cytosolic bacteria to the lysosomes for clearance.5,6 Furthermore, components of the autophagy pathway can also contribute to immunity by mechanisms independent of the formation of autophagosomes7,8 (recently reviewed by Cemma et al.).9 One notable example is LC3-associated phagocytosis (LAP), a pathway that is involved in promoting phagosome maturation, killing of microbes (e.g., S. cerevisiae), as well as suppressing proinflammatory signals.4,10,11 LAP requires some essential components of the autophagy pathway (ATG5 ATG7, BECN1) but not others (ULK1).11 While the mechanisms regulating LAP are not clear, reactive oxygen species production by the CYBB/NOX2 NADPH oxidase is required.10 LC3 recruitment to phagosomes containing the BopA mutant of Burkholderia pseudomallei is suggested to promote killing of bacteria trapped in phagosomes.12 Whether LAP targets other bacteria during infection, and whether this pathway can be subverted/exploited by intracellular pathogens, is not known.
Listeria monocytogenes (L. monocytogenes) is the causative agent of listeriosis, a gastroenteritis that is self-limiting in healthy individuals but may become life-threatening for neonates or young children, elderly and immunocompromised individuals.13L. monocytogenes can colonize host cells, including macrophages, during infection.14 Upon invasion, a population of bacteria is able to escape from the phagosome and colonize the nutrient-rich cytosol where they replicate rapidly. Phagosomal escape is mediated by the bacterial pore forming toxin Listeriolysin O (LLO) and two phospholipase C enzymes (PLCs). PLCs are thought to not only degrade the phagosome to promote escape but can also activate signal transduction cascades within the host cell as a result of their catalytic activity.14,15 Upon entry to the cytosol, the bacteria use a cell surface protein, ActA, to drive actin-based motility in the host cytosol, and eventual cell-to-cell transfer.14
Previous studies have shown that L. monocytogenes strain 10403S colocalizes with LC3 at 1 h postinfection (p.i.).16-18 This population of bacteria is thought to give rise to spacious Listeria-containing phagosomes (SLAPs), a membrane-bound compartment harboring slow-growing bacteria that is associated with persistent L. monocytogenes infection.19,20 A number of questions remain regarding the mechanism of LC3 recruitment to bacteria at 1 h p.i. Given the recent findings that LC3 can be involved in other degradative pathways that are independent of autophagy, it is unclear which pathway is activated upon L. monocytogenes infection. Furthermore, since L. monocytogenes can escape from the phagosome, it is unclear whether bacteria are targeted by LC3 within phagosomes or in the cytosol at the peak of LC3 colocalization. Finally, it is also unknown which host and/or bacterial factor(s) mediate LC3 colocalization with L. monocytogenes. We have previously demonstrated that a nonmotile (ActAΔ) mutant of L. monocytogenes colocalizes with ubiquitinated proteins in the cytosol.21 Recent work by Sasakawa and colleagues suggests that this protein ubiquitination event mediates recruitment of adaptor proteins such as SQSTM1/p62 that leads to autophagic targeting of the ActAΔ mutant of L. monocytogenes inside the cytosol, a process initiated after 2 h p.i.6,22 However, it is not known if protein ubiquitination plays a role in LC3 colocalization with wild-type L. monocytogenes at the peak of LC3 recruitment at 1 h p.i..
In this study, we examined the mechanisms that regulate LC3 colocalization with L. monocytogenes strain 10403S in macrophages at early stages of infection. We demonstrated that these bacteria colocalize with LC3 within single-membrane phagosomes at 1 h p.i. in a manner requiring both DAG production and ROS generated by the CYBB/NOX2 NADPH oxidase. However, protein ubiquitination was not associated with LC3 recruitment to these bacteria. Our studies suggest that the LAP pathway targets L. monocytogenes at an early stage of infection and gives rise to a vacuolar niche (SLAPs) that can cause persistent infection. We also identified the critical host and bacterial factors that regulate LC3 recruitment to L. monocytogenes. Importantly, these signals differ from autophagic targeting of bacteria in the cytosol at later stages of infection.
Results
LC3 colocalizes with wild-type L. monocytogenes in single-membrane phagosomes
Consistent with previous work,16-18 we found that LC3 recruitment to L. monocytogenes peaks at 1 h p.i. in an LLO-dependent manner (Fig. 1A and B). However, based on published reports of L. monocytogenes phagosome-escape kinetics,23-25 it is unclear whether L. monocytogenes is in the phagosome or cytosol during the peak of LC3 targeting. To determine whether L. monocytogenes is present inside phagosomes or the cytosol at 1 h p.i., we first quantified the percentage of L. monocytogenes that were both LC3+ and LAMP1+-associated. Previously, we have reported that roughly 70% to 80% of LC3+-associated L. monocytogenes are also LAMP1+-associated.16 Here, we found that the majority (78%) of LAMP1+ bacteria were also LC3+ (Fig. 1C and D). Together, these observations suggest that LC3 is targeted to L. monocytogenes within an intracellular compartment and not the cytosol. Furthermore, our data suggest that LC3 recruitment is a common fate for bacteria trapped in phagosomes.

Figure 1. LC3 is recruited to wild-type L. monocytogenes strain 10403S in macrophage phagosomes during early stages of infection. (A) Confocal images of RAW 264.7 macrophages transfected with GFP-LC3 and infected for 1 h with wild-type L. monocytogenes. (B) Quantification of LC3 colocalization with intracellular wild-type or LLOΔ bacteria over time. (C) Confocal images of RAW 264.7 macrophages transfected with GFP-LC3 and infected for 1 h with wild-type L. monocytogenes. Cells were then stained for LAMP1. (D) Quantification of the percentage of LAMP1+ bacteria that are LC3+ or percentage of LAMP1- bacteria that are LC3+. (E) Confocal images of RAW 264.7 macrophages transfected with CBD-YFP and infected for 1 h with wild-type L. monocytogenes. Cells were then stained with phalloidin to label F-actin. (F) Quantification of the percentage of CBD-YFP+ bacteria that are actin+. (G) Confocal images of RAW 264.7 macrophages expressing RFP-LC3 and CBD-YFP and infected for 1 h with wild-type L. monocytogenes. (H) Quantification of the percentage of LC3+L. monocytogenes that are CBD-YFP+. The inner panels represent a higher magnification of the boxed areas. Size bars: 5 µm. The brightness and contrast for the image (but not the inset) was enhanced to allow visualization of fluorescence inside the cell. (I) Correlative imaging of RAW 264.7 macrophage expressing GFP-LC3 infected with NHS-647 labeled wild-type L. monocytogenes. Images of the same cell were captured using either electron microscopy (upper row) or fluorescence microscopy (lower row). Size bars as indicated. Arrowheads indicate single membrane while arrows indicate double membrane. The asterix indicates a multivesicular body.
