

Abstract
Stem cell repair shows substantial translational potential for neurological injury, but the mechanisms of action remain unclear. This study aimed to investigate whether transplanted stem cells could induce comprehensive functional remyelination. Subventricular zone (SVZ)-derived adult neural precursor cells (aNPCs) were injected bilaterally into major cerebral white matter tracts of myelin-deficient shiverer mice on postnatal day (P) 0, P7, and P21. Tripotential NPCs, when transplanted in vivo, integrated anatomically and functionally into local white matter and preferentially became Olig2+, Myelin Associated Glycoprotein-positive, Myelin Basic Protein-positive oligodendrocytes, rather than Glial Fibrillary Acidic Protein-positive astrocytes or Neurofiliment 200-positive neurons. Processes interacted with axons and transmission electron microscopy showed multilamellar axonal ensheathment. Nodal architecture was restored and by quantifying these anatomical parameters a computer model was generated that accurately predicted action potential velocity, determined by ex vivo slice recordings. Although there was no obvious phenotypic improvement in transplanted shi/shis, myelinated axons exhibited faster conduction, lower activation threshold, less refractoriness, and improved response to high-frequency stimulation than dysmyelinated counterparts. Furthermore, they showed improved resilience to ischemic insult, a promising finding in the context of perinatal brain injury. This study describes, for the first time mechanistically, the functional characteristics and anatomical integration of nonimmortalized donor SVZ-derived murine aNPCs in the dysmyelinated brain at key developmental time points.
Introduction
Complex interactions between demyelination, axonal dysfunction and disruption, and subsequent neuromotor impairment, are characteristic of a myriad of neurological conditions, including cerebral palsy (CP), stroke, spinal cord injury (SCI), and multiple sclerosis (Lazzarini, 2004). Neural precursor cells (NPCs) show great potential for cellular replacement and remyelination in models of subcortical injury and dysmyelination. Specifically, the highly plastic neonatal microenvironment seems particularly amenable to cell survival. However, significant and thorough description of the functional impact of NPC-induced myelin remains unknown. The most common pediatric neurological and developmental disability, CP, typically presents with selective subcortical white matter death, due to the intrinsic vulnerability of the preoligodendrocyte to ischemic processes. This provides an ideal substrate for translational cell transplantation paradigms. In addition, most studies involving perinatal cell transplant exclusively use perinatal day (P) 0 injection, which offers immune privilege, but very little clinical relevance, as CP is not usually diagnosed until ∼2 years of age (with murine equivalent P21), when affected children fail to reach developmental milestones (Shevell and Bodensteiner, 2004). Therefore, P7, which is equivalent to human birth, is the earliest feasible translational time point, with P21 the most clinically relevant. A knowledge gap currently exists surrounding the ideal therapeutic window for cell transplantation strategies and, indeed, it is uncertain whether this window overlaps with feasible clinical transplant time points.
Although some preliminary evidence of functional remyelination by various cell types has been shown in previous studies, the functional impact of myelination has never been fully elucidated. Some groups have shown increased signal velocity in transplanted animals; however, none has investigated whether new myelin can normalize other essential biophysical signaling properties, such as stimulation threshold, functional ensheathment, and response to high-frequency stimulation (HFS), which are essential for normal neurological function. Furthermore, the application of a computer simulation model that can accurately predict these signaling profiles represents a first in the field and the first direct link between these structural profiles and active signaling.
The corpus callosum (CC) plays a significant role in interhemispheric transfer and integration of sensorimotor and cognitive information (Caillé et al., 2005) and is impacted early in the course of demyelinating conditions. Due to small axon size and slow action potential propagation in this structure, it is relatively easy to obtain distinctive profiles from myelinated and dysmyelinated axons (Crawford et al., 2009a) at a population level for direct functional comparison.
