

Abstract
Beta-thalassemia and sickle cell anemia (SCD) represent the most common hemoglobinopathies caused, respectively, by deficient production or alteration of the beta chain of hemoglobin (Hb). Patients affected by the most severe form of thalassemia suffer from profound anemia that requires chronic blood transfusions and chelation therapies to prevent iron overload. However, patients affected by beta-thalassemia intermedia, a milder form of the disease that does not require chronic blood transfusions, eventually also show elevated body iron content due to increased gastrointestinal iron absorption. Even SCD patients might require blood transfusions and iron chelation to prevent deleterious and painful vaso-occlusive crises and complications due to iron overload. Although definitive cures are presently available, such as bone marrow transplantation (BMT), or are in development, such as correction of the disease through hematopoietic stem cell beta-globin gene transfer, they are potentially hazardous procedures or too experimental to provide consistently safe and predictive clinical outcomes. Therefore, studies that aim to better understand the pathophysiology of the hemoglobinopathies might provide further insight and new drugs to dramatically improve the understanding and current treatment of these diseases. This review will describe how recent discoveries on iron metabolism and erythropoiesis could lead to new therapeutic strategies and better clinical care of these diseases, thereby yielding a much better quality of life for the patients.
Keywords: Hemoglobinopathies, iron metabolism, erythropoiesis, mouse models, hemochromatosis, hepcidin, ferroportin
GLOBIN GENES AND HEMOGLOBINOPATHIES
The human alpha and beta-globin genes and loci have been studied extensively for their important roles in human biology and pathophysiology [1]. The globin genes were among the small group of human genes first cloned [2–4] and the structure of the corresponding proteins was described at the early stages of human functional biology studies [5]. The alpha and beta-globin genes and the loci in which they are embedded were, and still are, utilized for understanding tissue and developmental stage-specific expression of gene families [6, 7]. Mutations that affect the corresponding genes lead, altogether, to the most frequent inherited group of human diseases: hemoglobinopathies [6]. Hemoglobinopathies are a heterogeneous group of diseases in which genetic defects result in an abnormal structure of one of the globin chains of the hemoglobin molecule or in the production of an abnormally low quantity of a given hemoglobin chain or chains.
In order to better understand the relationship between genes, mutations, phenotypes, and the mouse models utilized to study the hemoglobinopathies, a brief description of the human and mouse globin genomic loci and the corresponding ontogenic gene profile expression is required. The human alpha-globin locus is located near the telomere of the short arm of chromosome 16 and spans 70 kb, encompassing both the three expressed and translated globin genes and the cis-acting elements that direct their stage–specific expression during ontogeny [6, 8]. The genes are in the same transcriptional orientation and are arranged according to the order in which they are expressed during development, with the embryonic zeta gene located upstream from the fetal/adult alpha1- and alpha2-globin genes. In addition to regulation by promoters and enhancer elements, expression of the alpha-like globin genes is also dependent on a regulatory sequence called HS-40, which is located 40 kb upstream from the zeta-globin gene (Fig. (1A)). The human beta-globin locus is characterized by five expressed and translated genes, spans 80 kb, and is located on chromosome 11. The organization of the beta-globin locus reflects the common evolutionary origin of the a_ lpha- and beta-like globin genes. The embryonic epsilon-globin gene is located at the 5′ end, followed by the fetal-gammaG- and gammaA-globin genes and the adult delta- and beta-globin genes that define the 3′ end of the locus. Expression of the beta-like globin genes is also dependent on a regulatory sequence called locus control region, or LCR, which is located 8 to 22 kbs upstream of the epsilon-globin gene. The mouse alpha- and beta-globin loci are located, respectively, on chromosome 11 and 7. Their structure is similar to the human globin loci (Fig. (1A)). The alpha-globin locus is characterized by three expressed globin genes (the embryonic zeta-globin gene and the two fetal/adult genes, alpha1- and alpha2-globin) and the cis-acting elements (HS-26) that direct their stage–specific expression during ontogeny [6]. Four expressed globin genes and the LCR characterize the mouse beta-globin locus: two embryonic genes, beta-h1 and epsilon-y2, and two adult genes b1, or beta-globin major, and b2, or beta-globin minor [6, 8, 9] (Fig. (1A)). In humans, the switch from epsilon- to gamma-globin production begins very early in gestation, as fetal hemoglobin (HbF) is readily detected in five-week old human embryos and is complete by the tenth week of gestation (Fig. (1B) and (1C)). Beta-chain synthesis increases to approximately 10 percent of total hemoglobin (Hb) by 30 to 35 weeks of gestation (Fig. (1B)). At birth, HbF comprises 60–80 percent of total Hb. It takes about two years for the level of HbF to decrease to 0.5–1 percent, the level characteristic of adult red blood cells [6]. On the contrary, in mice, which do not harbor fetal globin genes, the embryonic beta-h1 and epsilon-y2 genes are expressed during the embryonic phase of development, and the b1 and b2 genes are switched on in utero around 11.5 days of gestation as opposed to the embryonic genes that are completely silenced by 14–15 days of gestation [8, 9] (Fig. (1B)).
Fig. 1. Schematic chromosomal organization of the globin-loci.

Genes of the human and mouse alpha-and beta-globin_ loci are represented, respectively, on the left and right of the diagram (A). Time course of developmental globin gene regulation (B). Hemoglobin tetramers produced at different stages of life (C).
The first group of hemoglobinopathies can be due to substitution of one amino acid for another (as with sickle cell Hb or HbS [10]), deletion of a portion of the amino acid sequence (Hb Gun Hill [11]), abnormal hybridization between two chains (Hb Lepore [12, 13]), or abnormal elongation of the globin chain (Hb Constant Spring [14]). These abnormal Hbs can have a variety of physiologically significant effects, although our review will focus on the most common variant, HbS. An adenine (A) to thymidine (T) substitution in codon 6 (GAG-GTG) results in the insertion of valine in the place of glutamic acid in the gene for beta-globin, causing HbS, which has the unique property of polymerizing when deoxygenated [6]. When the polymer becomes abundant, the red cells “sickle”, forming stiff rods that stretch and distort the red cells. These distorted cells can obstruct blood flow through the small vessels, affecting many organs and tissues. The restricted oxygen delivery to the tissues damages cells, injures organs, and produces pain. Red cell sickling results in acute, recurrent, and chronic complications, causing SCD. Vaso-occlusion within the microcirculation, and at times, in larger vessels, can occur almost anywhere blood flows and is responsible for most of the severe complications of the disease. Notably, sickle cells interact with other blood cells and with the vascular endothelium, causing vascular injury, tempering vascular tone, and occluding small and sometimes large blood vessels [6, 15, 16].
The second group of hemoglobinopathies is due to a large number of heterogeneous mutations causing abnormal globin gene expression and resulting in total absence or quantitative reduction of globin chain synthesis. Mutations in the alpha- or beta-globin gene lead to alpha- and beta-thalassemia, respectively [6]. Alpha-thalassemia is usually due to deletions within the alpha-globin gene cluster, leading to loss of function of one or both alpha-globin genes in each locus [17]. However, non-deletion mutations have been described, although they are much less frequent [6]. Depending on the number of genes that are unable to synthesize the alpha-globin protein, different clinical manifestations can be observed. If one or two alpha-globin genes are mutated (in cis or trans), normally no or minimal hematological effects are associated with these mutations, and individuals are normally silent thalassemia carriers or show alpha-thalassemia trait [6]. If three out of four genes are mutated, the condition is called hemoglobin H (HbH) disease, resulting in a hemolytic anemia that can worsen with febrile illness or exposure to certain drugs, chemicals, or infectious agents. Hemoglobin H disease is characterized by moderate to severe anemia, hepatosplenomegaly, and jaundice. Transfusion may occasionally be required and, if repeated frequently, can lead to iron overload. If all four alpha-globin genes are deleted, the resulting condition is called alpha-thalassemia major, which is so severe that death often occurs in utero. Children rescued through intrauterine transfusions will remain dependent on red blood cell transfusions for survival [18]. In contrast to alpha-thalassemia, the majority of the molecular defects associated with beta-thalassemia are usually point mutations involving only one or a limited number of nucleotides, but result in a major defect of beta-globin gene expression either at the transcriptional or post-transcriptional levels [6]. Notably, more than 200 different mutations have been associated with the beta-thalassemia condition [6, 19]. The hallmark of beta-thalassemia is ineffective erythropoiesis (IE) which leads to erythroid marrow expansion to as much as 30 times the normal level. Extramedullary erythropoietic tissues, primarily in the thorax and the paraspinal regions, may be stimulated to expand and can lead to characteristic deformities of the skull and face, osteopenia, and demineralization of the bones, which are then prone to fractures. Despite excessive erythropoietic activity, the affected person suffers from anemia, which is exacerbated by progressive splenomegaly and an increase in plasma volume as a result of shunting through the expanded marrow.
Interestingly, SCD and thalassemia can coexist with other hemoglobinopathies. A sickle cell trait can coexist with alpha- or beta-thalassemias and various other hemoglobinopathies, such as hemoglobin C or HbC, in which a lysine has replaced glutamic acid at position 6, and hemoglobin E or HbE, in which glutamic acid has replaced lysine at position 26 [6]. Beta-thalassemia is often co-inherited with alpha-thalassemia and/or sickle cell trait. Coinheritance of alpha- and beta-thalassemia may reduce the severity of the globin-chain imbalance [6, 20]. Coinheritance of HbS trait and severe beta-thalassemia may result in a clinical disorder indistinguishable from sickle cell anemia. By contrast, coinheritance of HbS trait and mild beta-thalassemia will result in a milder sickling disorder. Coinheritance of HbE and beta-thalassemia results in a wide spectrum of clinical disorders ranging from mild anemia to a severe disorder indistinguishable from thalassemia major [6].
SCD and the thalassemias are quite common, not only in Mediterranean, but also in African, African-American, and Asian populations. However, specific sets of mutations are associated with different ethnic groups [6]. It has been estimated that approximately 7% of the world population are carriers of such disorders, and that 300,000–400,000 babies with severe forms of these diseases are born each year [21]. Better understanding of the molecular biologic aspects of the hemoglobinopathies has led to improvements in population screening and prenatal diagnosis, which, in turn, have led to dramatic reductions in the number of births of children with beta-thalassemia major and SCD in the Mediterranean littoral [21]. However, as a consequence of decreases in neonatal and childhood mortality in other geographical areas, the severe hemoglobinopathies have become worldwide clinical problems. Therefore, severe forms of hemoglobinopathies remain increasingly important clinical problems in much of the world [21, 22]. Despite recent discoveries concerning the genetic abnormalities that lead to the beta-thalassemia syndrome, a number of unsolved pathophysiological issues remain or are being investigated, such as IE, extra-medullary hematopoiesis (EMH) [23, 24], abnormal iron absorption [25, 26], reactive oxygen species (ROS), and radical formation [27], bone fragility [28] and endocrinological disorders [29]. Better understanding of the pathophysiologic features of the disease could result in measures that might raise hemoglobin values, reduce splenomegaly, prevent iron overload, heart and liver failure, as well as bone abnormalities and endocrine disorders, thereby yielding a much better quality of life.
CLINICAL IMPACT OF IRON OVERLOAD
The same properties that make iron essential for basic biological processes such as the transport of oxygen and transfer of electrons also make it toxic, mainly because iron can promote oxidative damage to vital biological structures. Iron homeostasis needs to be tightly regulated through iron uptake, utilization, and storage. Because iron is essential for the homeostasis of all cells in the human body and the formation of heme and Hb, no physiological mechanism to aid in its excretion exists. In non-transfused patients, iron absorption builds, resulting in increases in body iron burden. Transfusions accelerate the pace of iron accumulation. The resulting iron overload leads to the most damaging effects of SCD and thalassemias, making iron chelation a major focus in the management of these diseases. Iron is progressively deposited in parenchymal tissues, where it may cause significant toxicity as compared to that within reticuloendothelial cells [30]. As iron loading continues, the capacity of serum transferrin, the main transport protein of iron, to bind and detoxify iron may be exceeded. Thereafter, the non-transferrin-bound fraction of iron within plasma may promote generation of free hydroxyl radicals, propagators of oxygen-related damage [27, 30]. As iron loading progresses, the accumulation may lead to progressive dysfunction of the liver, endocrine glands, and heart [31, 32].