To further examine LC3 recruitment to L. monocytogenes, we employed a recently described probe constructed from the cell wall binding domain (CBD) of a Listeria bacteriophage protein.24 A yellow fluorescent protein fusion to CBD (CBD-YFP) was developed by Swanson and colleagues to track phagosome escape by L. monocytogenes.24 CBD-YFP expressed within host cells binds to cytosolic bacteria, resulting in an accumulation of YFP fluorescence around the bacteria.24 Upon escape from the phagosome, L. monocytogenes also polymerizes actin via the ActA virulence factor. Thus, we first confirmed the ability of the CBD-YFP probe to detect cytosolic L. monocytogenes by quantifying the number of CBD-YFP+ bacteria that were also positive for polymerized actin at 4 h p.i. when bacteria are known to be localized in the cytosol. As expected, the majority (60%) of CBD-YFP+ L. monocytogenes were also actin+, confirming that the CBD-YFP can detect cytosolic L. monocytogenes after their escape from phagosomes (Fig. 1E and F). In contrast, we did not observe any accumulation of CBD-YFP to the LC3+ population of L. monocytogenes at 1 h p.i. (Fig. 1G and H). Together, these data demonstrate that LC3 is recruited to L. monocytogenes within phagosomes at early stages of infection.
Next, we examined LC3+ L. monocytogenes using correlative light and electron microscopy. We observed that LC3+ bacteria at 1 h p.i. were present in phagosomes that have a single membrane (Fig. 1I), consistent with our previous analysis of L. monocytogenes infected cells analyzed at 1 h p.i. by conventional transmission electron microscopy.16 This is in contrast to double-membrane autophagosomes observed during autophagy of nonmotile (ActAΔ) bacteria from the cytosol, that were observed by Webster and colleagues.6 We also did not observe multilammelar (3 to 4 membranes) structures observed when bacteria in disrupted phagosomes are targeted by autophagy.26,27
Protein ubiquitination is not a signal for recruitment of LC3 to L. monocytogenes at 1 h p.i
Previous studies suggest that protein ubiquitination mediates autophagic targeting of L. monocytogenes in the cytosol via the ubiquitin-binding adaptor SQSTM1.22 Autophagy of damaged phagosomes containing Salmonella enterica serovar Typhimurium and S. flexneri has also been shown to involve protein ubiquitination and ubiquitin-binding adaptors.2,27-29 These observations suggest the possibility that protein ubiquitination may also play a role in LC3 recruitment to L. monocytogenes within phagosomes. However, we observed that the majority (98%) of the LC3+ population of L. monocytogenes did not colocalize with ubiquitinated proteins at 1 h p.i. (Fig. 2A and B). Consistent with this finding, expression of the ubiquitin-binding adaptor SQSTM1 was not required for LC3 recruitment to L. monocytogenes at 1 h p.i. as siRNA-mediated silencing of Sqstm1 did not alter LC3 colocalization with bacteria (Fig. 2C). However, siRNA-mediated silencing of Atg12 showed an expected drop in LC3 recruitment.
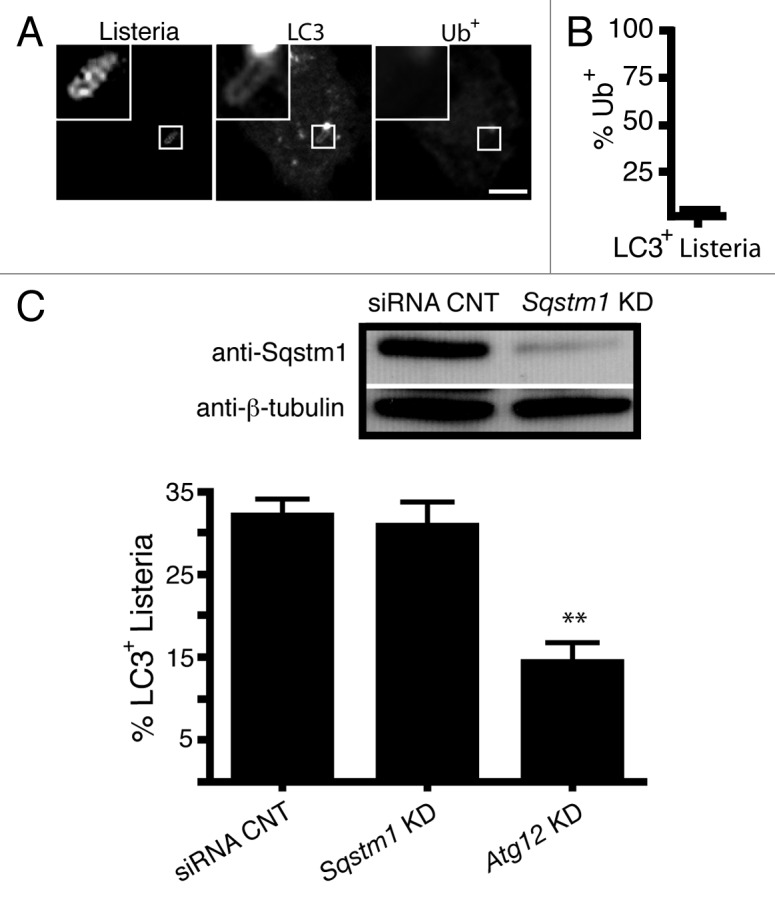
Figure 2. Protein ubiquitination and the ubiquitin-adaptor protein p62/SQSTMI do not mediate LC3 recruitment to L. monocytogenes at 1 h p.i. (A) Confocal images of RAW 264.7 macrophages transfected with GFP-LC3 and infected for 1 h with wild-type L. monocytogenes. Cells were then stained for ubiquitinated proteins (Ub+). (B) Quantification of the percentage of LC3+L. monocytogenes that are Ub+. The inner panels represent a higher magnification of the boxed areas. Size bars: 5 µm. (C) Quantification of LC3 colocalization to wild-type L. monocytogenes in RAW 264.7 macrophages with siRNA silencing of Sqstm1 or Atg12 (positive control). Knockdown was confirmed using sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE).
Since LC3+ L. monocytogenes localized to single-membrane compartments that were formed independently of protein ubiquitination and SQSTM1 targeting, autophagy is not likely to be responsible for LC3 localization to the bacteria. Instead, our data imply that LC3 recruitment to L. monocytogenes is likely to be mediated via the LAP pathway. Our data are consistent with a model whereby a population of bacteria become trapped in LC3+ phagosomes that do not undergo normal maturation steps (e.g., fusion with lysosomes) but rather become SLAPs through continued low level expression of LLO. In support of this model, we previously showed that bacteria in SLAPs do not colocalize with ubiquitinated proteins, indicating they were not exposed to the cytosol.20 Furthermore, we showed that monomeric red fluorescent protein expressed in the cytosol was not delivered to the lumen of SLAPs.20 Together, these findings suggested that targeting of L. monocytogenes by the LAP pathway at 1 h p.i. gives rise to the formation of SLAPs. Our model predicted that host factors promoting LAP targeting of bacteria at 1 h p.i. would also impact on the formation of SLAPs at subsequent stages of infection.
ROS production by the CYBB/NOX2 NADPH oxidase is required for recruitment of LC3 to L. monocytogenes at early stages of infection
We previously showed that reactive oxygen species (ROS) production by the CYBB/NOX2 NADPH oxidase is required for the LAP pathway when macrophages are fed IgG-coated latex beads or zymosan particles.10 Therefore, we examined the role of ROS in LC3 recruitment to L. monocytogenes. We inhibited ROS production via the addition of the NADPH oxidase inhibitor, diphenyliodonium (DPI), which at 1 h p.i. significantly reduced LC3 colocalization with L. monocytogenes. LC3 colocalization was also reduced when antioxidants, resveratrol and α-tocopherol, were added (Fig. 3A). These findings are consistent with a role for ROS in regulating LAP of L. monocytogenes.