Here, we explore the knowledge gap between myelination and function by (1) investigating the myelinating capacity of adult cells in the CC, (2) detailing the impact of clinically relevant developmental time points on myelinating capacity, and (3) elucidating in a cogent and thorough manner the mechanistic improvements in conduction that accompany myelination in a shi/shi mouse model. Shi/shi mice possess a deletion mutation in exons III–VII of the gene responsible for compact myelin: Myelin Basic Protein (MBP; Bird et al., 1978; Roach et al., 1985). Resulting CNS axonal dysmyelination makes shi/shi animals an ideal model in which to investigate the myelination potential of transplanted stem cells; since these animals lack intrinsic MBP, its presence can be attributed to enhanced yellow fluorescence protein (eYFP)+ transplanted adult-derived NPCs (aNPCs).
The majority of scientific literature up to this point has observed one or two phenotypic changes with myelination, but has yet to elucidate a causal relationship between the re-establishment of myelin-associated architecture and function. Our validated computer simulation makes this connection and provides evidence that myelination alone is sufficient to cause the functional improvement indicated by compound action potential (CAP) recording. We further present novel findings that such myelination results in the formation of a new axonal group characterized by improved conduction, normalized pharmacological response, and increased resilience to ischemia.
Materials and Methods
Breeding and animal husbandry
A total number of 53 shi/shi mice and 19 wild-type (WT) mice (C3Fe.SWV-Mbpshi/J and C3HeB/FeJ respectively; The Jackson Laboratory), from either sex, were used in this study. Mice were produced from an inbred colony of shi/shi mice, maintained in our facility on a normal light cycle, fed ad libitum, and kept in separately ventilated cages. Parental shiverer animals were bred and filial genotypes were verified by a shivering phenotype and genotype. All experimental protocols were approved by the animal care committee of the University Health Network in accordance with Canadian Council on Animal Care guidelines.
aNPC isolation and processing
aNPCs were obtained from the periventricular, subependymal layer of eYFP-expressing transgenic adult mouse brains [129-Tg (ACTB-EYFP) 2Nagy/J; The Jackson Laboratory], as described previously (Tropepe et al., 1999). Briefly, mice were culled by cervical dislocation, brains were placed, under sterile conditions, into artificial CSF (aCSF; 2 m NaCl, 1 m KCl, 1 m MgCl2, 155 mm NaHCO3, 108 mm CaCl2, 1 m glucose, and 1% penicillin/streptomycin; Sigma-Aldrich), overlying meninges were removed, and subventricular zone was isolated. Tissue was transferred to low-calcium aCSF solution (10 ml) for 1 h in a 37°C incubator solution contained 40 mg of trypsin, 20 mg of hyaluronidase, and 4 mg of kynurenic acid. Tissue was then mechanically dissociated into single cells, viability was measured, and cells were plated at 10 cells/μl, on uncoated tissue culture flasks, in serum-free media (200 ml) containing 20 ml of DMEM/F-12 (10×), 4 ml of 30% glucose, 3 ml of 7.5% NaHCO3, 1 ml of 1 m HEPES, 200 mg of transferrin, 50 mg of insulin, 19.25 mg of putrescine, 20 μl of selenium, 20 μl of progesterone, 1 μg of fibroblast growth medium 2, 2 μg of epidermal growth factor, and 1% penicillin/streptomycin. Cells formed nonadherent “neurosphere” colonies, which were dissociated and passaged weekly for 3–6 passages.
Transplantation of aNPCs
P0, P7, and P21 pups were injected with 250,000 aNPCs [passage 3–6, suspended in serum-free media (SFM)] distributed over five injection sites (50,000 cells per site in 1 μl of SFM). Under ice anesthesia (P0) or isoflurane anesthesia (P7, P21; 5% to induce, 2% to maintain, oxygen at 1 L/min), cells were introduced, using a glass pulled pipette, attached to a 10 μl Hamilton syringe (Hamilton), bilaterally into the anterior and posterior aspects of the CC, as well as into the cerebellar peduncle, or anlagens thereof (as appropriate), using stereotactic co-ordinates from Paxinos et al. (2006). P0 pups were injected through the skin, whereas an incision was made in P7 and P21 animals to expose the skull. Five holes were drilled into the calcified P21 skull, above injection points, to enable needle insertion, whereas the malleable skull of P7 animals allowed for direct needle insertion through the skull. Skin was closed using sutures or tissue glue (Vetbond). After surgery, P0 and P7 pups were returned to either the dam or an ICR foster mother (Jackson) until weaning at P21. Littermate controls were injected with SFM only. Buprenorphine was administered as a postoperative analgesic (0.05–0.1 mg/kg) every 8 h for 48 h.