Iron-induced liver disease is a common cause of death in transfused patients [33]. Within two years following the start of transfusions, collagen formation and portal fibrosis are observed, and in the absence of chelating therapy, cirrhosis may develop [34]. Iron-induced liver disease may be complicated by transfusion-related hepatitis [35]. Chronic iron deposition also damages the thyroid, parathyroid, adrenal glands, and exocrine pancreas. Iron loading within the anterior pituitary may cause disturbances in sexual maturation. Both hyperinsulinism, from the inability of the liver to extract insulin, and diabetes mellitus, from pancreatic beta-cell exhaustion and/or pancreatic destruction, may occur. Iron-induced myocardial dysfunction may be the most important factor determining the survival of patients with beta-thalassemia. Extensive iron deposits are associated with cardiac hypertrophy and dilatation, myocardial fiber degeneration and, though rarely, fibrosis. In transfused, unchelated patients, symptomatic cardiac disease is observed after about ten years following the start of transfusions and may be aggravated by pulmonary hypertension [36, 37]. Therefore, even if the goal of transfusion therapy is the correction of anemia, important efforts need to be undertaken in order to limit the damage derived from transfused iron overload [38].
MOUSE MODELS OF HEMOGLOBINOPATHIES
Murine Models of Beta-Thalassemia Intermedia
In mice, the b1 and b2 genes are responsible, respectively, for 80% and 20% of total adult Hb production. Normal adult mice produce Hb levels in the range of 13 to 15 g/dL of blood. No naturally occurring mutations or deletions that completely inhibit expression of the beta-globin genes have ever been observed, as mice, homozygous for mutations that prevent expression of the beta-globin genes, die perinatally due to the lack of expression of any Hb. Limited by this genetic constraint, only three adult mouse models of beta-thalassemia intermedia have been described and generated so far. In the first (th1), a spontaneous DNA deletion was found that included the b1 gene and its adjacent upstream sequence, including the promoter [39]. Mice that are homozygous (th1/th1) for this deletion show Hb levels in the range of 9 to 11 g/dL, despite the fact that the remaining b2 gene normally accounts for only 20% of beta-globin production. In these mice, relatively high Hb levels are obtained through a translational compensatory mechanism that increases b2 globin synthesis [40]. The second mouse model (th2) was generated by targeted deletion of the b1 gene, introducing a non-globin promoter into the locus of the disrupted b1 locus. Mice homozygous for this deletion (th2/th2) are severely anemic and do not survive more than a few hours after birth, while heterozygous mice only show a very mild phenotype [41]. It has been speculated that the presence of the newly introduced promoter in the disrupted b1 locus can compete with the b2 promoter for transcription factors brought to the globin genes by the LCR, thereby reducing transcription from the b2 gene. The third model, th3, was generated by deletion of both the b1 and b2 genes [42, 43]. Mice homozygous for this deletion die late in gestation, as expected by the simultaneous absence of the b1 or b2 genes. Heterozygotes (th3/+), however, are viable and thalassemic. Adult th3/+ mice exhibit the most severe anemia (8 to 10 g/dL of Hb) of the three mouse models of beta-thalassemia intermedia. Moreover they show strong abnormal red cell morphology, splenomegaly, and develop spontaneous hepatic iron deposition similar to that found in humans with beta-thalassemia intermedia.
Murine Models of Beta-Thalassemia Major or Cooley’s Anemia
Generation of an adult mouse model with features of Cooley’s anemia, including lethal anemia, has been hampered by the fact that in mice, in contrast to humans, gamma-like globin genes are not present. Lack of an animal model that can reproduce Cooley’s anemia has limited the full characterization of the critical pathophysiologic events of this disease, has prevented a full investigation of the biological mechanisms underlying the disease, and has hampered the evaluation of both pharmacological and genetic treatments. To overcome the embryonic lethal outcome, an adult mouse model of beta-thalassemia major was generated [44] by engrafting wt animals, after myelo-ablation, with hematopoietic fetal liver cells (HFLCs) harvested from alive th3/th3 embryos at 14.5 days of gestation. The embryo genotypes obtained from heterozygote matings were readily identified by Hb electrophoresis prior to transplantation [44]. After 6–7 weeks, these mice exhibited a severe anemia, with 1–3 g Hb/dL, low red blood cell (RBC) counts, low hematocrit values, and low reticulocyte counts together with very high levels of serum erythropoietin. Profound anemia settled in after 50 days, consistent with the clearance rate of the recipient’s normal RBCs, and the mice succumbed to ineffective erythropoiesis within 60 days [44]. Platelet and neutrophil counts were comparable in experimental and control groups, excluding aplasia or graft failure as the cause of death [44]. Moreover, these mice presented severe body mass reduction with massive splenomegaly due to major erythroid hyperplasia, as indicated by pathological analyses, as well as extensive hepatic EMH and iron overload [44]. These animals could be rescued by lentiviral mediated beta-globin gene transfer [44] or by blood transfusion [26], supporting the notion that their phenotype is specifically due to erythroid impairment.
Murine Models of Sickle Cell Disease
Initial efforts focused on the generation of transgenic animals expressing the mutant betaS-globin gene. However, chimeric Hb consisting of murine alpha-globin and human betaS-globin did not polymerize efficiently [45, 46]. Even with the addition of a human alpha-globin transgene, only a small fraction of the cells sickled in vivo because of the disruption of HbS by murine alpha-globin proteins [47]. In addition, under hypoxic conditions, there is more extensive deoxygenation of murine Hb than HbS, as mouse Hb has a lower O2 affinity than HbS [48].
To generate additional mouse models that were more faithful to the human disorder, two alternative strategies were devised. In order to obtain Hb that would polymerize more readily, Trudel and collaborators [49, 50] introduced two additional mutations into the human betaS-globin gene: a second mutation in codon 23 to reproduce the betaS-Antilles allele, and a third mutation to yield betaS-Antilles-D-Punjab or HbSAD. The beta SAD gene and the human alpha-2-globin gene, each linked to the beta-globin LCR, were co-introduced into the mouse germ line. In this transgenic line (SAD-1), red blood cells contained 19% Hb-SAD (human alpha/betaSAD tetramer) and mouse-human hybrids in addition to mouse hemoglobin. Adult SAD-1 transgenic mice were not anemic but had some abnormal features of erythrocytes and slightly enlarged spleens. In order to generate adult mice with a more severe sickle cell syndrome, a deletion in heterozygosity of the mouse beta-globin genes was introduced (beta-thal/SAD-1). In this model, the Hb-SAD was increased to 26%. The mice exhibited abnormal erythrocytes with regard to shape and density, and to a certain extent, anemia, a larger spleen, higher reticulocytes, and IE. In addition, they exhibited mortality upon hypoxia and polymerization of hemolysate similar to that found in human SCD. The SAD mice displayed a greater propensity for red cell sickling under hypoxic conditions, providing data that furthers the understanding of the physiopathology of SCD and test compounds that could be beneficial in treating the disease [51, 52]. However, attempts to generate a SAD mouse model completely lacking mouse Hbs failed due to perinatal lethality [49].
The second approach has been to generate mouse models in which the human alpha- and betaS-globin were expressed in the absence of murine globin genes. This was achieved through a series of crossing and interbreeding that included the transgenic founders and alpha- and beta-globin knock-out (KO) mice. Four groups have reported sickle transgenic mouse models in which all the murine globin genes were knocked out. To create a mouse model that produces sickle human Hb, Paszty et al. co-injected three fragments of human DNA into fertilized mouse eggs to generate transgenic founders expressing human alpha- and betaS-globin (Berkeley model) [53]. Since gamma-globin has anti-sickling properties, they included the gammaG- and gammaA-globin genes to decrease the likelihood that erythrocytes would sickle during gestation and cause fetal death. Recently the Berkeley mice have been thoroughly analyzed, showing a wide spectrum of hematologic and histopathologic findings that are very similar to those found in humans with SCD. However, notable differences were observed, such as higher anemia (6.1 g/dL) than SCD patients (on average 8.2 g/dL), as well as splenomegaly [54, 55]. Ryan et al. generated the second mouse model (Birmingham). They created transgenic animals carrying a 22-kb DNA fragment encompassing the human beta-globin LCR and a 9.7-kb DNA fragment containing the gammaA-globin and betaS-globin genes. The LCR was also linked to a 3.8-kb DNA fragment containing the human alpha-1-globin gene [56]. This mouse model is extremely anemic with a decrease in Hb almost double that seen in SCD patients and markedly reduced survival. The third murine model of SCD, created by Chang et al., recapitulates not only the phenotype but also the genetic locus encompassing betaS-globin gene in patients with SCD. For this purpose, they transferred a 240-kb betaS-globin YAC in which members of the human beta-globin gene cluster are present in their native genomic context [57]. However, even this mouse exhibits severe anemia and short lifespan. Fabry et al. have generated the fourth model [58]. These mice carry human LCR-alpha-2-globin and human LCR-betaS-globin constructs and produced three different levels of human HbF (<3%, 20% and 40%). Mice with the least HbF expression had the most severe pathology and markedly reduced survival. At least 20% of HbF was required to rescue animals with high HbS levels that simultaneously showed SCD features.
Thus, in all these mouse models, either co-expression of mouse beta-globin, as in the SAD mice, a mild thalassemic phenotype, as in the Berkeley mice, or an increased hemoglobin F level, as in the NY1KO mice, appears to be required for survival beyond several months of age. Therefore, the severity of anemia is compounded by the suboptimal expression of these genes, which adds thalassemia features, or by additional normal hemoglobins, thereby complicating the pathophysiology observed in the mice. Each of these lines may contribute to our understanding of SCD, although none of them fully recapitulates the relationship between hemolysis, IE, and iron metabolism observed in human SCD.
IRON METABOLISM
Dietary nonheme iron is absorbed though the enterocytes of the intestinal epithelium in the duodenum and proximal jejenum (Fig. (2)). Duodenal cytochrome-b (DCYTB) [59] reduces iron from the ferric (Fe3+) to the ferrous form (Fe2+), which is carried into epithelial cells by the divalent metal ion transporter 1 (DMT1). Targeted mutation of the murine DMT1 gene (Slc11a2−/− mice) determined that the main role for DMT1 is intestinal iron absorption and erythroid iron uptake [60]. Heme iron is absorbed through the heme carrier protein 1 (HCP1) [61]. The intracellular metabolism of heme is not fully understood, although inside the enterocytes, heme appears in membrane-bound vesicles within the cytoplasm [62] where it is likely degraded by heme oxygenase to yield ferrous iron. Egress of iron from these cells involves ferroportin (FPN) or iron-regulatory protein-1 [63, 64]. Similar to DMT1, FPN likely conducts Fe2+ ions. Cellular iron export, therefore, requires an associated ferroxidase activity. In the intestine, ceruloplasmin, a circulating multicopper oxidase, and hephaestin (HEPH), a homolog of ceruloplasmin, supply this activity [65–67], whereas hepatocytes and macrophages probably use ceruloplasmin exclusively. In the bloodstream, iron is bound to apotransferrin (apoTF) to form holotransferrin (holoTF) for delivery to other organs via transferrin receptor 1 (TFR1). In non-pathological conditions, very small amounts of iron may be loosely associated with albumin or alternative molecules.
Fig. 2. Simplified schema describing iron metabolism.
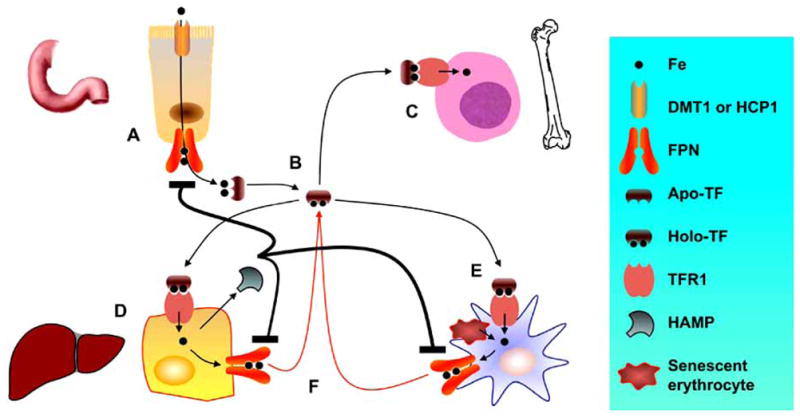
Iron is absorbed by duodenal enterocytes by DMT1 or HCP1 while FPN releases it to the plasma to bind apo-TF (A). Iron circulates as holo-TF (B). Most of the iron in the body is incorporated into erythroid precursors in the bone marrow, where Hb is synthesized (C). Iron is also stored in parenchymal cells of the liver (D) and reticuloendothelial macrophages (E). Parenchymal cells, mobilizing FT-bound iron, and macrophages cells, by degrading Hb from senescent erythrocytes, release iron to the plasma onto TF for delivery to other cells (F). When iron concentration increases, HAMP is secreted by parenchymal cells, targets FPN blocking iron absorption by duodenal enterocytes and iron egress from macrophages.