Figure 3. ROS production by the CYBB/NOX2 NADPH oxidase is required for LC3 recruitment to L. monocytogenes during early stages of infection. (A) RAW 264.7 cells were infected with wild-type L. monocytogenes for 1 h in the presence or absence of DPI, resveratrol or α-tocopherol, as indicated. The percentage of intracellular bacteria that are LC3+ upon treatment with each agent is shown. (B) Confocal images of wild-type or cybb−/− bone marrow-derived macrophages transfected with GFP-LC3 and infected for 1 h with wild-type bacteria. Size bar: 5 µm. (C) Quantification at 1 h p.i. of the percentage of intracellular L. monocytogenes that are LC3+ in bone marrow-derived macrophages isolated from wild-type or cybb-/- mice. Wild-type bacteria were compared with LLOΔ bacteria (negative control), as well as a LLOΔ mutant complemented with LLO. (D) Quantification of the percentage of L. monocytogenes that are LC3+ in RAW 264.7 macrophages infected with either wild-type or ActAΔ bacteria for: 8 h (GM); 3 h, followed by 5 h Cm treatment (+Cm); or 3 h, followed by 5 h Cm and DPI treatment (+Cm +DPI).
Next, we examined LC3 recruitment to L. monocytogenes in bone-marrow macrophages deficient in CYBB/NOX2 NADPH oxidase activity (cybb−/−) (Fig. 3B and C). We found that cybb−/− macrophages displayed a significant reduction in LC3 colocalization with wild-type L. monocytogenes (Fig. 3B and C). Furthermore, consistent with previous studies,16-18 LC3 recruitment to bacteria was dependent on LLO expression. Colocalization of LC3 with LLOΔ mutant bacteria was restored upon LLO complementation (Fig. 3C). LLOΔ mutant with or without LLO expression did not colocalize with LC3 in cybb−/− macrophages.
Previous studies by Webster and colleagues showed that ActA deficient L. monocytogenes (ActAΔ) are targeted by autophagy in the cytosol after the addition of chloramphenicol (Cm), an agent that blocks bacterial protein synthesis.6 Cells were infected with ActAΔ mutant bacteria for 3 h, followed by a 5 h treatment with Cm, after which autophagy of bacteria was detected by electron microscopy.6 Following a similar protocol of infection and Cm treatment, we observed significant colocalization of LC3 with the ActAΔ L. monocytogenes, consistent with autophagy of bacteria (Fig. 3D). However, this LC3 colocalization was unaltered when cells were treated with DPI and Cm concomitantly. Thus, ROS production by the CYBB/NOX2 NADPH oxidase plays a key role in LC3 recruitment to L. monocytogenes at early stages of infection (1 h p.i.) presumably via LAP, but not in autophagy of ActAΔ L. monocytogenes in the cytosol at later stages of infection.
ROS production by the CYBB/NOX2 NADPH oxidase is required for spacious Listeria-containing phagosomes (SLAPs) formation
Previously, we showed that LC3 recruitment to L. monocytogenes at 1 h p.i. is associated with the formation of SLAPs, L. monocytogenes-containing compartments that are spacious, neutral pH, nondegradative, LAMP1+ and LC3+, and appearing after 4 h p.i.20 Formation of SLAPs was found to require bacterial expression of low levels of LLO, insufficient to allow bacterial escape into the cytosol.20 We proposed that SLAPs represent a “stalemate” between the host and bacteria, allowing slow bacterial replication in SLAPs that may allow persistent L. monocytogenes infection in a host.20 Indeed, compartments resembling SLAPs have been observed in a mouse model of L. monocytogenes persistent infection.19 Consistent with the notion that early LC3 recruitment to bacteria leads to SLAP formation, we observed that DPI treatment reduced the formation of SLAPs (Fig. 4A and B). SLAP formation was also decreased in cybb−/− bone marrow-derived macrophages compared with wild-type macrophages (Fig. 4C and D). These findings suggest that ROS-dependent LC3 recruitment to L. monocytogenes at 1 h p.i. may be required for the later formation of SLAPs. Thus, signals that mediate early LC3 recruitment to L. monocytogenes may also be important in determining events that lead to establishment of persistent infection in a host.

Figure 4. ROS production and LC3-associated phagocytosis is required for SLAP formation during later stages of infection. ROS production by the CYBB/NOX2 NADPH oxidase is required for the generation of SLAPs. (A) Confocal images of RAW 264.7 macrophages infected for 4 h with wild-type L. monocytogenes, with or without DPI. Cells were stained with LAMP1 antibodies. Size bar: 5 µm. White arrowheads indicate spacious Listeriaphagosomes (SLAPs) while the white arrow indicates LAMP1 colocalization with bacteria that are not in SLAPs (B) Quantification at 4 h p.i. of the percentage of infected macrophages that form SLAPs in the presence or absence of DPI, resveratrol or α-tocopherol, as indicated. (C) Confocal images of wild-type or cybb−/− bone marrow-derived macrophages infected for 8 h with wild-type L. monocytogenes. Cells were stained with LAMP1 antibodies. Size bar: 5 µm. White arrowheads indicate SLAPs while the white arrows indicate LAMP1 colocalization and not SLAPs. (D) Quantification at 8 h p.i. of the percentage of infected wild-type or cybb-/- bone marrow-derived macrophages that form SLAPs from (C).
Host and bacterial factors promote DAG accumulation on phagosomes containing L. monocytogenes
Next, we examined the signals that are upstream of ROS production by the CYBB/NOX2 NADPH oxidase. Previous studies have shown that L. monocytogenes infection induces diacylglycerol (DAG) production30,31 in a manner dependent on the activity of LLO,30 bacterial PLCs31 and host phospholipases.32 Generation of DAG is upstream of ROS production via the CYBB/NOX2 NADPH oxidase33 (reviewed by Lam et al.).34 Therefore, it is conceivable that DAG production on the phagosome may play a role in mediating LAP of L. monocytogenes.
To visualize DAG during L. monocytogenes infection, we employed a fluorescent probe constructed from the DAG binding C1 domain of PKCδ (PRKCD) fused to green fluorescent protein (PRKCD-C1-GFP). The majority (90%) of LC3+ L. monocytogenes were observed to be DAG+ (Fig. 5A). DAG colocalization with L. monocytogenes was observed prior to the peak of LC3 recruitment (Fig. 5B and C), suggesting it may serve as an upstream signal for LAP.