Animal perfusion for immunohistochemistry
After 45 d, animals were killed with an overdose of pentobarbital and then transcardially perfused with an initial wash using 60 ml of 0.1 m PBS, followed by fixation with 60 ml 4% PFA in 0.1 m PBS, pH 7.4. Brains were immediately extracted and postfixed in 4% PFA in 0.1 m PBS plus 10% sucrose overnight at 4°C and then transferred to 30% sucrose in PBS for minimum of 8 h at 4°C for cryoprotection. Brains were cut in half coronally with a straight razor ∼1 mm anterior of the interaural line, mounted flat side down, embedded in optimum cutting temperature mounting medium (HistoPrep, Fisher Scientific) on dry ice. Brain slices collected after electrophysiological recordings were also postfixed, embedded, and sliced (20 μm) for immunohistochemistry, as described in Crawford et al. (2009a). Tissue was then cut into 20 μm sections onto superfrost slides (Fisher Scientific), three sections per slide, using a Leica CM 3050 cryostat and stored at −80°C for future use.
Immunohistochemistry
Frozen slides were removed from −80°C and tissue sections, at similar levels within the CC, were spread in distilled water. Sections were allowed to dry at an angle in an indirect breeze for 15 min and outlined with liquid blocker (Dako). Tissue was treated as described previously (Eftekharpour et al., 2007). Briefly, slides were washed in PBS for 10 min, blocked in 1% BSA, 5% nonfat milk, and 0.3% Triton X-100 in PBS for 1 h at room temperature and incubated with primary antibody in blocking solution overnight or for a minimum of 8 h at 4°C. Primary antibody concentrations were 1:400 for rabbit anti-MBP (AB980, Millipore), 1:1000 for Mouse anti-Myelin Associated Glycoprotein (anti-MAG; MAB1567, Millipore Bioscience Research Reagents), 1:200 for goat anti-Glial Fibrillary Acidic Protein (anti-GFAP; SAB2500462, Sigma-Aldrich), 1:500 for mouse anti-Neurofilament 200 (anti-NF200; N0142, Sigma-Aldrich), 1:2000 for rabbit anti-contactin associated protein (Caspr; ab34151, Abcam), 1: 200 for mouse anti-Kv1.2 [University of California (UC) Davis/National Institutes of Health (NIH) NeuroMab Facility, Davis, California], 1:200 for mouse anti-Nav1.6 (UC Davis/NIH NeuroMab Facility), and 1:400 for chicken anti-GFP (GFP-1020, Aves Labs). Slides were then washed three times in PBS and incubated with appropriate fluorescent secondary antibody (anti-goat or anti-donkey Alexa 350, 488, 594, or 647, 1:300; Invitrogen; or anti-chicken 488; Jackson ImmunoResearch) for 1 h. For multiple labeling experiments, slides were reblocked following secondary application and the aforementioned protocol was repeated. Before applying mouse monoclonal primary antibodies, slides were blocked for at least 2 h at room temperature with 1:10 mouse IgG in blocking serum. Slides were washed three times with 0.1 m PBS and mounted in Mowiol mounting medium (Polysciences). Images were captured on a Zeiss LSM 510 laser confocal microscope and analyzed using LSM510 software and StereoInvestigator (with associated multimeasure jar, http://www.optinav.com/Multi-Measure.htm).