Spontaneous mutations and targeted mutations disrupting the Tf and transferring receptor-1 (Tfr1) genes demonstrate the importance of these proteins in iron metabolism and, in particular erythropoiesis, both in animals and humans. Erythroid precursors take up holoTF through TFR1 by receptor-mediated endocytosis. Profound deficiency of TF results in severe anemia, and iron deficiency occurs only in hematopoietic cells, while other tissues develop massive iron overload [68, 69]. Targeted disruption of the murine Tfr1 gene causes severe anemia and embryonic death [70]. Following endocytosis and vesicle acidification, iron is reduced to its ferrous form prior to being transferred across the endosomal membrane. The endosomal ferrireductase activity is mediated by the six-transmembrane epithelial antigen of the prostate-3 gene or STEAP3 and, to a lesser extent, by other STEAP-like genes (STEAP1, 2 and 4) [71, 72]. STEAP3 is highly expressed in the liver, and its product is a protein which co-localises in endosomes with TFR1 and DMT1. However, different members are expressed at higher level in some tissues than others. Once the iron leaves the endosome, it must move to the mitochondrion for incorporation into protoporphyrin IX by ferrochelatase to form heme. Mitoferrin carries out mitochondrial iron import, as determined by experiments in which absence of mitoferrin was associated with failure to incorporate erythroid iron into heme [73]. Another protein capable of cellular heme export, FLVCR, likely plays a key role in erythropoiesis [74]. If FLVCR is deleted neonatally, leads to the development of a severe macrocytic anemia with proerythroblast maturation arrest. In addition, it has been shown that FLVCR mediates heme export from macrophages. Altogether these observations suggest that erythroid precursors export excess heme to ensure survival and that trafficking of heme facilitates erythropoiesis and systemic iron balance [75].
Hepatocytes are the main depots for iron storage, and iron is predominantly stored in the form of ferritin (FT), which can be mobilized when needed elsewhere in the body. The liver can also clear from the circulation other forms of iron associated to proteins. Circulating ferritin contains very small amounts of iron [76] and, as such, it is not a major source of iron in normal conditions. Nevertheless, the liver clears ferritin by a method involving binding to a specific ferritin receptor [77]. In addition, plasma Hb and heme can be elevated in hemolytic states and be an important source of toxic iron. Hemopexin and haptoglobin are scavenging proteins that respectively bind heme and Hb [78]. Uptake of the heme-hemopexin and Hb-haptoglobin complexes are mediated by their specific receptor, respectively CD91 and CD163 [79, 80]. Hepatocytes can also acquire their iron load in a different fashion which is not mediated by the previously described mechanism: non–transferrin-bound iron (NTBI) uptake. This pathways become particularly important when serum iron levels exceed TF binding capacity [81]. The identity of the hepatocyte NTBI uptake molecules has not been yet clarified. DMT1 is a potential candidate in view of the fact that NTBI uptake is increased in cells in which DMT1 mRNA and protein expression are upregulated [82, 83]. A few additional candidates have been proposed, such as ZIP14, originally described as a zinc transporter [84, 85], and neutrophil gelatinase-associated lipocalin (NGAL), which is elevated in beta-thalassemia [86], and it has been described as a siderophore-iron-binding protein as well as a growth factor [87]. Moreover, other tissues have NTBI uptake activities and load iron when NTBI is present in the plasma. In particular, it has been proposed that cardiomyocytes, in condition of iron overload, use L-type calcium channels for ferrous iron intake [88]. However, for all these potential NTBI transporters, the information is still inadequate to draw any conclusion.
FPN, the iron exporter on the surface of absorptive intestinal enterocytes, hepatocytes, placental cells and macrophages, releases iron into the plasma [64, 89]. MON1A, a membrane trafficking protein, controls the amount of FNP to the surface of iron-recycling macrophages. This was discovered in an elegant study in mice, in which an allelic variant of Mon1a was associated with lower amounts of Fpn molecules on the macrophage surface membrane and these correlated with increased intracellular iron content in enterocytes, hepatocytes and macrophages [90]. FPN can be internalized and degraded by association with hepcidin (HAMP), an iron sensor protein (Fig. (2)) [91–94]. In particular, degradation of FPN interrupts cellular iron export in at least two sites: the intestinal epithelium and tissue macrophages (Fig. (2)). In particular, FPN degradation in the duodenum leads to decreased iron absorption from the diet. After HAMP binding, FPN is tyrosine phosphorylated at the plasma membrane. Mutants of human FPN that do not get internalized or that are internalized slowly show either absent or impaired phosphorylation. Once internalized, FPN is dephosphorylated and subsequently ubiquitinated. Ubiquitinated FPN is trafficked through the multivesicular body pathway en route to degradation in the late endosome/lysosome [64, 95, 96]. HAMP, a cysteine-rich 25-amino acid peptide produced by hepatocytes and secreted into the bloodstream, derives from the C-terminus of an 84–amino acid prepropeptide, originally isolated from urine and blood ultrafiltrate [97]. Hepatic iron homeostatically regulates HAMP levels [98], which in turn controls the concentration of FPN on the cell surface. HAMP is also induced during inflammation, in which HAMP’s effect on iron transport causes the characteristic decrease in blood iron (hypoferremia of inflammation). Hypoferremia is thought to increase host resistance not only to microbial infection [99], but it also leads to the anemia of inflammation (often referred to as the anemia of chronic disease) [100]. In these conditions, HAMP transcription is likely to be activated by IL-6 and its receptor through STAT-3 [101–103], (Fig. (3)). In addition, it has been demonstrated that p53 binds the HAMP promoter activating its expression, suggesting that up-regulation of HAMP by p53 is part of a defense mechanism against cancer through iron deprivation, and that HAMP induction by p53 might be involved in the pathogenesis of anemia accompanying cancer [104] (Fig. (3)). In addition, Cebpa-KO mice showed low Hamp expression. It has been suggested that this transcriptional factor controls Hamp expression [105] through the Epo/EpoR pathway [106]. Therefore, it seems that expression and synthesis of this peptide involves multiple pathways that require further studies.
Fig. 3. Transcriptional regulation of HAMP expression.

Basal HAMP expression depends upon signaling by interaction between BMPs, BMPRs through the downstream SMAD factors. HJV acts as a BMP co-receptor. HoloTF also induces HAMP expression through the same pathway. It has been proposed that the HFE/β2M/TFR2 and the BMP/BMPR/HJV complexes interact to synergistically increase HAMP expression through the SMAD factors. HAMP induction in inflammation results, at least in part, from the signaling of IL-6 through its receptor and STAT3. P53 also activates HAMP transcription. Erythropoiesis represses HAMP transcription directly, by GDF15; indirectly, by hypoxia or by other unknown mechanisms. The question mark indicates that some of these pathways are not completely elucidated yet.
Increased plasma iron, from macrophage recycling of aged red blood cells or from intestinal absorption of iron, stimulates hepatocytes by an as of yet not completely understood mechanism to produce more HAMP. Recently it has been proposed that holoTF concentrations regulate HAMP expression in primary hepatocytes and cell lines [107] by activating an iron homeostatic regulatory pathway that was previously characterized by a series of studies in vitro, use of animal models and characterization of related human pathological disorders termed hereditary hemochromatosis (HH). HH refers to the clinical disorder that results from an excess of total body iron and leads to organ failure due to iron toxicity. In HH, hyper-absorption of dietary iron leads to hyper-ferremia, tissue iron deposition, and a paucity of iron in intestinal epithelial cells and tissue macrophages. Complications include cirrhosis; hepatocellular carcinoma; heart disease; and neuroendocrinopathies, particularly diabetes. It is worthwhile to note that patients affected by hemoglobinopathies and iron overload share many clinical manifestations with HH. HH is a heterogeneous genetic disease that may result from mutations in at least 4 genes: HFE, transferrin receptor 2 (TFR2), hemojuvelin (HJV) and HAMP. The majority of patients affected by HH [108] have a mutation in HFE at position 282 (tyrosine for cysteine at position 282: C282Y). HFE is a HLA class I atypical protein, which binds beta2-microglobulin [109], TFR1 [110, 111] and TFR2 [112]. Targeted mutagenesis was used to produce two mutations in the murine hemochromatosis gene (Hfe) locus. The first mutation deleted a large portion of the coding sequence, generating a null allele [113]. The second mutation introduced a missense mutation C294Y (corresponding to C282Y in humans) [114] into the Hfe locus, but otherwise left the gene intact. Homozygosity for either mutation results in postnatal iron loading, although the effects of the null mutation are more severe than the effects of the C294Y/C282Y mutation [113, 115]. Patients with HH disorder have low/normal HAMP, measured either as RNA on liver biopsies or as the amount of peptide excreted in the urine [116–118]. However, HAMP levels are inappropriately low if related to the degree of iron loading [117, 118]. Hfe-KO mice had similar features, showing high iron liver content and inappropriately low expression of Hamp [113]. However, the actual amount of Hamp secreted in the blood stream has never been quantified and it cannot be excluded that, directly or indirectly, Hfe mediates Hamp synthesis and/or secretion more than Hamp expression. In fact, posttranslational processing of HAMP it is a controlled mechanism that requires the prohormone convertase furin [119], but it is not known if Hfe is involved in this process. Nevertheless, the previous observations indicated that Hfe has a role in sensing iron and regulating Hamp synthesis and/or activity. In fact, when Hfe-KO mice were crossed with transgenic mice over-expressing Hamp [93], Hamp inhibited the iron accumulation normally observed in the Hfe-KO mice [120].
Similar findings characterize TFR2, which was cloned based on the sequence homology with TFR1 and its ability to bind holoTF and HFE, although its function is different from TFR1 [121, 122]. HH associated with mutations in TFR2 [123] is often reported as a disorder similar to HH associated-HFE, since the pattern of liver iron storage and the response to phlebotomy are identical in the two disorders [124]. TFR2 RNA is not iron-regulated since it has no iron-responsive (IRE) elements. IREs are RNA stem-loops found in the untranslated regions (UTR) of mRNAs involved in iron transport and storage and are recognized by iron regulatory proteins (IRPs). IRPs, whose activity is modulated by iron, interfere with ribosome assembly or determine the stability of IRE-mRNAs in the function of iron demand or surplus [125]. Therefore, the main function of TFR2 is probably to monitor the amount of iron in the form of holoTF and competing with TFR1 to bind HFE [126]. Release of HFE from TFR1 likely modulates the ability of HFE to activate HAMP expression [112]. In fact, low Hamp liver RNA was documented in Tfr2-deficient mice [127], and low HAMP urinary levels and low HAMP/ferritin ratios were found in patients with TFR2 mutations [128]. The importance of the interaction of Hfe with Tfr1 in controlling Hamp expression was shown utilizing murine mutant lines that interfered or favored a constitutive Hfe/Tfr1 interaction. In the former case mice developed iron deficiency associated with inappropriately high Hamp expression, while in the latter case mice developed iron overload attributable to inappropriately low expression of Hamp. These data suggest that Hfe induces Hamp expression when it is not in complex with Tfr1 [126].
HJV is highly expressed in the liver, in addition to the skeletal muscle and the heart [129]. The gene is transcribed in several isoforms because 5 alternative splicing variants have been recognized. The larger transcript encodes a 426 amino acid, glycosylphosphatidylinosytol (GPI)-linked protein, characterized by an RGD motif and a von-Willebrand type D domain. The GPI anchor suggests that HJV can be present in either a soluble (HJV.Fc) or a cell-associated form. The finding of extremely low HAMP levels in patients carrying mutations in HJV indicates that this protein controls HAMP production [129]. In fact, HJV signals through the bone morphogenic proteins (BMP), namely BMP-2 (but also BMP-4 to a lesser extent) and BMP-receptors, to activate a downstream transcriptional factor called SMAD-4, which triggers transcription of HAMP [130, 131]. BMP-2 administration increases HAMP expression and decreases serum iron levels in vivo, whereas HJV.Fc selectively inhibits BMP-2 induction of HAMP expression [132]. In cells engineered to express HJV, increasing iron concentrations progressively inhibited HJV.Fc release [133]. Thus, soluble and cell-associated HJV reciprocally regulate HAMP expression in response to changes in extracellular iron concentration.