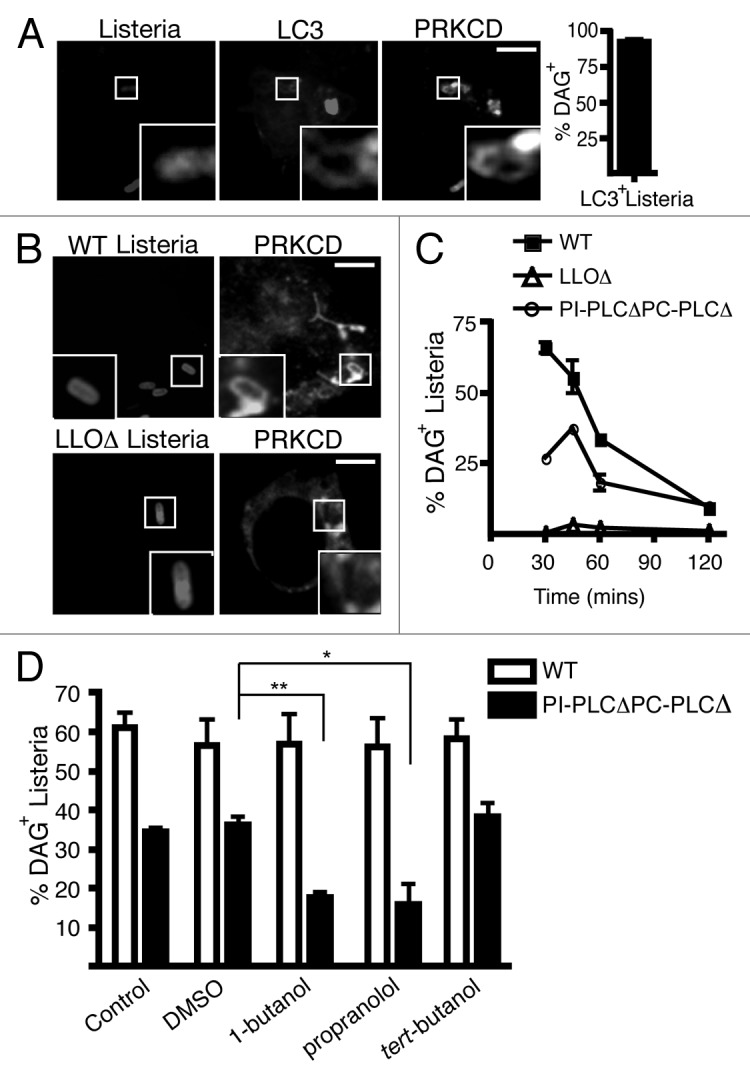
Figure 5. Host and bacterial factors promote DAG accumulation on phagosomes containing L. monocytogenes. (A) Confocal images of RAW 264.7 macrophages co-transfected with RFP-LC3 and PRKCD-C1-GFP and infected for 1 h with wild-type L. monocytogenes. PRKCD-C1-GFP is a specific probe for DAG. Size bar: 5 µm. Quantification at 1 h p.i. of the percentage of LC3+L. monocytogenes that are PRKCD-C1-GFP+ (B) Confocal images of RAW 264.7 macrophages transfected with PRKCD-C1-GFP and infected for 1 h with wild-type, LLOΔ or PI-PLCΔ PC-PLCΔ bacteria. Size bar: 5 µm. (C) Quantification of the percentage of intracellular wild-type, LLOΔ or PI-PLCΔ PC-PLCΔ L. monocytogenes that are DAG+ over time. (D) Quantification at 45 min p.i. of intracellular wild-type or PI-PLCΔ PC-PLCΔ L. monocytogenes that are PRKCD-C1-GFP + upon treatment with DMSO, 1-butanol, propranolol or tert-butanol.
The kinetics of DAG colocalization with L. monocytogenes lacking both PLCs (PI-PLCΔ PC-PLCΔ) was also examined. As shown in Figure 5C, the percentage of DAG+ PI-PLCΔ PC-PLCΔ bacteria was approximately 50% less than wild-type bacteria at 30 min and 45 min p.i.. This suggests that bacterial PLCs contribute to DAG production on phagosomes containing L. monocytogenes. However, the decrease in DAG localization to PI-PLCΔ PC-PLCΔ bacteria was not complete, raising the possibility that host factors may also contribute to DAG production. Indeed, host phospholipases were previously shown to contribute to DAG production during L. monocytogenes infection.31
We determined the effect of inhibiting host enzymes that produce DAG. Recent studies of Salmonella enterica serovar Typhimurium infection indicated that DAG is produced by host phospholipase D (PLD) and phosphatidic acid phosphatase (PPAP2A).35 We therefore employed inhibitors of PLD (1-butanol) and PPAP2A (propranolol hydrochloride) to examine changes to DAG production on phagosomes containing wild-type or PI-PLCΔ PC-PLCΔ bacteria. Inhibition of PLD or PPAP2A resulted in a decrease in DAG colocalization to the PI-PLCΔ PC-PLCΔ mutants (Fig. 5D). This effect was not observed with tert-butanol, an isomer of 1-butanol that has no inhibitory effect on PLD. This suggests that host factors PLD and PPAP2A contribute to the accumulation of DAG at L. monocytogenes-containing phagosomes. Surprisingly, DAG colocalization to phagosomes containing wild-type L. monocytogenes was not altered upon addition of PLD or PPAP2A inhibitors. This suggests that bacterial PLCs compensate for inhibition of host DAG production. These results indicate that both host and bacterial factors contribute to DAG accumulation on phagosomes containing L. monocytogenes.
DAG production is required for LC3 recruitment to L. monocytogenes at early stages of infection
To investigate whether DAG plays a role in mediating LC3 recruitment to L. monocytogenes, we examined LC3 recruitment to both wild-type and PI-PLCΔPC-PLCΔ mutants upon treatment with PLD and PPAP2A inhibitors (Fig. 6A). Similar to what we observed with DAG localization in Figure 5D, LC3 recruitment to wild-type L. monocytogenes did not change upon PLD or PPAP2A inhibition. In contrast, LC3 recruitment to the PI-PLCΔ PC-PLCΔ mutant was significantly inhibited upon treatment with either the PLD inhibitor (90% decrease) or the PPAP2A inhibitor (73% decrease). This suggests that DAG is important LC3 recruitment to L. monocytogenes. Consistent with these findings, inhibition of DAG turnover via inhibition of DAG kinase (DGK) results in increased DAG and LC3 colocalization to L. monocytogenes confirming that the presence of DAG on L. monocytogenes containing phagosomes is correlated with LC3 recruitment (Fig. 6B).