YFP quantification
Brains into which cells had successfully incorporated (YFP+ cell presence; n = 40 of 53) were used for immunohistochemistry and electron microscopy. Only samples containing >50 cells per coronal section in two sections at least 100 μm apart were used for quantification. To characterize the differentiation capacity and phenotype of transplanted cells, we gauged GFAP (astrocyte), MAG [immature oligodendrocyte (OL)], MBP (mature, myelinating OL), and NF200 (neuronal) marker presence and compared them to YFP immunoreactivity, quantifying double-labeled cells using confocal microscopy.
Quantification of cell differentiation was performed in an unbiased stereological manner according to the stereological principals originally described by Konigsmark and Murphy (Konigsmark and Murphy, 1970) and used previously by our group (Eftekharpour et al., 2007; Karimi-Abdolrezaee et al., 2006, 2010). Briefly, we chose the brain sections with the highest number of YFP-positive cells from three animals (n = 3 per group). For each cell marker, we immunostained every tenth slide per mouse (for example, slides 1, 11, 21, 31 for MBP/NF200, and slides 2, 12, 22, 32, etc. for MAG/GFAP). Slides were then analyzed for the area of GFP within the CC (takes into account the processes) divided by the total area of the CC to give the area of GFP+ cells found within the CC. Sections were analyzed at 20× magnification. For quantification of nodes of Ranvier, the number of node-like structures double labeled with Nav1.6 and Caspr or Caspr and Kv1.2 subunits in WT, control shi/shi, and transplanted shi/shi (5 fields per animal; n = 3 per group) were counted using confocal images obtained from the dorsal and lateral white matter tracts. Data were presented as nodal density (number of nodes/100 μm2).
Quantification of NPC differentiation
To quantify the differentiation pattern of engrafted cells (n = 9), we immunostained coronal 20 μm sections containing YFP+ cells. For each cell marker (NeuN for neurons, GFAP for activated astroglia, and Olig2 for OLs), we randomly selected three tissue sections per animal. Using images taken by confocal microscopy at 60× primary magnification, we counted the number of YFP+/DAPI+ cells in five random fields per section. We also counted the YFP+ cells that were colabeled with the cell marker. We only quantified the YFP+ cell bodies that contained a nucleus (identified with DAPI). Then, we calculated the percentage of colabeled YFP+/cell marker+ cells.
Electron microscopy
For electron microscopy, animals were transcardially perfused with 60 ml of 4% PFA and 0.15% glutaraldehyde in phosphate buffer (PB). Brains were dissected and stored in this solution. Sections of CC were isolated from transplanted animals by presence of eYFP fluorescent signal (to isolate cell-specific sections), and equivalent sections were taken from control shiverer and WT brains. Isolated tissue was treated with 1% osmium tetroxide in 0.1 m PB for 1 h at room temperature and then dehydrated in graded ethanol solutions. Sections were embedded in Araldite 502/Embed-812 embedding media (Electron Microscopy Sciences) and ultrathin (80 nm) sections were cut using a ultramicrotome, counterstained with uranyl acetate and lead citrate, and examined with a transmission electron microscope (model 7000, Hitachi).
Computer modeling
To estimate the effect of myelin layer change after aNPC transplantation, the number of myelin layers was measured from transmission electron microscopy (TEM) photomicrographs by calculating the total myelin thickness of n = 3 animals per group at 2–3 fields per animal. This was done by measuring the total myelin thickness using ImageJ software and dividing by the thickness of a single lamellae for each EM image. McIntyre et al.'s (McIntyre et al., 2002) model of a mammalian myelinated axon, adapted from the database archive of the NEURON simulation environment (http://senselab.med.yale.edu/senselab/modeldb/), was implemented to estimate the effects of observed changes in myelin thickness. Briefly, the axon is modeled as a multicompartmental double cable, with separate representations of the axolemma and the myelin sheath. Nodes of Ranvier, paranodal myelin attachment areas, paranodal main segments, and internodal regions are included as separate compartments with different geometry and electrical properties. Action potentials are generated at the nodes with modified Hodgkin–Huxley equations that incorporate nonlinear fast Na+ conductance, persistent Na+ conductance, slow K+ conductance, a linear leak conductance, and membrane capacitance. Internodes have no active conductance and the axolemma and myelin sheath in paranodal and internodal segments each have a passive linear conductance in parallel with membrane capacitance. Myelin capacitance is represented as a function of capacitance per unit area and myelin thickness. Model axons propagated an action potential in response to a suprathreshold, depolarizing current step delivered to the node at one end. Model parameters were modified, using published data, to represent axons in the CC (Tables 1, 2). Node-to-node distance was modified to be 79.1 μm (Bakiri et al., 2011) and the overall length of simulated axon is 7.91 mm (including 101 nodes). Conduction velocity (CV) was measured as the distance between the first and 101st nodes, divided by the action potential conduction time between those nodes. All simulations were run in NEURON (Hines and Carnevale, 1997).