The majority of the mutations in HAMP that cause HH are due to either a complete inactivation of the protein [134] or a substitution of one of the invariant cysteines of the peptide [135]. In addition, Hamp-deficient mice accumulate iron in parenchymal cells due to greater intestinal absorption, increased circulating iron levels, impaired retention of iron by reticuloendothelial cells, and hepatic iron overload [91]. In contrast, transgenic mice over-expressing Hamp have markedly lower iron stores, resulting in severe anemia [93]. Mutations in the HJV and HAMP are responsible for a subset of HHs called juvenile-HH. The disease shares numerous features with HH associated-HFE, but all the clinical manifestations develop earlier because intestinal iron absorption is greater, and the rate of iron accumulation faster, than in the classic form. In addition, juvenile-HH often leads to hypogonadism and premature death due to cardiac arrest.
Although mutations in FNP were previously described as additional forms of HH (type IV), they are now grouped under ferroportin diseases which have a dominant genetic pattern of inheritance and heterogeneous clinical presentation [96, 136, 137]. Based on their clinical manifestations, some patients show iron loading of Kupffer cells with relatively low saturation of plasma TF, but others with high TF saturation and iron-loaded hepatocytes. Mutations in FPN are either defective in cell surface localization or have a decreased ability to be internalized and degraded in response to HAMP [138–141]. Since FNP forms multimeric complexes, the mutant FNP affects the localization of the wild-type FNP [138]. The first subset of mutations causes a loss of iron export function, causing haploinsufficiency. By contrast, a second set of mutations [138–140, 142], which can be associated with greater TF saturation and more prominent iron deposition in liver parenchyma in vivo, resist HAMP inhibition, resulting in a permanently “turned on” iron exporter.
In contrast to all other types of HHs, which are characterized by HAMP deficiency, patients with ferroportin disease showed increased HAMP levels [143]. Except for iron overload in the ferroportin diseases in which HAMP synthesis is of no consequence to prevent iron absorption, the insufficient production of HAMP is now considered the key pathogenetic feature of all forms of HH. Given the essential role of HAMP in iron homeostasis and the severity of juvenile-HH that results from mutations in HAMP, it is likely that HFE, TFR2, and HJV work cooperatively together to control HAMP expression. Recently, a model for the regulation of iron homeostasis that includes all these proteins has been proposed [126]. HFE and TFR2, upon their interaction, would associate with the HJV/BMP complex, synergistically inducing HAMP transcription through the SMAD-4 pathway [112].
IRON ABSORPTION AND INEFFECTIVE ERYTHROPOIESIS
Iron balance must be carefully regulated to provide iron as needed but avert the toxicity associated with iron excess. Tissue iron overload is a primary focus of beta-thalassemia and SCD management, and if not prevented or adequately treated, is fatal in both transfused and non-transfused patients. Iron metabolism shows similar features regardless if SCD and thalassemia patients require blood transfusion. In transfused and non-transfused patients, iron overloaded in absence of chelation therapy causes elevated serum TF saturation and FT levels. NTBI increases when serum iron levels exceed TF binding capacity [81, 144]. NTBI and excess iron are primarily deposited in liver parenchymal cells and other organs. The heart and endocrine tissues are particularly susceptible.
The expression level of genes that regulate iron absorption and metabolism is different depending on whether the iron burden is derived from increased iron absorption or through blood transfusion, as it will be described later in our review. In beta-thalassemia intermedia, where regular transfusion therapy is unnecessary, there is evidence of iron overload [145–148]. Iron absorption studies show that the rate of iron loading from the gastrointestinal tract is approximately three to four times greater than the normal rate [32]. Increased iron absorption also plays a role in beta-thalassemia major, where its importance is inversely related to Hb levels. In non-transfused patients with severe thalassemia, abnormally regulated iron absorption results in increases in body iron burden (which may vary between 2 and 5 g per year) depending on the severity of erythroid expansion [149]. Regular transfusions may double this rate of iron accumulation. Increased iron absorption was observed in three non-transfused SCD patients [150]. In the same study, patients affected by beta-thalassemia major were compared before and after transfusion therapy. The results clearly showed that in the absence of a transfusion, iron absorption was elevated, whereas it was decreased or normalized after transfusion therapy. In addition, in the presence of transfusion regimens, endogenous erythropoiesis was suppressed [150]. This study and previous investigations led to the conclusion that iron absorption in humans is regulated by the combined influences of hypoxia, the body’s iron stores [91, 92, 98], and the erythropoietic demand for iron [26, 94].
Therefore, IE would further increase iron absorption. IE is triggered by a combination of several factors: premature destruction or elimination of abnormal non-nucleated erythroid cells (e.g. hemolytic anemia and macrophagocytosis), cell death of erythroid precursor by apoptosis, and limited erythroid cell differentiation. The most important features of IE in thalassemias and SCD are, respectively, the excess of the complementary functional globin chains and the abnormal betaS-globin that causes premature destruction of erythroid cells in the bone marrow (BM) and at extramedullary sites. In addition, our studies suggest that IE in thalassemia is also characterized by reduced erythroid differentiation [151], which would further exacerbate anemia. In beta-thalassemia and alpha-thalassemia major, as well as in HbH disease, IE is supported by ferrokinetic reports, as well as additional studies assessing plasma levels of EPO and the soluble transferrin receptor [18, 152–154]. In SCD, the data is scarcer and less straightforward. Iron deficiency, evaluated as the absence of BM iron stores, rather than iron overload, is very common among non-transfused patients with sickle cell anemia [55]. In addition, iron therapy given to SCD patients with low MCV, serum FT and TF saturation levels was successful increasing the Hb level more than 1g/dL, suggesting that these patients were iron deficient [155]. However, Erlandson et al. [150] documented increased iron absorption in three non-transfused SCD patients showing 6.5 to 8.7 g/dL of Hb. Increased reticulocytosis was observed together with anemia, which suggested that IE occurred in those patients. It is thus likely that all the hemoglobinopathies associated with IE have increased iron absorption.
How does erythropoiesis control iron demand? The mechanisms that control iron homeostasis predict signaling through iron absorbing duodenal cells, iron storing hepatocyte and iron recycling macrophages by regulators, historically defined as “store” and “erythroid” regulators [156]. To date, the only iron store candidate is the circulating peptide hormone, HAMP [91, 157]. Normally, intestinal absorption accounts for only a fraction of iron circulating bound to TF, but this can be dramatically increased if HAMP synthesis is decreased. The effect of decreasing the level of HAMP also affects iron concentration in the macrophages. Circulating iron is mostly derived from the recovery of iron from senescent erythrocytes, through phagocytosis by tissue macrophages, particularly in the spleen. The cells are lysed, and Hb is catabolized, presumably by heme oxygenase, to liberate iron. FPN is critical for macrophage iron export. Therefore, different HAMP levels can change the ratio between stored and released iron. In the presence of IE, iron absorption and the amount of iron released from macrophages is expected to increase.
Although several mechanisms have been proposed to explain how erythropoiesis affects iron absorption, to date, the characterization of the erythroid regulator has been more elusive. The function of the erythroid regulator is to maintain the production of erythrocytes irrespective of the body’s iron balance. While increased amounts of the erythroid regulator are extremely helpful in resolving transient blood loss, in hemoglobinopathies, this process will end in iron overload due to the inadequacy of IE in addressing anemia. After phlebotomy, hemolysis, or EPO administration, HAMP production is decreased, [94, 158, 159], indicating that the erythroid regulator exerts its activity, at least in part, by suppressing production of this peptide. Two major lines of thought identify the erythroid regulator either with a diffusible factor or an intrinsic cellular process, both of which repress HAMP expression. Increased erythropoiesis through secretion of soluble mediators could directly influence HAMP production. Erythropoiesis also causes a fall in tissue iron content and serum levels and they too could down-regulate HAMP. Alternatively, anemia could cause hypoxia, triggering EPO expression or activation of transcription factors that repress HAMP synthesis.
Pak and colleagues [160] administered inhibitors of erythropoiesis after phlebotomy to disassociate the effects of anemia, hypoxia, and Epo from the effects of increased erythropoiesis and iron use. Phlebotomized mice developed anemia, tissue hypoxia, increased EPO, increased erythropoiesis, decreased serum iron, and hepatic Hamp mRNA levels. When erythropoietic inhibitors were administered, serum and tissue iron as well as Hamp mRNA rose dramatically, even though the mice were anemic. These experiments suggest that the dominant regulators of Hamp during increased erythropoiesis include a signal arising from the erythropoietic activity in the bone marrow or the effects of increased iron use on plasma or tissue iron (Fig. (3)). The existence of an erythroid factor is also supported by studies in which the serum of patients affected by beta-thalassemia or HH associated with HFE, were compared in terms of their ability to induce the expression of HAMP and other iron metabolism-related genes. Sera from beta-thalassemia major and intermedia patients down-regulated HAMP expression in the HepG2 cell line. In contrast, the majority of sera from hereditary hemochromatosis patients induced an increase in HAMP expression, which correlated with TF saturation [161]. Recently, a candidate erythroid regulator has been proposed called GDF15. Compared to controls, GDF15 is elevated in the serum of beta-thalassemic patients and suppresses HAMP expression in vitro [162]. GDF15 is a member of the TGF-beta superfamily of proteins, which are known to control cell proliferation, differentiation, and apoptosis in numerous cell types. Interestingly GDF15, also called MIC-1, can be modulated by p53 and conditioned medium from cells expressing this protein can suppress the growth of certain tumor cells provided they express TGF-beta receptors and SMAD4 [163–165]. However, the mechanism of function and efficacy of GDF15 in repressing HAMP expression still needs to be elucidated. Alternatively, a direct involvement of EPO in Hamp regulation can be hypothesized. In fact it has been reported that EPO, through EPOR signaling and in a dose-increasing fashion, inversely mediates Hamp expression in freshly isolated mouse hepatocytes and in the HepG2 cells. Interestingly, chromatin Immunoprecipitation experiments showed a significant decrease of C/EBPalpha binding to the Hamp promoter after EPO supplementation, suggesting the involvement of this transcription factor in the transcriptional response of Hamp to EPO [106].
Alternatively, studies by Yoon et al. and Peyssonnaux et al. [166, 167] emphasized the role of hypoxia as a mechanism to control iron absorption and erythropoiesis. Using several conditional transgenic lines to modify expression of the hypoxic response, they demonstrated that the hypoxia-inducible transcription factor Hif1alpha binds the Hamp promoter, down-regulating its expression in hypoxic conditions (Fig. (3)). Their studies also suggested a more general role for the hypoxia pathway in coordinating iron metabolism and oxygen transport by the simultaneous coordinate expression of Hamp, Fpn, and Epo.
All these studies might also indicate that in the simultaneous condition of increased erythropoiesis and hypoxia, several factors synergistically lead to HAMP down-regulation. This model does not fail to appreciate the individual role of erythroid soluble factors, hypoxia and iron levels, although their separate effect on HAMP synthesis would be of a lower magnitude. It is also possible that other genes might be influenced by erythroid demand, further increasing the rate of iron absorption. This was illustrated by using mouse models of beta-thalassemia, in which the effect of IE on Hamp synthesis was investigated. The first studies indicated that both mice affected by beta-thalassemia major (th3/th3) and intermedia (th3/+ and th1/th1) had, respectively, low or relatively low Hamp expression levels compared to the amount of iron overload [168–171]. A more extensive analysis indicated that, while in 1 year-old th3/+ mice, Hamp expression was similar to or higher than that of control mice, it was rather the up-regulation of Fpn in the duodenum that caused iron overload [26]. In addition, analysis of the mRNA level of several iron related genes revealed that Hfe and Cebpa were reduced in the liver of all the animals that expressed low Hamp levels. In particular, Cebpa decreased in th3/th3 mice that showed the lowest level of Hamp expression. Therefore, our data suggest that Hfe could play a direct role in Hamp regulation in beta-thalassemia, while low Cebpa might further decrease Hamp expression in conditions of extreme IE. In transfused mice Hamp, Hfe, and Cebpa expression augmented probably due to the combined effect of reduced anemia, increased iron content, and suppression of erythropoiesis [26].