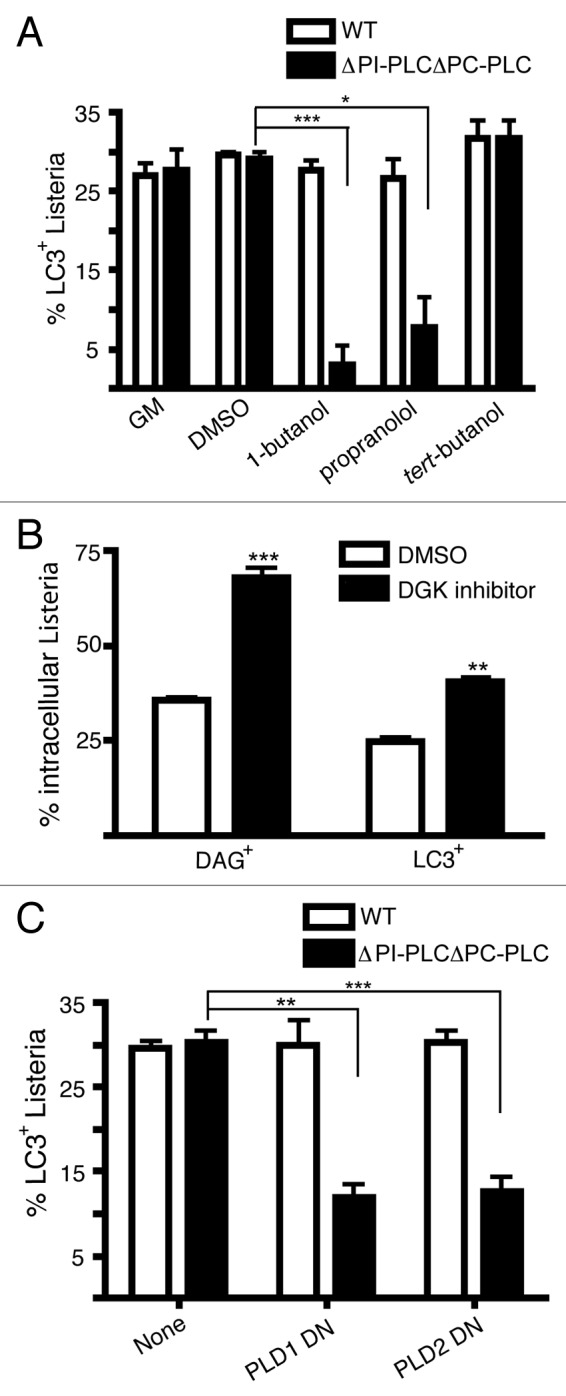
Figure 6. DAG production is required for LC3-associated phagocytosis of L. monocytogenes during early stages of infection. (A) Quantification at 1 h p.i. of intracellular wild-type or PI-PLCΔ PC-PLCΔ L. monocytogenes that are LC3+ following treatment with DMSO, 1-butanol, propranolol, or tert-butanol. (B) Quantification at 1 h p.i. of DAG (PRKCD-C1-GFP+) or LC3 colocalization with wild-type L. monocytogenes with or without treatment with the DAG kinase I inhibitor. (C) Quantification at 1 h p.i. of LC3+ intracellular wild-type or PI-PLCΔ PC-PLCΔ L. monocytogenes in RAW 264.7 macrophages that were cotransfected with LC3-GFP and either PLD1 dominant negative (DN) or PLD2 DN constructs.
To confirm the contribution of PLD-mediated generation of DAG to LC3 targeting, dominant negative constructs for both isoforms of PLD (PLD1 and PLD2) were employed (Fig. S1). Similar to the pharmacological evidence, dominant negative forms of PLD reduced DAG recruitment (40 to 62% decrease) as well as LC3 recruitment (60% to 61% decrease) to PI-PLCΔ PC-PLCΔ mutants (Fig. 6C). Taken together, both bacterial and host mediated production of DAG can contribute to LC3 recruitment to L. monocytogenes.
Discussion
In this study, we examined LC3 recruitment to L. monocytogenes during the early stages of infection (1 h p.i.) and how this affects later stages of infection. Our data suggest that LC3 colocalizes with bacteria not as a result of autophagy as previously thought,16,18 but rather by the LAP pathway. This conclusion is supported by the following evidence: (1) the LC3+ population of L. monocytogenes at 1 h p.i. was confined to single-membrane, LAMP1+ phagosomes; (2) LC3+ bacteria were not exposed to the cytosol as judged by a lack of colocalization with F-actin or the cytosolic probe CBD-YFP; (3) ubiquitinated proteins did not colocalize with LC3+ bacteria, as was previously observed for other bacteria targeted by autophagy;2 (4) the ubiquitin-binding autophagy adaptor SQSTM1 was not required for LC3 recruitment to L. monocytogenes though it is required for autophagy of these bacteria within the cytosol;22 (5) ROS production by the CYBB/NOX2 NADPH oxidase was required for LC3 recruitment to bacteria, consistent with previous studies of LAP using model phagosomes containing latex beads or zymosan particles.10 Our study provides a strong argument for LAP as the main pathway for LC3 recruitment to L. monocytogenes at 1 h p.i., although we do not discount the possibility that autophagy may also contribute to LC3 colocalization with bacteria at this early time of infection.
Our data suggests that L. monocytogenes can be targeted by two potentially degradative pathways involving components of the autophagy pathway during infection (see model in Fig. 7), thus serving as a reminder that LC3 must be used carefully as a marker for studies of autophagy. We propose that in the phagosome, bacteria are targeted by the LAP pathway via localized DAG enrichment and downstream ROS production via the CYBB/NOX2 NADPH oxidase (Fig. 7, left-hand pathway). In contrast, cytosolic ActAΔ L. monocytogenes strain 10403S treated with Cm is targeted by autophagy via protein ubiquitination and the recruitment of autophagy adaptors such as SQSTM1 (Fig. 7B, right-hand pathway). This model is consistent with other studies that indicate mammalian cells have evolved multiple pathways that utilize autophagy components to contribute to host immunity (reviewed by Cemma et al.).9 While our study focused on L. monocytogenes infection in macrophages, we expect that both LAP and autophagy will be capable of targeting other bacteria during infection in other cell types. Nonphagocytic cells can also produce ROS but via expression of other members of the NOX/DUOX family (reviewed by Nauseef36) and thus, LAP may be relevant in bacterial infections of all cell types.

Figure 7. Two pathways mediate LC3 recruitment to L. monocytogenes strain 10403S during infection. Model shows the two pathways that can mediate LC3 recruitment to bacteria. First, LC3-associated phagocytosis (LAP) can mediate LC3 localization to phagosomes containing L. monocytogenes at 1 h p.i. (left-hand side pathway). In this study we showed that diacylglycerol (DAG) accumulates on the phagosome as the result of bacterial (PLC) and host (PLD and PPAP2A) enzymatic activity. DAG accumulation promotes activation of the CYBB/NOX2 NADPH oxidase and the production of reactive oxygen species (ROS). ROS production mediates LAP of L. monocytogenes, a process that is characterized by LC3 recruitment to single-membrane phagosomes containing bacteria. While LAP has been shown to promote phagosome maturation and killing of certain microbes, our data indicates that LAP promotes the formation of spacious Listeria-containing phagosomes (SLAPs), a compartment that has been associated with persistent infection. Second, LC3 can be recruited to L. monocytogenes in the cytosol under specific conditions. When treated with the bacteristatic agent, chloramphenicol (Cm), the ActAΔ mutant of L. monocytogenes can be targeted by autophagy into double-membrane autophagosomes via protein ubiquitination and ubiquitin-binding adaptor proteins including SQSTM1 (right-hand side pathway). In contrast, wild-type L. monocytogenes evades autophagy in the cytosol via expression of ActA and other factors, allowing them to replicate rapidly and undergo actin-based motility for cell-to-cell spread.
Previous studies showed that LAP targets B. pseudomallei mutants lacking its type III secretion system or the type III secreted effector BopA.12 In this case, LC3 recruitment to LAMP1+ phagosomes correlated with enhanced killing of bacteria. In the case of L. monocytogenes, LC3 recruitment to wild-type bacteria strain 10403S correlates with slow growth of bacteria with SLAPs. Our studies imply that bacteria may exploit the LAP pathway and SLAPs to cause persistent infection,20 a model supported by the observation of structures resembling SLAPs in the liver of SCID mice after 28 d of infection.19 The finding that the CYBB/NOX2 NADPH oxidase contributes to early LC3 recruitment to bacteria and the subsequent formation of SLAPs is intriguing, pointing to a role for this enzyme (and possible other NADPH oxidases) in the establishment and/or maintenance of persistent infections. It is noteworthy that Swanson and colleagues suggest a role for the CYBB/NOX2 NADPH oxidase in restricting phagosome escape by L. monocytogenes, possibly by limiting LLO activity.37 Therefore, ROS production by the CYBB/NOX2 NADPH oxidase may serve a dual function in limiting the activity of bacterial virulence factors and also promoting phagosome maturation. Whether other pathogens exploit the LAP pathway to cause persistent infection is an important question for future studies.