Table 1.
Model geometric parametersa
| Axon diameter | 0.36 μm |
| Myelin diameter | 0.48 μm |
| Node length | 1 μm |
| Internode length | 79.1 μm |
| Number of myelin layers | 1–40 (set to 1 for demyelinated axon) |
| Node diameter | 0.36 μm |
| MYSA length | 3 μm |
| MYSA diameter | 0.36 μm |
| MYSA periaxonal space width | 0.002 μm |
| FLUT length | 16 μm |
| FLUT diameter | 0.36 μm |
| FLUT periaxonal space width | 0.004 μm |
| STIN diameter | 0.36 μm |
| STIN periaxonal space width | 0.004 μm |
a Based on McIntyre et al. (2002), and Bakiri et al. (2011).
MYSA, Myelin sheath attachment axon segment; FLUT, fluted paranodal axon segment; STIN, stereotype intermodel region.
Table 2.
Model electrophysiological parametersa
| Nodal Na+ conductance | 3000 mS/cm−2 |
| Nodal K+ conductance | 80 mS/cm−2 |
| Node leakage conductance | 80 mS/cm−2 |
| Myelin membrane conductance | 0.1 mS/cm−2 |
| Axon membrane capacitance | |
| Node | 0.9 μF/cm−2 |
| Internode | 0.9 μF/cm−2 |
| Myelin membrane capacitance | 0.9 μF/cm−2 |
| Axoplasmic resistivity | 70 Ω/cm |
| Periaxonal resistivity | 70 Ω/cm |
| Resting potential | 82 mV |
| Leakage potential | 83.38 mV |
| Na+ reversal potential | 50 mV |
| K+ reversal potential | −84 mV |
aBased on Bakiri et al. (2011).
Transcallosal stimulation
Brain slice preparation.
Coronal brain slices from WT, shi/shi, and aNPC-transplanted shi/shi groups were obtained for electrophysiological recording of CAPs. Mice were terminally anesthetized intraperitoneally by ketamine (10 mg/kg) administration. Transcardial perfusion was performed with ice-cold oxygenated (95% O2 plus 5% CO2) glucose-based aCSF, since hypothermia can reduce traumatic and hypoxic perturbations that are associated with dissection/decapitation. This normal aCSF contained (in mm) the following: 120 NaCl, 2.5 KCl, 2 CaCl2, 2 MgCl2, 25 NaHCO3, and 10 d-glucose, pH 7.4. The animals were then decapitated and the brain was quickly removed, hemisected, and placed into ice-cold high-sucrose aCSF for ∼3 min. The high-sucrose dissection solution contained (in mm) the following: 210 sucrose, 26 NaHCO3, 2.5 KCL, 1 CaCl2, 4 MgCl2, 1.25 NaH2PO4, and 10 d-glucose, pH 7.4 (295 mOsmol), and was saturated with 95% O2 plus 5% CO2. This solution has been shown to enhance tissue viability, permitting long-term recordings of synaptically evoked currents (Moyer and Brown, 1998). A coronal cut was made ∼1–2 mm from the rostral end of the brain, as well as at the fissure between the cerebrum and cerebellum. Then brain was placed and glued to the specimen disc of the vibratome cutting chamber with a drop of cyanoacrylate glue. The brain was oriented such that the caudal end was placed down onto the drop of glue, with the dorsal surface facing the cutting blade. Brain slices (400 μm), corresponding approximately to Plates 29–48 in the Paxinos and Franklin atlas (Paxinos et al., 2006), were obtained and incubated in normal aCSF at room temperature for a minimum of 1 h before electrophysiological recording. After recording, brains were collected and recut for immunohistochemistry analysis.