The importance of the physiological role of HAMP in patients affected by hemoglobinopathies has been underscored by studies in which low HAMP levels corresponded with iron burden and anemia. In a study by Kattamis, liver HAMP mRNA levels correlated with hemoglobin concentration and inversely correlated with serum transferrin receptor, erythropoietin, and non-transferrin-bound iron [172]. In beta-thalassemic patients, they did not correlate with indices of iron load, and urinary HAMP levels were disproportionably suppressed with regard to iron burden. In parallel studies, the HAMP to FT ratio was used as a marker of the appropriateness of HAMP expression relative to the degree of iron burden. In thalassemic syndromes, urinary HAMP was suppressed relative to the degree of iron burden, whereas transfusion led to its increase. In addition, SCD patients were analyzed, showing that urinary HAMP was suppressed and inversely associated with erythropoietic drive [143, 173, 174]. These studies illustrate that low HAMP levels contribute to iron overload and suggest the potential therapeutic role of HAMP or HAMP agonists in these disorders.
(UN)SOLVED QUESTIONS AND FUTURE DIRECTIONS
Although hemoglobinopathies are monogenic disorders, they are complex diseases displaying many unexplained secondary clinical manifestations and pleiotropic aspects. Very few other traumas can be as rapidly life-threatening as major blood loss and tissue oxygen deprivation. Therefore, it is conceivable that the nature of these diseases requires extreme physiological responses in order to limit anemia and sustain life. In hemoglobinopathies, the majority of the body’s physiological responses are likely re-directed or focused to optimize blood production at the expense of other cellular homeostatic circuitry systems, triggering pathological effects if these conditions chronically persist. One example that supports this concept involves iron homeostasis. As one of the main functional components of Hb, iron is absolutely required for RBC production. Iron absorption is dramatically altered in th3/th3 mice to the extent that the concentration of iron in tissue and serum may be as high as 10 times the normal level [26]. These levels far exceed erythroid demands, as shown by parenchymal iron deposition and suggest that, in conditions of extreme anemia, the circuitry that controls iron homeostasis is lost. Under normal conditions, the iron levels observed in beta-thalassemia major would trigger an elevated production of Hamp; however, the extreme level of IE in these animals prevents Hamp from sensing the iron burden and keeps its expression very low. In contrast, in th3/+ mice, which have less erythropoietic drive compared to th3/th3, Hamp expression is likely to be determined by the relative ratio between anemia and iron overload, which varies with age. In fact, although the level of expression of Hamp escalated in aging mice, the net rate at which iron content increased was maintained due to a persistent up-regulation of both Fpn mRNA and protein in the duodenum, thus underscoring this iron transporter’s fundamental role in the iron overload of beta-thalassemia (Fig. (4)). Altogether, these observations indicate that in beta-thalassemia, iron absorption fails to obey normal physiological rules and is increased despite the tissue damage that accompanies iron overload.
Fig. 4.

Schematic representation of iron metabolism mediated by HAMP and FPN, from the top, in condition of normal erythropoiesis, beta-thalassemia intermedia in early and late stage and in beta-thalassemia major. Discussed in the text.
If it is now clear why Hamp is decreased in the condition of IE, it is unknown why Fpn is increased in the duodenum in th3/+ mice. As previously noted [166, 167], increasing levels of hypoxia over time could trigger up-regulation of Fpn in the duodenum, although the same has not been observed for th3/th3 mice. Haptoglobin is a glycoprotein produced mainly by the liver and secreted into the blood. Haptoglobin, by virtue of its high affinity for hemoglobin, protects the tissues against hemoglobin-induced oxidative damage and allows heme iron recycling. It has been shown that iron induces a dramatic post-transcriptional decrease of liver and serum haptoglobin in C57BL/6 mice [175], the same strain of thalassemic mice that showed increased duodenal Fpn [26]. In addition, it has been shown that haptoglobin-KO mice export significantly more iron from the duodenal mucosa to plasma compared to their control counterparts. This is due to increased duodenal expression of Fpn, both at the protein and mRNA levels, whereas hepatic Hamp expression remains unchanged [176]. In the same study, it was also suggested that increased free Hb levels might activate Fpn expression. Therefore, due to the underlying IE in th3/+ mice, high free Hb and low haptoglobin serum levels could be responsible for increased Fpn expression in the duodenum. This hypothesis could be easily verified.
As in thalassemic patients, the organs most affected in HH patients are the liver and the heart. Therefore, HH and beta-thalassemia might share clinical manifestations with the same molecular origin: dysregulation of gene(s) that sense and control iron absorption. In thalassemic mice, it was shown that when Hamp was down-regulated, the expression of the Hfe gene was low as well. This is the first observation that one of the pleiotropic effects observed in beta-thalassemia (iron overload) might share the same molecular mechanism observed in another disorder. Is it possible, hence, that the low penetrance observed in HH and the variability in the clinical features triggered by iron overload observed in thalassemia might be due to similar modifier genes? Comparing thalassemic mice with other mouse models of hemochromatosis (e.g. Hfe-KO mice), in addition to their interbreeding, could greatly expand our knowledge of these disorders, simplifying the isolation of the genes that contribute to these pathologies.
A few studies have emphasized the possibility that administration of Hamp or control of its level of expression could be utilized to control iron absorption [26, 132, 168, 177–179]. Rivera at al. showed that HAMP injection reduced iron absorption in normal animals [178], while Roy and colleagues demonstrated that mice over-expressing Hamp developed a mild-to-moderate anemia associated with iron deficiency and iron-restricted erythropoiesis [179]. In addition, it has been shown that BMP-2 administration increases HAMP expression and decreases serum iron levels in vivo [132]. All these studies support a role for HAMP administration or modulators of the BMP signaling pathway for treating diseases of iron overload. Future studies should address whether abnormal iron absorption could be prevented by administration of Hamp. Mouse models of beta-thalassemia and SCD, as a first step to evaluate the potential therapeutic approach of Hamp administration in these diseases, could be utilized. Anemia, liver, and macrophage iron metabolism will need to be carefully monitored. However, the decreased levels of HAMP observed in the urine of thalassemic patients [143] indicate that these new potential therapeutic approaches are worthwhile to investigate for their potential to reduce iron overload in thalassemia and other hemoglobinopathies.
Acknowledgments
Gideon Rechavi: G. Rechavi holds the Djerassi Chair in Oncology at the Sackler Faculty of Medicine, Tel-Aviv University, Tel-Aviv, Israel. We thank the Kahn Family Foundation for their support.
Stefano Rivella: This work was supported by grants from NIH-NIDDK R21DK071575, the Carlo and Micol Schejola Foundation, the Roche Foundation for Anemia Research (RoFar), Associazione per la Lotta alla Talassemia di Rovigo (AVLT), Associazione Regionale Sarda per la Lotta Contro la Thalassemia e per l’Assistenza dei Talassemici, the Cooley’s Anemia Foundation, the Anova foundation and the Children’s Cancer and Blood Foundation. The authors would like to thank Mirella Misiaszek, Sara Gardenghi, Pedro Ramos and Kimberly Young for reading the manuscript and for helpful discussions, as well as Scott A. Kerns for technical support. S.R. especially thanks Ms. Gina Cioffi and Mr. Frank Somma (New York), Mr. Elio Zago (Ferrara), Mr. Michele Lippucci (Pisa), Mr. Giorgio Vargiu and Mr. Carlo Deligia (Cagliari) for their continued support of beta-thalassemia research.
References
- 1.Itano HA, Pauling L. Nature. 1961;191:398–9. doi: 10.1038/191398a0. [DOI] [PubMed] [Google Scholar]
- 2.Fritsch EF, Lawn RM, Maniatis T. Cell. 1980;19:959–72. doi: 10.1016/0092-8674(80)90087-2. [DOI] [PubMed] [Google Scholar]
- 3.Lauer J, Shen CK, Maniatis T. Cell. 1980;20:119–30. doi: 10.1016/0092-8674(80)90240-8. [DOI] [PubMed] [Google Scholar]
- 4.Lawn RM, Efstratiadis A, O’Connell C, Maniatis T. Cell. 1980;21:647–51. doi: 10.1016/0092-8674(80)90428-6. [DOI] [PubMed] [Google Scholar]
- 5.Perutz MF, Rossmann MG, Cullis AF, Muirhead H, Will G, North ACT. Nature. 1960;185:416–422. doi: 10.1038/185416a0. [DOI] [PubMed] [Google Scholar]
- 6.Steinberg MH, Forget BG, Higgs DR, Nagel RL. Disorders of Hemoglobin: Genetics, Pathophysiology and Clinical Management. Cambridge University Press; Cambridge, UK: 2001. [Google Scholar]
- 7.Tolhuis B, Palstra RJ, Splinter E, Grosveld F, de Laat W. Mol Cell. 2002;10:1453–65. doi: 10.1016/s1097-2765(02)00781-5. [DOI] [PubMed] [Google Scholar]
- 8.Breda L, Rivella S. Minerva Biotecnol. 2003;15:107–121. [Google Scholar]
- 9.Trimborn T, Gribnau J, Grosveld F, Fraser P. Genes Dev. 1999;13:112–24. doi: 10.1101/gad.13.1.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pauling L, Itano HA, Singer SJ, Wells IC. Science. 1949;110:543–46. doi: 10.1126/science.110.2865.543. [DOI] [PubMed] [Google Scholar]