Materials and Methods
Reagents and antibodies
The following drugs were used as indicated: 0.3% v/v 1-butanol (Sigma, B7906), 0.3% v/v tert-butanol (Sigma, 360538), 250 µM propranolol hydrochloride (Biomol International, AR107-0100), 10 µM DGK inhibitor I (Sigma, R59022) and 10 µM DPI (Sigma, D2926). Rabbit polyclonal antibodies against L. monocytogenes were a gift from Dr. Pascale Cossart (Institut Pasteur), mouse monoclonal antibodies against GFP were from Invitrogen (A-11120); rat monoclonal antibodies against LAMP1 (clone ID4B) was from Developmental Studies Hybridoma Bank (University of Iowa); and antibodies against mono- and poly-ubiquitinated protein were from Biomol International (FK2; BML-PW8810-0500). All fluorescent secondary antibodies—goat anti-rabbit 405, goat anti-rat Cy3, goat anti-mouse 568, goat anti-mouse 488—were AlexaFluor conjugates from Molecular Probes (Invitrogen, A31556, A10522, A11004, A11029).
Bacterial strains and tissue culture
Bacterial strains used in this study were as follows: wild-type L. monocytogenes 10403S38 and isogenic mutants lacking hly (LLOΔ; DP-L2161),39 and plcA, plcB (PI-PLCΔ PC-PLCΔ; DP-L1936).31 The LLO complementation mutant, LLOΔ + LLO (DP-L4818) was also used.40
RAW 264.7 macrophages were from American Type Culture Collection (TIB-71). RAW 264.7 cells were maintained in DMEM (HyClone, SH30271.01) supplemented with 10% FBS (Wisent, 090-510) at 37°C in 5% CO2 without antibiotics. Macrophages were seeded at 1.25 × 105 cells/well 48 h prior to infection.
Bone marrow-derived macrophage generation and culture conditions
All experimental protocols involving mice were approved by the Animal Care Committee of The Hospital for Sick Children. Mice were euthanized by cervical dislocation. The femur and tibia were removed, cleansed of muscle fibers and cut distally. The bone marrow was then removed via a 10 sec pulse of centrifugation at 2000 rpm. The resulting cells were centrifuged at 1500 rpm for 5 min, washed with growth media and plated on 10 cm tissue culture dishes. Media was replaced with fresh RPMI growth media (see below) every 3 d. 108 bone marrow-derived macrophages (BMDM) were typically recovered after 7 d. Murine macrophages were maintained in RPMI-1640 medium (Wisent, SH3002701) supplemented with 10% FBS (Wisent, 090-510), 5% sodium pyruvate (Invitrogen, 11360-070), 5% antibiotics (Invitrogen, 15140122), 5% nonessential amino acids (Invitrogen, 11140050) and 0.5 µM β-mercaptoethanol (Invitrogen, 21985023). BMDMs were differentiated in 30% L929 conditioned media. L929 conditioned medium was generated by growing L929 cells (CCL-1; ATCC) in 150-cm2 flasks at an initial density of 1 × 108 cells per flask in growth media as described above. After 3 d, confluency was reached and the growth media was substituted with DMEM alone. After 7 to 10 d, culture supernatant was collected and centrifuged at 1,500 rpm for 5 min, aliquotted and stored at −20°C.
Plasmids, transfections and siRNA silencing
Transfections were performed 24 h prior to infection. Transfection reagents FuGene 6 and FuGene 6 HD (Roche Applied Sciences, 11 815 091 001; 04 709 691 001) were used according to the manufacturer’s instructions. BMDM were transfected using the Amaxa Primary Murine Macrophage Transfection kit according to the manufacturer’s instructions (Amaxa, VPA-1009). Constructs used were GFP-LC3 (provided by Tamotsu Yoshimori, Osaka University, Japan),41 RFP-LC3 (provided by Walter Beron, Universidad Nacional de Cuyo, Argentina), PRKCD-C1-GFP (provided by Sergio Grinstein, Hospital for Sick Children, Canada)42 and HA-PLD1 K898R and HA-PLD2 K758R (provided by Michael Frohman, SUNY, USA).43 siGenome SMARTpool siRNA reagents (Dharmacon) were used for targeting murine Atg12 (GUGGGCAGUAGAGCGAACA) and Sqstm1 (Dharmacon, D-010230-02) as well as scrambled control siRNA (Dharmacon, M-011020-01)
Bacterial infection conditions
Bacteria were grown in Brain-Heart Infusion (BHI) broth for 14 to 16 h at 30°C in a standing incubator. A 1:10 dilution of the culture was grown for 2 h at 37°C in a shaking incubator prior to infection. Both RAW 264.7 macrophages and BMDM were infected at a multiplicity of infection (MOI) of 10 as described.16
Immunofluorescence and microscopy
Immunostaining was conducted as previously described.44 In brief, after infections, cells were fixed using 2.5% paraformaldehyde for 10 min at 37°C. Extracellular L. monocytogenes were detected by immunostaining prior to permeabilization. Cells were then permeabilized and blocked using 0.2% saponin with 10% normal goat serum for 14 to 16 h at 4°C. All colocalization quantifications were done using a Leica DMIRE2 epifluorescence microscope equipped with a 100× oil objective, 1.4 numerical aperture. 100 intracellular bacteria were examined in each experiment. Images are single confocal z-slices taken using a Zeiss Axiovert confocal microscope and LSM 510 software. Volocity software (Improvision) was used to analyze images. Images were imported into Adobe Photoshop and assembled in Adobe Illustrator.
Light and electron microscopy
RAW 264.7 cells were seeded on gridded coverslips and transfected with GFP-LC3. Twenty-four h later, cells were infected with NHS-647 labeled wild-type L. monocytogenes for 60 min as described and cells were washed with 0.2M sodium cacodylate buffer and fixed with 2.5% paraformaldehyde in sodium cacodylate buffer (all reagents were from Sigma-Aldrich unless otherwise specified). Cells were imaged using the Zeiss Axiovert confocal microscope as described above. Cells were subsequently fixed in 2.5% glutaraldehyde in sodium cacodylate buffer for 14 to 16 h, postfixed in 1% OsO4, stained with 1% aqueous uranyl acetate, dehydrated in graded series of ethanols and embedded in epoxy resin. Samples were then sectioned, stained with 2% uranyl acetate, then 0.2% lead citrate, and examined on a Tecnai 20 transmission electron microscope (FEI) operating at 200 kV.
Statistical analysis
Statistical analyses were conducted using GraphPad Prism v4.0a. In all figures, data are expressed as the mean ± standard error of the mean (s.e.m) from three separate experiments. p values were calculated using two-tailed two-sample equal variance Student’s t-test. A p-value of less than 0.05 was considered statistically significant and is denoted by “*”. p < 0.01 is denoted by “**” and p < 0.005 is denoted by “***”.