CAP recordings and analysis.
Slices were transferred to a recording chamber and perfused continuously with aCSF at a rate of 1 ml/min. Additionally, humidified 95% O2 plus 5% CO2 gas was superfused over the slices during all experiments. In vitro oxygen–glucose deprivation (OGD) was conducted by exposing brain slices to a glucose-free medium aerated with 95% N2 plus 5% CO2 for 15 min. The sucrose concentration in this solution was raised to 10 mm to maintain appropriate osmolarity. For extracellular recordings of CAPs from CC axons, fibers were stimulated with a bipolar electrode made of polyimide-insulated stainless steel wires (outside diameter 0.125 mm; Plastics One). Two recording borosilicate glass electrodes, filled with 150 mm NaCl, were placed in the CC. To elucidate electrophysiological evidence of myelination, positioning of both the stimulation and recording electrodes was adjusted before recording. The distance between each of the three electrodes was recorded for offline analysis of CV. Constant current pulses of 0.1 ms duration were generated by a Grass S88 stimulator (Grass Instruments) and delivered through an isolation unit. Pulse polarity was selected to produce the largest potentials with a clearly distinguishable artifact. Responses were filtered (low-pass filter at 5 kHz), amplified, and recorded with an Axoclamp 700 amplifier (Molecular Devices), and acquisition of data was performed using pClamp (v.10) software (Molecular Devices). CAPs were recorded at room temperature (21–23°C) to allow for better separation between the stimulus artifact, CAPs generated by the fast-conducting myelinated axons, and CAPs generated by the slow-conducting dysmyelinated axons, respectively. This distinct signal separation may be lost if recordings are performed at 35–37°C (Reeves et al., 2005; Crawford et al., 2009a). For computing CV, peak latency of the fast (N1) and slow (N2) components recorded by two recording electrodes was measured and divided by their distance.
Successful CAP recordings were confirmed by the size, width, and refractory period of the CAPs, and also by their sensitivity to the voltage-gated, fast sodium channel blocker tetrodotoxin (TTX, 1.0 μm). Stimulus–response relationships were recorded by varying stimulus intensity from 1 to 16 mA and by measuring the amplitude of corresponding graded CAPs. For computing CV, peak latency of the fast (N1) and slow (N2) components recorded by two recording electrodes was measured and divided by their distance. To examine the refractoriness of the axons, paired stimuli with varied intervals (from 0.5 to 16 ms with a 0.5 ms step increment) were delivered to the tissue, and the ratio between the second CAPs to the first CAP (CAP2/CAP1) was derived to assess relative recovery of the fibers. A low CAP2/CAP1 ratio corresponds to high refractoriness in the fibers. To examine the ability of axons to conduct signals at high frequency, tissue was stimulated at 20, 50, 100 Hz frequency (100 pulses in total). The amplitude of the last CAP was expressed as a percentage of the first CAP in the train.
Pharmacology.
A wide-range blocker of voltage-dependent K channels, 4-aminopyridine (4-AP, 200 μm), was bath-applied to the tissue during CAP recording. The effects of 4-AP were quantified by measuring the changes in the amplitude and area of the CAPs. The area of CAPs was defined as the area under the curve from beginning to 10 ms after the peak of the CAP (Sinha et al., 2006). The sodium channel blocker TTX (1.0 μm) was used to confirm the successful recording of CAPs and to block initiation and propagation of action potentials. All drugs were purchased from Sigma-Aldrich.