- 11.Murari J, Smith LL, Wilson JB, Schneider RG, Huisman TH. Hemoglobin. 1977;1:267–82. doi: 10.3109/03630267709003409. [DOI] [PubMed] [Google Scholar]
- 12.Silvestroni E, Bianco I. Policlinico (Prat) 1963;70:1513–7. [PubMed] [Google Scholar]
- 13.Silvestroni E, Bianco I. Progr Med (Napoli) 1963;19:545–8. [PubMed] [Google Scholar]
- 14.Milner PF, Clegg JB, Weatherall DJ. Lancet. 1971;1:729–32. doi: 10.1016/s0140-6736(71)91992-1. [DOI] [PubMed] [Google Scholar]
- 15.Eldor A, Rachmilewitz EA. Blood. 2002;99:36–43. doi: 10.1182/blood.v99.1.36. [DOI] [PubMed] [Google Scholar]
- 16.Rodgers GP. Semin Hematol. 1997;34:2–7. [PubMed] [Google Scholar]
- 17.Vichinsky EP. Ann NY Acad Sci. 2005;1054:18–24. doi: 10.1196/annals.1345.003. [DOI] [PubMed] [Google Scholar]
- 18.Sohan K, Billington M, Pamphilon D, Goulden N, Kyle P. BJOG. 2002;109:1308–10. doi: 10.1046/j.1471-0528.2002.01051.x. [DOI] [PubMed] [Google Scholar]
- 19.Giardine B, van Baal S, Kaimakis P, Riemer C, Miller W, Samara M, Kollia P, Anagnou NP, Chui DH, Wajcman H, Hardison RC, Patrinos GP. Hum Mutat. 2007;28:206. doi: 10.1002/humu.9479. [DOI] [PubMed] [Google Scholar]
- 20.Voon HP, Wardan H, Vadolas J. Blood Cells Mol Dis. 2007;39:184–188. doi: 10.1016/j.bcmd.2007.01.006. [DOI] [PubMed] [Google Scholar]
- 21.Weatherall DJ, Clegg JB. Bull World Health Organ. 2001;79:704–12. [PMC free article] [PubMed] [Google Scholar]
- 22.Weatherall DJ. Nat Rev Genet. 2001;2:245–55. doi: 10.1038/35066048. [DOI] [PubMed] [Google Scholar]
- 23.Schrier SL. Curr Opin Hematol. 2002;9:123–6. doi: 10.1097/00062752-200203000-00007. [DOI] [PubMed] [Google Scholar]
- 24.Thein SL. Hematology Am. Soc. Hematol. Educ. Prog. 2005:31–7. doi: 10.1182/asheducation-2005.1.31. [DOI] [PubMed] [Google Scholar]
- 25.Pippard MJ, Callender ST, Warner GT, Weatherall DJ. Lancet. 1979;2:819–21. doi: 10.1016/s0140-6736(79)92175-5. [DOI] [PubMed] [Google Scholar]
- 26.Gardenghi S, Marongiu MF, Ramos P, Guy E, Breda L, Chadburn A, Liu Y, Amariglio N, Rechavi G, Rachmilewitz EA, Breuer W, Cabantchik ZI, Wrighting DM, Andrews NC, de Sousa M, Giardina PJ, Grady RW, Rivella S. Blood. 2007;109:5027–35. doi: 10.1182/blood-2006-09-048868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rachmilewitz EA, Weizer-Stern O, Adamsky K, Amariglio N, Rechavi G, Breda L, Rivella S, Cabantchik ZI. Ann NY Acad Sci. 2005;1054:118–23. doi: 10.1196/annals.1345.014. [DOI] [PubMed] [Google Scholar]
- 28.Vogiatzi MG, Macklin EA, Fung EB, Vichinsky E, Olivieri N, Kwiatkowski J, Cohen A, Neufeld E, Giardina PJ. Bone. 2006;38:571–5. doi: 10.1016/j.bone.2005.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.De Sanctis V, Vullo C, Katz M, Wonke B, Hoffbrand VA, Di Palma A, Bagni B. Prog Clin Biol Res. 1989;309:77–83. [PubMed] [Google Scholar]
- 30.Pippard MJ, Callender ST, Finch CA. Blood. 1982;60:288–94. [PubMed] [Google Scholar]
- 31.Olivieri NF. Semin Hematol. 2001;38:57–62. doi: 10.1016/s0037-1963(01)90060-5. [DOI] [PubMed] [Google Scholar]
- 32.Olivieri NF, DJW . Clinicl Aspects of Beta-thalassemia. Cambridge University Press; Cambridge, UK: 2001. Book. [Google Scholar]
- 33.Propper RD, Cooper B, Rufo RR, Nienhuis AW, Anderson WF, Bunn HF, Rosenthal A, Nathan DG. N Engl J Med. 1977;297:418–23. doi: 10.1056/NEJM197708252970804. [DOI] [PubMed] [Google Scholar]
- 34.Berdoukas V, Bohane T, Tobias V, De Silva K, Fraser I, Aessopos A, Lindeman R. Hematol J. 2005;5:572–8. doi: 10.1038/sj.thj.6200569. [DOI] [PubMed] [Google Scholar]
- 35.Thursz M. Gut. 2007;56:613–4. doi: 10.1136/gut.2006.113076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Taher A, Aoun E, Sharara AI, Mourad F, Gharzuddine W, Koussa S, Inati A, Dhillon AP, Hoffbrand AV. Acta Haematol. 2004;112:179–83. doi: 10.1159/000081268. [DOI] [PubMed] [Google Scholar]
- 37.Taher A. Semin Hematol. 2005;42:S5–9. doi: 10.1053/j.seminhematol.2005.01.005. [DOI] [PubMed] [Google Scholar]
- 38.Vichinsky E, Butensky E, Fung E, Hudes M, Theil E, Ferrell L, Williams R, Louie L, Lee PD, Harmatz P. Am J Hematol. 2005;80:70–4. doi: 10.1002/ajh.20402. [DOI] [PubMed] [Google Scholar]
- 39.Skow LC, Burkhart BA, Johnson FM, Popp RA, Popp DM, Goldberg SZ, Anderson WF, Barnett LB, Lewis SE. Cell. 1983;34:1043–52. doi: 10.1016/0092-8674(83)90562-7. [DOI] [PubMed] [Google Scholar]
- 40.Curcio MJ, Kantoff P, Schafer MP, Anderson WF, Safer B. J Biol Chem. 1986;261:16126–32. [PubMed] [Google Scholar]
- 41.Shehee WR, Oliver P, Smithies O. Proc Natl Acad Sci USA. 1993;90:3177–81. doi: 10.1073/pnas.90.8.3177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yang B, Kirby S, Lewis J, Detloff PJ, Maeda N, Smithies O. Proc Natl Acad Sci USA. 1995;92:11608–12. doi: 10.1073/pnas.92.25.11608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ciavatta DJ, Ryan TM, Farmer SC, Townes TM. Proc Natl Acad Sci USA. 1995;92:9259–63. doi: 10.1073/pnas.92.20.9259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rivella S, May C, Chadburn A, Riviere I, Sadelain M. Blood. 2003;101:2932–9. doi: 10.1182/blood-2002-10-3305. [DOI] [PubMed] [Google Scholar]
- 45.Ryan TM, Townes TM, Reilly MP, Asakura T, Palmiter RD, Brinster RL, Behringer RR. Science. 1990;247:566–8. doi: 10.1126/science.2154033. [DOI] [PubMed] [Google Scholar]
- 46.Greaves DR, Fraser P, Vidal MA, Hedges MJ, Ropers D, Luzzatto L, Grosveld F. Nature. 1990;343:183–5. doi: 10.1038/343183a0. [DOI] [PubMed] [Google Scholar]
- 47.Fabry ME, Bouhassira EE, Suzuka SM, Nagel RL. Methods Mol Med. 2003;82:213–41. doi: 10.1385/1-59259-373-9:213. [DOI] [PubMed] [Google Scholar]
- 48.D’Surney SJ, Popp RA. Genetics. 1992;132:545–51. doi: 10.1093/genetics/132.2.545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Trudel M, Saadane N, Garel MC, Bardakdjian-Michau J, Blouquit Y, Guerquin-Kern JL, Rouyer-Fessard P, Vidaud D, Pachnis A, Romeo PH, Beuzard Y, Costantini F. EMBO J. 1991;10:3157–65. doi: 10.1002/j.1460-2075.1991.tb04877.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Trudel M, De Paepe ME, Chretien N, Saadane N, Jacmain J, Sorette M, Hoang T, Beuzard Y. Blood. 1994;84:3189–97. [PubMed] [Google Scholar]
- 51.De Franceschi L, Saadane N, Trudel M, Alper SL, Brugnara C, Beuzard Y. J Clin Invest. 1994;93:1670–6. doi: 10.1172/JCI117149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rust MB, Alper SL, Rudhard Y, Shmukler BE, Vicente R, Brugnara C, Trudel M, Jentsch TJ, Hubner CA. J Clin Invest. 2007;117:1708–17. doi: 10.1172/JCI30630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Paszty C, Brion CM, Manci E, Witkowska HE, Stevens ME, Mohandas N, Rubin EM. Science. 1997;278:876–8. doi: 10.1126/science.278.5339.876. [DOI] [PubMed] [Google Scholar]
- 54.Manci EA, Hillery CA, Bodian CA, Zhang ZG, Lutty GA, Coller BS. Blood. 2006;107:1651–8. doi: 10.1182/blood-2005-07-2839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rao KR, Patel AR, McGinnis P, Patel MK. J Lab Clin Med. 1984;103:792–7. [PubMed] [Google Scholar]
- 56.Ryan TM, Ciavatta DJ, Townes TM. Science. 1997;278:873–6. doi: 10.1126/science.278.5339.873. [DOI] [PubMed] [Google Scholar]
- 57.Chang JC, Lu R, Lin C, Xu SM, Kan YW, Porcu S, Carlson E, Kitamura M, Yang S, Flebbe-Rehwaldt L, Gaensler KM. Proc Natl Acad Sci USA. 1998;95:14886–90. doi: 10.1073/pnas.95.25.14886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Fabry ME, Suzuka SM, Weinberg RS, Lawrence C, Factor SM, Gilman JG, Costantini F, Nagel RL. Blood. 2001;97:410–8. doi: 10.1182/blood.v97.2.410. [DOI] [PubMed] [Google Scholar]
- 59.McKie AT, Barrow D, Latunde-Dada GO, Rolfs A, Sager G, Mudaly E, Mudaly M, Richardson C, Barlow D, Bomford A, Peters TJ, Raja KB, Shirali S, Hediger MA, Farzaneh F, Simpson RJ. Science. 2001;291:1755–9. doi: 10.1126/science.1057206. [DOI] [PubMed] [Google Scholar]
- 60.Gunshin H, Fujiwara Y, Custodio AO, Direnzo C, Robine S, Andrews NC. J Clin Invest. 2005;115:1258–66. doi: 10.1172/JCI24356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Shayeghi M, Latunde-Dada GO, Oakhill JS, Laftah AH, Takeuchi K, Halliday N, Khan Y, Warley A, McCann FE, Hider RC, Frazer DM, Anderson GJ, Vulpe CD, Simpson RJ, McKie AT. Cell. 2005;122:789–801. doi: 10.1016/j.cell.2005.06.025. [DOI] [PubMed] [Google Scholar]
- 62.Wyllie JC, Kaufman N. Lab Invest. 1982;47:471–6. [PubMed] [Google Scholar]
- 63.Donovan A, Brownlie A, Zhou Y, Shepard J, Pratt SJ, Moynihan J, Paw BH, Drejer A, Barut B, Zapata A, Law TC, Brugnara C, Lux SE, Pinkus GS, Pinkus JL, Kingsley PD, Palis J, Fleming MD, Andrews NC, Zon LI. Nature. 2000;403:776–81. doi: 10.1038/35001596. [DOI] [PubMed] [Google Scholar]
- 64.Nemeth E, Tuttle MS, Powelson J, Vaughn MB, Donovan A, Ward DM, Ganz T, Kaplan J. Science. 2004;306:2090–3. doi: 10.1126/science.1104742. [DOI] [PubMed] [Google Scholar]
- 65.Harris ZL, Durley AP, Man TK, Gitlin JD. Proc Natl Acad Sci USA. 1999;96:10812–7. doi: 10.1073/pnas.96.19.10812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Vulpe CD, Kuo YM, Murphy TL, Cowley L, Askwith C, Libina N, Gitschier J, Anderson GJ. Nat Genet. 1999;21:195–9. doi: 10.1038/5979. [DOI] [PubMed] [Google Scholar]
- 67.Cherukuri S, Potla R, Sarkar J, Nurko S, Harris ZL, Fox PL. Cell Metab. 2005;2:309–19. doi: 10.1016/j.cmet.2005.10.003. [DOI] [PubMed] [Google Scholar]
- 68.Craven CM, Alexander J, Eldridge M, Kushner JP, Bernstein S, Kaplan J. Proc Natl Acad Sci USA. 1987;84:3457–61. doi: 10.1073/pnas.84.10.3457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Beutler E, Gelbart T, Lee P, Trevino R, Fernandez MA, Fairbanks VF. Blood. 2000;96:4071–4. [PubMed] [Google Scholar]
- 70.Levy JE, Jin O, Fujiwara Y, Kuo F, Andrews NC. Nat Genet. 1999;21:396–9. doi: 10.1038/7727. [DOI] [PubMed] [Google Scholar]