Supplementary Material
Acknowledgments
We wish to thank members of the Brumell Lab and Dr. Mathew Estey for critical reading of this manuscript. We are grateful to Drs. D. Portnoy, S. Grinstein and J. Swanson for generously providing bacterial strains, constructs and helpful advice. We also thank Mike Woodside and Paul Paroutis for help with fluorescence microscopy as well as Dr. David Bazett-Jones, Doug Holmyard and Yew-Ming Heng for assistance with electron microscopy. J.H.B. is the recipient of an Investigators in Pathogenesis of Infectious Disease Award from the Burroughs Wellcome Fund. Infrastructure for the Brumell Laboratory was provided by a New Opportunities Fund from the Canadian Foundation for Innovation and the Ontario Innovation Trust. G.Y.L. was supported by a M.D/Ph.D Studentship and Canadian Graduate Scholarship Doctoral Research Award from the Canadian Institutes of Health Research. M.C. was supported by NSERC PGS-D scholarship and CIHR Training Fellowship (TGF-53877). This work was supported by operating grants from the Canadian Institutes of Health Research (MOP#97756), The Arthritis Society of Canada (#RG11/013) and a United States Public Health Service grant (AI053669) from the National Institutes of Health (D.E.H.).
Glossary
Abbreviation
- list
LAP, LC3-associated phagocytosis: ROS, reactive oxygen species
- SLAPs
spacious Listeria-containing phagosomes
- LLO
Listeriolysin O
- PLC
phospholipase C
- CBD
cell wall binding domain
- SQSTM1
sequestome-1
- DPI
diphenyliodonium
- Cm
chloramphenicol
- DAG
diacylglycerol
- PLD
phospholipase D
- PPAP2A
phosphatidic acid phosphatase
- DGK
DAG kinase
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplemental Materials
Supplemental materials may be found here: www.landesbioscience.com/journals/autophagy/article/24406
Footnotes
Previously published online: www.landesbioscience.com/journals/autophagy/article/24406
References
- 1.Levine B, Mizushima N, Virgin HW. Autophagy in immunity and inflammation. Nature. 2011;469:323–35. doi: 10.1038/nature09782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Birmingham CL, Smith AC, Bakowski MA, Yoshimori T, Brumell JH. Autophagy controls Salmonella infection in response to damage to the Salmonella-containing vacuole. J Biol Chem. 2006;281:11374–83. doi: 10.1074/jbc.M509157200. [DOI] [PubMed] [Google Scholar]
- 3.Gutierrez MG, Master SS, Singh SB, Taylor GA, Colombo MI, Deretic V. Autophagy is a defense mechanism inhibiting BCG and Mycobacterium tuberculosis survival in infected macrophages. Cell. 2004;119:753–66. doi: 10.1016/j.cell.2004.11.038. [DOI] [PubMed] [Google Scholar]
- 4.Sanjuan MA, Dillon CP, Tait SW, Moshiach S, Dorsey F, Connell S, et al. Toll-like receptor signalling in macrophages links the autophagy pathway to phagocytosis. Nature. 2007;450:1253–7. doi: 10.1038/nature06421. [DOI] [PubMed] [Google Scholar]
- 5.Nakagawa I, Amano A, Mizushima N, Yamamoto A, Yamaguchi H, Kamimoto T, et al. Autophagy defends cells against invading group A Streptococcus. Science. 2004;306:1037–40. doi: 10.1126/science.1103966. [DOI] [PubMed] [Google Scholar]
- 6.Rich KA, Burkett C, Webster P. Cytoplasmic bacteria can be targets for autophagy. Cell Microbiol. 2003;5:455–68. doi: 10.1046/j.1462-5822.2003.00292.x. [DOI] [PubMed] [Google Scholar]
- 7.Hwang S, Maloney NS, Bruinsma MW, Goel G, Duan E, Zhang L, et al. Nondegradative role of Atg5-Atg12/ Atg16L1 autophagy protein complex in antiviral activity of interferon gamma. Cell Host Microbe. 2012;11:397–409. doi: 10.1016/j.chom.2012.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shintani T, Klionsky DJ. Cargo proteins facilitate the formation of transport vesicles in the cytoplasm to vacuole targeting pathway. J Biol Chem. 2004;279:29889–94. doi: 10.1074/jbc.M404399200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cemma M, Brumell JH. Interactions of pathogenic bacteria with autophagy systems. Curr Biol. 2012;22:R540–5. doi: 10.1016/j.cub.2012.06.001. [DOI] [PubMed] [Google Scholar]
- 10.Huang J, Canadien V, Lam GY, Steinberg BE, Dinauer MC, Magalhaes MA, et al. Activation of antibacterial autophagy by NADPH oxidases. Proc Natl Acad Sci U S A. 2009;106:6226–31. doi: 10.1073/pnas.0811045106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Martinez J, Almendinger J, Oberst A, Ness R, Dillon CP, Fitzgerald P, et al. Microtubule-associated protein 1 light chain 3 alpha (LC3)-associated phagocytosis is required for the efficient clearance of dead cells. Proc Natl Acad Sci U S A. 2011;108:17396–401. doi: 10.1073/pnas.1113421108. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 12.Gong L, Cullinane M, Treerat P, Ramm G, Prescott M, Adler B, et al. The Burkholderia pseudomallei type III secretion system and BopA are required for evasion of LC3-associated phagocytosis. PLoS One. 2011;6:e17852. doi: 10.1371/journal.pone.0017852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rocourt J, Bille J. Foodborne listeriosis. World Health Stat Q. 1997;50:67–73. [PubMed] [Google Scholar]
- 14.Portnoy DA, Auerbuch V, Glomski IJ. The cell biology of Listeria monocytogenes infection: the intersection of bacterial pathogenesis and cell-mediated immunity. J Cell Biol. 2002;158:409–14. doi: 10.1083/jcb.200205009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Goldfine H, Wadsworth SJ. Macrophage intracellular signaling induced by Listeria monocytogenes. Microbes Infect. 2002;4:1335–43. doi: 10.1016/S1286-4579(02)00011-4. [DOI] [PubMed] [Google Scholar]
- 16.Birmingham CL, Canadien V, Gouin E, Troy EB, Yoshimori T, Cossart P, et al. Listeria monocytogenes evades killing by autophagy during colonization of host cells. Autophagy. 2007;3:442–51. doi: 10.4161/auto.4450. [DOI] [PubMed] [Google Scholar]
- 17.Meyer-Morse N, Robbins JR, Rae CS, Mochegova SN, Swanson MS, Zhao Z, et al. Listeriolysin O is necessary and sufficient to induce autophagy during Listeria monocytogenes infection. PLoS One. 2010;5:e8610. doi: 10.1371/journal.pone.0008610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Py BF, Lipinski MM, Yuan J. Autophagy limits Listeria monocytogenes intracellular growth in the early phase of primary infection. Autophagy. 2007;3:117–25. doi: 10.4161/auto.3618. [DOI] [PubMed] [Google Scholar]
- 19.Bhardwaj V, Kanagawa O, Swanson PE, Unanue ER. Chronic Listeria infection in SCID mice: requirements for the carrier state and the dual role of T cells in transferring protection or suppression. J Immunol. 1998;160:376–84. [PubMed] [Google Scholar]