Statistical analysis
Cell differentiation data are presented using descriptive statistics, based on proportions expressing cell-specific markers. One-way ANOVA was used, followed by a Tukey's post hoc analysis to compare TEM and ionic profiling data. Electrophysiological data were calculated using paired and unpaired t tests, or two-way ANOVA, as appropriate, using SigmaStat software (v. 3.0, Aspire Software International). Data are presented as means ± SEM, unless otherwise stated.
Results
Transplanted aNPCs survive, migrate, and localize to CC, hippocampal fimbria, and periventricular parenchyma in an age-dependent manner
Appropriate cellular localization is paramount to successful biological therapy. As such, the first parameter investigated in this study was cell tracking. Transplanted aNPCs were visible 45 d post-transplant and localized to white matter tracts (Fig. 1A), such as the CC (Fig. 1B) and fimbria of the hippocampus (Fig. 1C), as well as to the periventricular white matter (Fig. 1D, shown 14 d post-transplant).
Figure 1.

Transplanted aNPCs localize to the perinatal white matter, incorporating into the CC, fimbria of the hippocampus, and periventricular parenchyma. A, Postinjection, aNPCs anatomically incorporate into major white matter tracts of P0-injected animals. B–D, Cells are present in the CC (B), hippocampal fimbria (C), and periventricular parenchyma (D).
aNPCs differentiate preferentially into myelinating OLs that associate with axons
Next, using common progenitor and neural lineage markers, we investigated cellular differentiation. In vivo, these cells have demonstrated tripotential lineage differentiation (Eftekharpour et al., 2007). However, in the in vivo spinal cord microenvironment, they show oligodendroglial differentiation. Here, Olig2, one of the earliest-expressed OL markers; MAG, a periaxonal oligodendrocytic marker, expressed later during development; and MBP, an indicator of mature, myelinating, transplanted OLs (congenitally absent in shi/shi animals), were used to identify oligodendroglial lineage cells; GFAP was used as a marker of activated astroglia; and NF200 was used to identify neuronal axons. eYFP was transgenically present in all transplanted cells (driven by the Actin promoter). When costained for these markers, the majority of YFP+ transplanted cells (Fig. 2A,B,E,F,I,J,M,N,Q,R,U,V,Y) differentiated into myelinating MBP+ (Fig. 2B,C,F,G,J,K), MAG+ (Fig. 2N,O,R,S,V,W), or Olig2+ (Fig. 2Z,AA) OLs. Distribution of YFP fluorescence and MBP immunoreactivity was concordant with MBP-positive processes aligned as a bundle oriented parallel to the longitudinal axis of the axonal tract. These MBP-positive cells were closely associated with ∼20–30% of NF200-positive axons (Fig. 2A,C,E,G,I,K), but only rarely showed GFAP immunoreactivity (Fig. 2M,O,Q,S,U,W). Quantification of NeuN (neuronal marker), GFAP, and Olig2 revealed 1.3 ± 0.6% neuronal differentiation, 2.0 ± 0.4% astrocytic differentiation, and 96.5 ± 0.6% oligodendroglial differentiation among transplanted cells (Fig. 2AB; n = 9, 3 mice per age group at transplant).
Figure 2.

Transplanted stem cell fate favors oligodendrocytic lineage and cells associate with axons in the CC. A, E, I, YFP+ NPCs extend processes that associate with NF200+ axons in the CC. B, C, F, G, J, K, N, R, V, AA, Complete eYFP colocalization is present with OL markers MBP (B, F, J, late myelin marker), MAG (N, R, V; in extruded processes, mid-developmental marker), and Olig2 (AA, early lineage marker), while MBP immunoreactive projections only interact with the NF200+ axons (C, G, K). M, O, Q, S, U, W, Conversely, eYFP (M, Q, U) and MAG (O, S, W) expression does not overlap with astrocytic marker GFAP. D, H, L, P, T, X, AA, Merged images. Y, Z, Singly stained eYFP (Y) and Olig2 (Z) immunopositivity. AB, Quantification of neuronal marker NeuN+, GFAP+, and Olig2+ cells shows 96.5 ± 0.6% oligodendroglial differentiation, with 1.3 ± 0.6% and 2.0 ± 0.4% neuronal and astroglial differentiation respectively (n = 9). Data are presented as means ± SEM.