- 71.Ohgami RS, Campagna DR, McDonald A, Fleming MD. Blood. 2006;108:1388–94. doi: 10.1182/blood-2006-02-003681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ohgami RS, Campagna DR, Greer EL, Antiochos B, McDonald A, Chen J, Sharp JJ, Fujiwara Y, Barker JE, Fleming MD. Nat Genet. 2005;37:1264–9. doi: 10.1038/ng1658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Shaw GC, Cope JJ, Li L, Corson K, Hersey C, Ackermann GE, Gwynn B, Lambert AJ, Wingert RA, Traver D, Trede NS, Barut BA, Zhou Y, Minet E, Donovan A, Brownlie A, Balzan R, Weiss MJ, Peters LL, Kaplan J, Zon LI, Paw BH. Nature. 2006;440:96–100. doi: 10.1038/nature04512. [DOI] [PubMed] [Google Scholar]
- 74.Quigley JG, Yang Z, Worthington MT, Phillips JD, Sabo KM, Sabath DE, Berg CL, Sassa S, Wood BL, Abkowitz JL. Cell. 2004;118:757–66. doi: 10.1016/j.cell.2004.08.014. [DOI] [PubMed] [Google Scholar]
- 75.Keel SB, Doty RT, Yang Z, Quigley JG, Chen J, Knoblaugh S, Kingsley PD, De Domenico I, Vaughn MB, Kaplan J, Palis J, Abkowitz JL. Science. 2008;319:825–8. doi: 10.1126/science.1151133. [DOI] [PubMed] [Google Scholar]
- 76.Pootrakul P, Josephson B, Huebers HA, Finch CA. Blood. 1988;71:1120–3. [PubMed] [Google Scholar]
- 77.Adams PC, Powell LW, Halliday JW. Hepatology. 1988;8:719–21. doi: 10.1002/hep.1840080402. [DOI] [PubMed] [Google Scholar]
- 78.Anderson GJ, Frazer DM. Semin Liver Dis. 2005;25:420–32. doi: 10.1055/s-2005-923314. [DOI] [PubMed] [Google Scholar]
- 79.Kristiansen M, Graversen JH, Jacobsen C, Sonne O, Hoffman HJ, Law SK, Moestrup SK. Nature. 2001;409:198–201. doi: 10.1038/35051594. [DOI] [PubMed] [Google Scholar]
- 80.Hvidberg V, Maniecki MB, Jacobsen C, Hojrup P, Moller HJ, Moestrup SK. Blood. 2005;106:2572–9. doi: 10.1182/blood-2005-03-1185. [DOI] [PubMed] [Google Scholar]
- 81.Pootrakul P, Breuer W, Sametband M, Sirankapracha P, Hershko C, Cabantchik ZI. Blood. 2004;104:1504–10. doi: 10.1182/blood-2004-02-0630. [DOI] [PubMed] [Google Scholar]
- 82.Chua AC, Olynyk JK, Leedman PJ, Trinder D. Blood. 2004;104:1519–25. doi: 10.1182/blood-2003-11-3872. [DOI] [PubMed] [Google Scholar]
- 83.Baker E, Baker SM, Morgan EH. Biochim Biophys Acta. 1998;1380:21–30. doi: 10.1016/s0304-4165(97)00120-7. [DOI] [PubMed] [Google Scholar]
- 84.Liuzzi JP, Lichten LA, Rivera S, Blanchard RK, Aydemir TB, Knutson MD, Ganz T, Cousins RJ. Proc Natl Acad Sci USA. 2005;102:6843–8. doi: 10.1073/pnas.0502257102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Liuzzi JP, Aydemir F, Nam H, Knutson MD, Cousins RJ. Proc Natl Acad Sci USA. 2006;103:13612–7. doi: 10.1073/pnas.0606424103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Roudkenar MH, Halabian R, Oodi A, Roushandeh AM, Yaghmai P, Najar MR, Amirizadeh N, Shokrgozar MA. Arch Med Res. 2008;39:402–7. doi: 10.1016/j.arcmed.2007.12.002. [DOI] [PubMed] [Google Scholar]
- 87.Schmidt-Ott KM, Mori K, Li JY, Kalandadze A, Cohen DJ, Devarajan P, Barasch J. J Am Soc Nephrol. 2007;18:407–13. doi: 10.1681/ASN.2006080882. [DOI] [PubMed] [Google Scholar]
- 88.Oudit GY, Sun H, Trivieri MG, Koch SE, Dawood F, Ackerley C, Yazdanpanah M, Wilson GJ, Schwartz A, Liu PP, Backx PH. Nat Med. 2003;9:1187–94. doi: 10.1038/nm920. [DOI] [PubMed] [Google Scholar]
- 89.Ganz T. Isr Med Assoc J. 2002;4:1043–5. [PubMed] [Google Scholar]
- 90.Wang F, Paradkar PN, Custodio AO, McVey Ward D, Fleming MD, Campagna D, Roberts KA, Boyartchuk V, Dietrich WF, Kaplan J, Andrews NC. Nat Genet. 2007;39:1025–32. doi: 10.1038/ng2059. [DOI] [PubMed] [Google Scholar]
- 91.Nicolas G, Bennoun M, Devaux I, Beaumont C, Grandchamp B, Kahn A, Vaulont S. Proc Natl Acad Sci USA. 2001;98:8780–5. doi: 10.1073/pnas.151179498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Fleming RE, Sly WS. Proc Natl Acad Sci USA. 2001;98:8160–2. doi: 10.1073/pnas.161296298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Nicolas G, Bennoun M, Porteu A, Mativet S, Beaumont C, Grandchamp B, Sirito M, Sawadogo M, Kahn A, Vaulont S. Proc Natl Acad Sci USA. 2002;99:4596–601. doi: 10.1073/pnas.072632499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Nicolas G, Chauvet C, Viatte L, Danan JL, Bigard X, Devaux I, Beaumont C, Kahn A, Vaulont S. J Clin Invest. 2002;110:1037–44. doi: 10.1172/JCI15686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.De Domenico I, Ward DM, Musci G, Kaplan J. Blood. 2007;109:2205–9. doi: 10.1182/blood-2006-06-032516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.De Domenico I, Ward DM, Langelier C, Vaughn MB, Nemeth E, Sundquist WI, Ganz T, Musci G, Kaplan J. Mol Biol Cell. 2007;18:2569–78. doi: 10.1091/mbc.E07-01-0060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Park CH, Valore EV, Waring AJ, Ganz T. J Biol Chem. 2001;276:7806–10. doi: 10.1074/jbc.M008922200. [DOI] [PubMed] [Google Scholar]
- 98.Pigeon C, Ilyin G, Courselaud B, Leroyer P, Turlin B, Brissot P, Loreal O. J Biol Chem. 2001;276:7811–9. doi: 10.1074/jbc.M008923200. [DOI] [PubMed] [Google Scholar]
- 99.Paradkar P, De Domenico I, Durchfort N, Zohn I, Kaplan J, Ward DM. Blood. 2008 doi: 10.1182/blood-2007-12-126854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Roy CN, Andrews NC. Curr Opin Hematol. 2005;12:107–11. doi: 10.1097/00062752-200503000-00001. [DOI] [PubMed] [Google Scholar]
- 101.Wrighting DM, Andrews NC. Blood. 2006;108:3204–9. doi: 10.1182/blood-2006-06-027631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Pietrangelo A, Dierssen U, Valli L, Garuti C, Rump A, Corradini E, Ernst M, Klein C, Trautwein C. Gastroenterology. 2007;132:294–300. doi: 10.1053/j.gastro.2006.10.018. [DOI] [PubMed] [Google Scholar]
- 103.Verga Falzacappa MV, Vujic Spasic M, Kessler R, Stolte J, Hentze MW, Muckenthaler MU. Blood. 2007;109:353–8. doi: 10.1182/blood-2006-07-033969. [DOI] [PubMed] [Google Scholar]
- 104.Weizer-Stern O, Adamsky K, Margalit O, Ashur-Fabian O, Givol D, Amariglio N, Rechavi G. Br J Haematol. 2007;138:253–62. doi: 10.1111/j.1365-2141.2007.06638.x. [DOI] [PubMed] [Google Scholar]
- 105.Courselaud B, Pigeon C, Inoue Y, Inoue J, Gonzalez FJ, Leroyer P, Gilot D, Boudjema K, Guguen-Guillouzo C, Brissot P, Loreal O, Ilyin G. J Biol Chem. 2002;277:41163–70. doi: 10.1074/jbc.M202653200. [DOI] [PubMed] [Google Scholar]
- 106.Pinto JP, Ribeiro S, Pontes H, Thowfeequ S, Tosh D, Carvalho F, Porto G. Blood. 2008 doi: 10.1182/blood-2007-08-106195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Lin L, Valore EV, Nemeth E, Goodnough JB, Gabayan V, Ganz T. Blood. 2007 doi: 10.1182/blood-2007-04-087593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Feder JN, Gnirke A, Thomas W, Tsuchihashi Z, Ruddy DA, Basava A, Dormishian F, Domingo R, Jr, Ellis MC, Fullan A, Hinton LM, Jones NL, Kimmel BE, Kronmal GS, Lauer P, Lee VK, Loeb DB, Mapa FA, McClelland E, Meyer NC, Mintier GA, Moeller N, Moore T, Morikang E, Prass CE, Quintana L, Starnes SM, Schatzman RC, Brunke KJ, Drayna DT, Risch NJ, Backon BR, Wolff RK. Nat Genet. 1996;13:399–408. doi: 10.1038/ng0896-399. [DOI] [PubMed] [Google Scholar]
- 109.de Almeida SF, Carvalho IF, Cardoso CS, Cordeiro JV, Azevedo JE, Neefjes J, de Sousa M. Blood. 2005;106:971–7. doi: 10.1182/blood-2004-12-4640. [DOI] [PubMed] [Google Scholar]
- 110.Roy CN, Penny DM, Feder JN, Enns CA. J Biol Chem. 1999;274:9022–8. doi: 10.1074/jbc.274.13.9022. [DOI] [PubMed] [Google Scholar]
- 111.Lebron JA, West AP, Jr, Bjorkman PJ. J Mol Biol. 1999;294:239–45. doi: 10.1006/jmbi.1999.3252. [DOI] [PubMed] [Google Scholar]
- 112.Goswami T, Andrews NC. J Biol Chem. 2006;281:28494–8. doi: 10.1074/jbc.C600197200. [DOI] [PubMed] [Google Scholar]
- 113.Zhou XY, Tomatsu S, Fleming RE, Parkkila S, Waheed A, Jiang J, Fei Y, Brunt EM, Ruddy DA, Prass CE, Schatzman RC, O’Neill R, Britton RS, Bacon BR, Sly WS. Proc Natl Acad Sci USA. 1998;95:2492–7. doi: 10.1073/pnas.95.5.2492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Tomatsu S, Orii KO, Fleming RE, Holden CC, Waheed A, Britton RS, Gutierrez MA, Velez-Castrillon S, Bacon BR, Sly WS. Proc Natl Acad Sci USA. 2003;100:15788–93. doi: 10.1073/pnas.2237037100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Levy JE, Montross LK, Cohen DE, Fleming MD, Andrews NC. Blood. 1999;94:9–11. [PubMed] [Google Scholar]
- 116.Gehrke SG, Kulaksiz H, Herrmann T, Riedel HD, Bents K, Veltkamp C, Stremmel W. Blood. 2003;102:371–6. doi: 10.1182/blood-2002-11-3610. [DOI] [PubMed] [Google Scholar]
- 117.Bridle KR, Frazer DM, Wilkins SJ, Dixon JL, Purdie DM, Crawford DH, Subramaniam VN, Powell LW, Anderson GJ, Ramm GA. Lancet. 2003;361:669–73. doi: 10.1016/S0140-6736(03)12602-5. [DOI] [PubMed] [Google Scholar]
- 118.Piperno A, Girelli D, Nemeth E, Trombini P, Bozzini C, Poggiali E, Phung Y, Ganz T, Camaschella C. Blood. 2007;110(12):4096–4100. doi: 10.1182/blood-2007-06-096503. [DOI] [PubMed] [Google Scholar]
- 119.Valore EV, Ganz T. Blood Cells Mol Dis. 2008;40:132–8. doi: 10.1016/j.bcmd.2007.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Nicolas G, Viatte L, Lou DQ, Bennoun M, Beaumont C, Kahn A, Andrews NC, Vaulont S. Nat Genet. 2003;34:97–101. doi: 10.1038/ng1150. [DOI] [PubMed] [Google Scholar]
- 121.Kawabata H, Yang R, Hirama T, Vuong PT, Kawano S, Gombart AF, Koeffler HP. J Biol Chem. 1999;274:20826–32. doi: 10.1074/jbc.274.30.20826. [DOI] [PubMed] [Google Scholar]
- 122.Zhang AS, Xiong S, Tsukamoto H, Enns CA. Blood. 2004;103:1509–14. doi: 10.1182/blood-2003-07-2378. [DOI] [PubMed] [Google Scholar]
- 123.Camaschella C, Roetto A, Cali A, De Gobbi M, Garozzo G, Carella M, Majorano N, Totaro A, Gasparini P. Nat Genet. 2000;25:14–5. doi: 10.1038/75534. [DOI] [PubMed] [Google Scholar]
- 124.Pietrangelo A. N Engl J Med. 2004;350:2383–97. doi: 10.1056/NEJMra031573. [DOI] [PubMed] [Google Scholar]
- 125.Rouault TA. Nat Chem Biol. 2006;2:406–14. doi: 10.1038/nchembio807. [DOI] [PubMed] [Google Scholar]