- 20.Birmingham CL, Canadien V, Kaniuk NA, Steinberg BE, Higgins DE, Brumell JH. Listeriolysin O allows Listeria monocytogenes replication in macrophage vacuoles. Nature. 2008;451:350–4. doi: 10.1038/nature06479. [DOI] [PubMed] [Google Scholar]
- 21.Perrin AJ, Jiang X, Birmingham CL, So NS, Brumell JH. Recognition of bacteria in the cytosol of Mammalian cells by the ubiquitin system. Curr Biol. 2004;14:806–11. doi: 10.1016/j.cub.2004.04.033. [DOI] [PubMed] [Google Scholar]
- 22.Yoshikawa Y, Ogawa M, Hain T, Yoshida M, Fukumatsu M, Kim M, et al. Listeria monocytogenes ActA-mediated escape from autophagic recognition. Nat Cell Biol. 2009;11:1233–40. doi: 10.1038/ncb1967. [DOI] [PubMed] [Google Scholar]
- 23.de Chastellier C, Berche P. Fate of Listeria monocytogenes in murine macrophages: evidence for simultaneous killing and survival of intracellular bacteria. Infect Immun. 1994;62:543–53. doi: 10.1128/iai.62.2.543-553.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Henry R, Shaughnessy L, Loessner MJ, Alberti-Segui C, Higgins DE, Swanson JA. Cytolysin-dependent delay of vacuole maturation in macrophages infected with Listeria monocytogenes. Cell Microbiol. 2006;8:107–19. doi: 10.1111/j.1462-5822.2005.00604.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Beauregard KE, Lee KD, Collier RJ, Swanson JA. pH-dependent perforation of macrophage phagosomes by listeriolysin O from Listeria monocytogenes. J Exp Med. 1997;186:1159–63. doi: 10.1084/jem.186.7.1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kageyama S, Omori H, Saitoh T, Sone T, Guan JL, Akira S, et al. The LC3 recruitment mechanism is separate from Atg9L1-dependent membrane formation in the autophagic response against Salmonella. Mol Biol Cell. 2011;22:2290–300. doi: 10.1091/mbc.E10-11-0893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zheng YT, Shahnazari S, Brech A, Lamark T, Johansen T, Brumell JH. The adaptor protein p62/SQSTM1 targets invading bacteria to the autophagy pathway. J Immunol. 2009;183:5909–16. doi: 10.4049/jimmunol.0900441. [DOI] [PubMed] [Google Scholar]
- 28.Dupont N, Lacas-Gervais S, Bertout J, Paz I, Freche B, Van Nhieu GT, et al. Shigella phagocytic vacuolar membrane remnants participate in the cellular response to pathogen invasion and are regulated by autophagy. Cell Host Microbe. 2009;6:137–49. doi: 10.1016/j.chom.2009.07.005. [DOI] [PubMed] [Google Scholar]
- 29.Thurston TL, Ryzhakov G, Bloor S, von Muhlinen N, Randow F. The TBK1 adaptor and autophagy receptor NDP52 restricts the proliferation of ubiquitin-coated bacteria. Nat Immunol. 2009;10:1215–21. doi: 10.1038/ni.1800. [DOI] [PubMed] [Google Scholar]
- 30.Sibelius U, Chakraborty T, Krögel B, Wolf J, Rose F, Schmidt R, et al. The listerial exotoxins listeriolysin and phosphatidylinositol-specific phospholipase C synergize to elicit endothelial cell phosphoinositide metabolism. J Immunol. 1996;157:4055–60. [PubMed] [Google Scholar]
- 31.Smith GA, Marquis H, Jones S, Johnston NC, Portnoy DA, Goldfine H. The two distinct phospholipases C of Listeria monocytogenes have overlapping roles in escape from a vacuole and cell-to-cell spread. Infect Immun. 1995;63:4231–7. doi: 10.1128/iai.63.11.4231-4237.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Goldfine H, Wadsworth SJ, Johnston NC. Activation of host phospholipases C and D in macrophages after infection with Listeria monocytogenes. Infect Immun. 2000;68:5735–41. doi: 10.1128/IAI.68.10.5735-5741.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Usatyuk PV, Gorshkova IA, He D, Zhao Y, Kalari SK, Garcia JG, et al. Phospholipase D-mediated activation of IQGAP1 through Rac1 regulates hyperoxia-induced p47phox translocation and reactive oxygen species generation in lung endothelial cells. J Biol Chem. 2009;284:15339–52. doi: 10.1074/jbc.M109.005439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lam GY, Huang J, Brumell JH. The many roles of NOX2 NADPH oxidase-derived ROS in immunity. Semin Immunopathol. 2010;32:415–30. doi: 10.1007/s00281-010-0221-0. [DOI] [PubMed] [Google Scholar]
- 35.Shahnazari S, Yen W-L, Birmingham CL, Shiu J, Namolovan A, Zheng YT, et al. A diacylglycerol-dependent signaling pathway contributes to regulation of antibacterial autophagy. Cell Host Microbe. 2010;8:137–46. doi: 10.1016/j.chom.2010.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nauseef WM. Biological roles for the NOX family NADPH oxidases. J Biol Chem. 2008;283:16961–5. doi: 10.1074/jbc.R700045200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Myers JT, Tsang AW, Swanson JA. Localized reactive oxygen and nitrogen intermediates inhibit escape of Listeria monocytogenes from vacuoles in activated macrophages. J Immunol. 2003;171:5447–53. doi: 10.4049/jimmunol.171.10.5447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bishop DK, Hinrichs DJ. Adoptive transfer of immunity to Listeria monocytogenes. The influence of in vitro stimulation on lymphocyte subset requirements. J Immunol. 1987;139:2005–9. [PubMed] [Google Scholar]
- 39.Jones S, Portnoy DA. Characterization of Listeria monocytogenes pathogenesis in a strain expressing perfringolysin O in place of listeriolysin O. Infect Immun. 1994;62:5608–13. doi: 10.1128/iai.62.12.5608-5613.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lauer P, Chow MY, Loessner MJ, Portnoy DA, Calendar R. Construction, characterization, and use of two Listeria monocytogenes site-specific phage integration vectors. J Bacteriol. 2002;184:4177–86. doi: 10.1128/JB.184.15.4177-4186.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, et al. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000;19:5720–8. doi: 10.1093/emboj/19.21.5720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tse SM, Mason D, Botelho RJ, Chiu B, Reyland M, Hanada K, et al. Accumulation of diacylglycerol in the Chlamydia inclusion vacuole: possible role in the inhibition of host cell apoptosis. J Biol Chem. 2005;280:25210–5. doi: 10.1074/jbc.M501980200. [DOI] [PubMed] [Google Scholar]
- 43.Denmat-Ouisse LA, Phebidias C, Honkavaara P, Robin P, Geny B, Min DS, et al. Regulation of constitutive protein transit by phospholipase D in HT29-cl19A cells. J Biol Chem. 2001;276:48840–6. doi: 10.1074/jbc.M104276200. [DOI] [PubMed] [Google Scholar]
- 44.Brumell JH, Rosenberger CM, Gotto GT, Marcus SL, Finlay BB. SifA permits survival and replication of Salmonella typhimurium in murine macrophages. Cell Microbiol. 2001;3:75–84. doi: 10.1046/j.1462-5822.2001.00087.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.