- 126.Schmidt PJ, Toran PT, Giannetti AM, Bjorkman PJ, Andrews NC. Cell Metab. 2008;7:205–14. doi: 10.1016/j.cmet.2007.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Kawabata H, Fleming RE, Gui D, Moon SY, Saitoh T, O’Kelly J, Umehara Y, Wano Y, Said JW, Koeffler HP. Blood. 2005;105:376–81. doi: 10.1182/blood-2004-04-1416. [DOI] [PubMed] [Google Scholar]
- 128.Nemeth E, Roetto A, Garozzo G, Ganz T, Camaschella C. Blood. 2005;105:1803–6. doi: 10.1182/blood-2004-08-3042. [DOI] [PubMed] [Google Scholar]
- 129.Papanikolaou G, Samuels ME, Ludwig EH, MacDonald ML, Franchini PL, Dube MP, Andres L, MacFarlane J, Sakellaropoulos N, Politou M, Nemeth E, Thompson J, Risler JK, Zaborowska C, Babakaiff R, Radomski CC, Pape TD, Davidas O, Christakis J, Brissot P, Lockitch G, Ganz T, Hayden MR, Goldberg YP. Nat Genet. 2004;36:77–82. doi: 10.1038/ng1274. [DOI] [PubMed] [Google Scholar]
- 130.Wang RH, Li C, Xu X, Zheng Y, Xiao C, Zerfas P, Cooperman S, Eckhaus M, Rouault T, Mishra L, Deng CX. Cell Metab. 2005;2:399–409. doi: 10.1016/j.cmet.2005.10.010. [DOI] [PubMed] [Google Scholar]
- 131.Babitt JL, Huang FW, Wrighting DM, Xia Y, Sidis Y, Samad TA, Campagna JA, Chung RT, Schneyer AL, Woolf CJ, Andrews NC, Lin HY. Nat Genet. 2006;38:531–9. doi: 10.1038/ng1777. [DOI] [PubMed] [Google Scholar]
- 132.Babitt JL, Huang FW, Xia Y, Sidis Y, Andrews NC, Lin HY. J Clin Invest. 2007;117:1933–9. doi: 10.1172/JCI31342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Lin L, Goldberg YP, Ganz T. Blood. 2005;106:2884–9. doi: 10.1182/blood-2005-05-1845. [DOI] [PubMed] [Google Scholar]
- 134.Roetto A, Papanikolaou G, Politou M, Alberti F, Girelli D, Christakis J, Loukopoulos D, Camaschella C. Nat Genet. 2003;33:21–2. doi: 10.1038/ng1053. [DOI] [PubMed] [Google Scholar]
- 135.Roetto A, Daraio F, Porporato P, Caruso R, Cox TM, Cazzola M, Gasparini P, Piperno A, Camaschella C. Blood. 2004;103:2407–9. doi: 10.1182/blood-2003-10-3390. [DOI] [PubMed] [Google Scholar]
- 136.Pietrangelo A. Blood Cells Mol Dis. 2004;32:131–8. doi: 10.1016/j.bcmd.2003.08.003. [DOI] [PubMed] [Google Scholar]
- 137.De Domenico I, McVey Ward D, Nemeth E, Ganz T, Corradini E, Ferrara F, Musci G, Pietrangelo A, Kaplan J. Haematologica. 2006;91:1092–5. [PMC free article] [PubMed] [Google Scholar]
- 138.De Domenico I, Ward DM, Nemeth E, Vaughn MB, Musci G, Ganz T, Kaplan J. Proc Natl Acad Sci USA. 2005;102:8955–60. doi: 10.1073/pnas.0503804102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Drakesmith H, Schimanski LM, Ormerod E, Merryweather-Clarke AT, Viprakasit V, Edwards JP, Sweetland E, Bastin JM, Cowley D, Chinthammitr Y, Robson KJ, Townsend AR. Blood. 2005;106:1092–7. doi: 10.1182/blood-2005-02-0561. [DOI] [PubMed] [Google Scholar]
- 140.Schimanski LM, Drakesmith H, Merryweather-Clarke AT, Viprakasit V, Edwards JP, Sweetland E, Bastin JM, Cowley D, Chinthammitr Y, Robson KJ, Townsend AR. Blood. 2005;105:4096–102. doi: 10.1182/blood-2004-11-4502. [DOI] [PubMed] [Google Scholar]
- 141.De Domenico I, Vaughn MB, Yoon D, Kushner JP, Ward DM, Kaplan J. Blood. 2007;110(10):3780.3. doi: 10.1182/blood-2007-07-100248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Corradini E, Montosi G, Ferrara F, Caleffi A, Pignatti E, Barelli S, Garuti C, Pietrangelo A. Blood Cells Mol Dis. 2005;35(3):315–318. doi: 10.1016/j.bcmd.2005.07.010. [DOI] [PubMed] [Google Scholar]
- 143.Papanikolaou G, Tzilianos M, Christakis JI, Bogdanos D, Tsimirika K, MacFarlane J, Goldberg YP, Sakellaropoulos N, Ganz T, Nemeth E. Blood. 2005;105:4103–5. doi: 10.1182/blood-2004-12-4844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Esposito BP, Breuer W, Sirankapracha P, Pootrakul P, Hershko C, Cabantchik ZI. Blood. 2003;102:2670–7. doi: 10.1182/blood-2003-03-0807. [DOI] [PubMed] [Google Scholar]
- 145.Bannerman RM, Keusch G, Kreimer-Birnbaum M, Vance VK, Vaughan S. Am J Med. 1967;42:476–86. doi: 10.1016/0002-9343(67)90276-8. [DOI] [PubMed] [Google Scholar]
- 146.Cossu P, Toccafondi C, Vardeu F, Sanna G, Frau F, Lobrano R, Cornacchia G, Nucaro A, Bertolino F, Loi A, De Virgiliis S, Cao A. Eur J Pediatr. 1981;137:267–71. doi: 10.1007/BF00443255. [DOI] [PubMed] [Google Scholar]
- 147.Fiorelli G, Fargion S, Piperno A, Battafarano N, Cappellini MD. Haematologica. 1990;75:89–95. [PubMed] [Google Scholar]
- 148.Piperno A, Mariani R, Arosio C, Vergani A, Bosio S, Fargion S, Sampietro M, Girelli D, Fraquelli M, Conte D, Fiorelli G, Camaschella C. Br J Haematol. 2000;111:908–14. [PubMed] [Google Scholar]
- 149.Hershko C, Rachmilewitz EA. Br J Haematol. 1979;42:125–32. doi: 10.1111/j.1365-2141.1979.tb03704.x. [DOI] [PubMed] [Google Scholar]
- 150.Erlandson ME, Walden B, Stern G, Hilgartner MW, Wehman J, Smith CH. Blood. 1962;19:359–78. [PubMed] [Google Scholar]
- 151.Libani IV, Guy EC, Melchiori L, Schiro R, Ramos P, Breda L, Scholzen T, Chadburn A, Liu Y, Kernbach M, Baron-Luhr B, Porotto M, de Sousa M, Rachmilewitz EA, Hood JD, Cappellini MD, Giardina PJ, Grady RW, Gerdes J, Rivella S. Blood. 2008;112:875–85. doi: 10.1182/blood-2007-12-126938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Finch CA, Sturgeon P. Blood. 1957;12:64–73. [PubMed] [Google Scholar]
- 153.Pootrakul P, Kitcharoen K, Yansukon P, Wasi P, Fucharoen S, Charoenlarp P, Brittenham G, Pippard MJ, Finch CA. Blood. 1988;71:1124–9. [PubMed] [Google Scholar]
- 154.Rees DC, Williams TN, Maitland K, Clegg JB, Weatherall DJ. Br J Haematol. 1998;103:365–9. doi: 10.1046/j.1365-2141.1998.00971.x. [DOI] [PubMed] [Google Scholar]
- 155.Vichinsky E, Kleman K, Embury S, Lubin B. Blood. 1981;58:963–8. [PubMed] [Google Scholar]
- 156.Finch C. Blood. 1994;84:1697–702. [PubMed] [Google Scholar]
- 157.Ganz T. Curr Top Microbiol Immunol. 2006;306:183–98. doi: 10.1007/3-540-29916-5_7. [DOI] [PubMed] [Google Scholar]
- 158.Nicolas G, Viatte L, Bennoun M, Beaumont C, Kahn A, Vaulont S. Blood Cells Mol Dis. 2002;29:327–35. doi: 10.1006/bcmd.2002.0573. [DOI] [PubMed] [Google Scholar]
- 159.Vokurka M, Necas E. Cas Lek Cesk. 2003;142:465–9. [PubMed] [Google Scholar]
- 160.Pak M, Lopez MA, Gabayan V, Ganz T, Rivera S. Blood. 2006;108:3730–5. doi: 10.1182/blood-2006-06-028787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Weizer-Stern O, Adamsky K, Amariglio N, Levin C, Koren A, Breuer W, Rachmilewitz E, Breda L, Rivella S, Cabantchik ZI, Rechavi G. Br J Haematol. 2006;135:129–38. doi: 10.1111/j.1365-2141.2006.06258.x. [DOI] [PubMed] [Google Scholar]
- 162.Tanno T, Bhanu NV, Oneal PA, Goh SH, Staker P, Lee YT, Moroney JW, Reed CH, Luban NL, Wang RH, Eling TE, Childs R, Ganz T, Leitman SF, Fucharoen S, Miller JL. Nat Med. 2007;13(9):1096–1101. doi: 10.1038/nm1629. [DOI] [PubMed] [Google Scholar]
- 163.Kannan K, Amariglio N, Rechavi G, Givol D. FEBS Lett. 2000;470:77–82. doi: 10.1016/s0014-5793(00)01291-6. [DOI] [PubMed] [Google Scholar]
- 164.Kannan K, Amariglio N, Rechavi G, Jakob-Hirsch J, Kela I, Kaminski N, Getz G, Domany E, Givol D. Oncogene. 2001;20:2225–34. doi: 10.1038/sj.onc.1204319. [DOI] [PubMed] [Google Scholar]
- 165.Tan M, Wang Y, Guan K, Sun Y. Proc Natl Acad Sci USA. 2000;97:109–14. doi: 10.1073/pnas.97.1.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Yoon D, Pastore YD, Divoky V, Liu E, Mlodnicka AE, Rainey K, Ponka P, Semenza GL, Schumacher A, Prchal JT. J Biol Chem. 2006;281:25703–11. doi: 10.1074/jbc.M602329200. [DOI] [PubMed] [Google Scholar]
- 167.Peyssonnaux C, Zinkernagel AS, Schuepbach RA, Rankin E, Vaulont S, Haase VH, Nizet V, Johnson RS. J Clin Invest. 2007;117:1926–32. doi: 10.1172/JCI31370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Adamsky K, Weizer O, Amariglio N, Breda L, Harmelin A, Rivella S, Rachmilewitz E, Rechavi G. Br J Haematol. 2004;124:123–4. doi: 10.1046/j.1365-2141.2003.04734.x. [DOI] [PubMed] [Google Scholar]
- 169.Weizer O, Adamsky K, Breda L, Cabantchik I, Rachmilewitz E, Breuer W, Harmelin A, Levin C, Koren A, Amariglio N, Rivella S, Rechavi G. The American Society of Hematology 46th Annual Meeting; San Diego. 2004. [Google Scholar]
- 170.De Franceschi L, Daraio F, Filippini A, Carturan S, Muchitsch EM, Roetto A, Camaschella C. Haematologica. 2006;91:1336–42. [PubMed] [Google Scholar]
- 171.Weizer-Stern O, Adamsky K, Amariglio N, Rachmilewitz E, Breda L, Rivella S, Rechavi G. Am J Hematol. 2006;81:479–83. doi: 10.1002/ajh.20549. [DOI] [PubMed] [Google Scholar]
- 172.Kattamis A, Papassotiriou I, Palaiologou D, Apostolakou F, Galani A, Ladis V, Sakellaropoulos N, Papanikolaou G. Haematologica. 2006;91:809–12. [PubMed] [Google Scholar]
- 173.Kearney SL, Nemeth E, Neufeld EJ, Thapa D, Ganz T, Weinstein DA, Cunningham MJ. Pediatr Blood Cancer. 2005;48(1):57–63. doi: 10.1002/pbc.20616. [DOI] [PubMed] [Google Scholar]
- 174.Origa R, Galanello R, Ganz T, Giagu N, Maccioni L, Faa G, Nemeth E. Haematologica. 2007;92:583–8. doi: 10.3324/haematol.10842. [DOI] [PubMed] [Google Scholar]
- 175.Faye A, Ramey G, Foretz M, Vaulont S. Blood Cells Mol Dis. 2007;39(3):229–237. doi: 10.1016/j.bcmd.2007.05.008. [DOI] [PubMed] [Google Scholar]
- 176.Morra S, Barisani D, Chiabrando D, Fagoonee S, Muckenthaler M, Stolte J, Meneveri R, Haile D, Silengo L, Altruda F, Tolosano T. Am J Hematol; Bio-Iron Abstracts. 2007:82. doi: 10.1053/j.gastro.2007.07.004. [DOI] [PubMed] [Google Scholar]
- 177.Breda L, Gardenghi S, Guy E, Rachmilewitz EA, Weizer-Stern O, Adamsky K, Amariglio N, Rechavi G, Giardina PJ, Grady RW, Rivella S. Ann NY Acad Sci. 2005;1054:417–22. doi: 10.1196/annals.1345.069. [DOI] [PubMed] [Google Scholar]
- 178.Rivera S, Nemeth E, Gabayan V, Lopez MA, Farshidi D, Ganz T. Blood. 2005;106:2196–9. doi: 10.1182/blood-2005-04-1766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Roy CN, Mak HH, Akpan I, Losyev G, Zurakowski D, Andrews NC. Blood. 2007;109:4038–44. doi: 10.1182/blood-2006-10-051755. [DOI] [PMC free article] [PubMed] [Google Scholar]