Myelination displays age-dependent effects
Furthermore, myelin+ cell integration decreased linearly with increasing age at transplant. P0-transplanted animals (n = 3; Fig. 3A–D) had greater NPC incorporation than their P7 (n = 4; Fig. 3E–H) or P21 (n = 4; Fig. 3I–L) counterparts, with significant differences between all groups (Fig. 3M). Average in YFP+ areas, as a percentage of the total CC area, were 26.7 ± 2.4% for P0-injected, 17.7 ± 3.5% for P7-injected, and 6.3 ± 0.9% for P21-injected animals. Although incorporation decreased with increasing age, as shown in Figure 3B–D,F–H,J–L, age at transplant did not affect differentiation capacity, as cells differentiated along an oligodendroglial lineage, expressing MBP. In addition to decreased incorporation, there were also several animals injected at P21 only that showed no evidence of cell incorporation (N = 3 of 16 animals; Table 3). All animals that received P0 injections had strong evidence of cell incorporation at endpoint (<50 cells), but a similar amount (44 and 43% respectively for P7-injected and P21-injected animals) showed low incorporation, defined as <50 cells/slice. This is summarized in Table 3. For quantifications of myelination capacity, 3–4 animals/group with the strongest YFP+ cell incorporation in each group were used.
Figure 3.
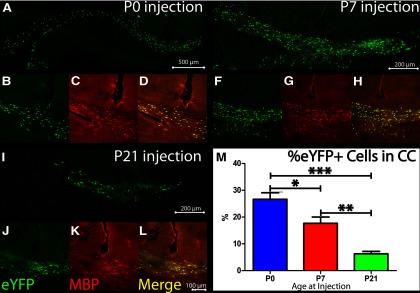
Transplanted cells incorporate in an age-dependent manner. A–L, Cell incorporation following transplant into neonatal P0 (A–D), P7 (E–H), and P21 (I–L) animals. A, E, I, Gross incorporation of YFP+ cells, after injection at P0, P7, and P21 respectively. B, F, J, Presence of YFP+ cells. C, G, K, Evidence that YFP+ cells can express MBP [P0-injected (C), P7-injected (G), and P21-injected (K) animals], the gene lacking in the Shiverer mutation. D, H, L, Merged images. M, Quantification shows age-dependent linearity in area of YFP+ incorporation. P0, n = 3; P7, n = 4, P21, n = 4. *p < 0.05, **p < 0.01, ***p < 0.001 respectively, in one-way ANOVA, followed by Tukey's post hoc test. Data are presented as means ± SEM.
Table 3.
Animal numbers
| Age | Total injected | No cells found | 1–50 Visible cells | >50 Visible cells |
|---|---|---|---|---|
| P0 | 6 | 0 | 0 | 6 |
| P7 | 10 | 0 | 3 | 7 |
| P21 | 26 | 3 | 7 | 16 |
Myelin formed from aNPCs compacts around axons
Although MBP+/YPF+ OL processes (Fig. 4A,B) colocalized with axons (Fig. 4B), this is suggestive, but not conclusive evidence for myelination. To definitively determine the presence of axon-associated compact myelin, TEM was employed. Shiverer mice possessed a congenital lack of compacted myelin by endogenous OLs, which was easily identifiable by a sheath of two to three layers with an average thickness of 2.7 ± 0.2 bands. In contrast, WT animals showed average myelin thickness of 5.8 ± 0.8 bands. At 45 d following aNPC transplantation, regions of multilayered, compacted myelin were visible within YFP+-selected sections of the CC (Fig. 4C,F,I). Compacted myelin, formed by YFP+ OLs, reached a maximum thickness of 40 layers (Fig. 4C). Transplanted shi/shi animals displayed an intermediate average myelin thickness (4.1 ± 0.3 bands), which did not reach significance, likely due to the large number of unmyelinated axons skewing the distribution. When only lamellar myelin structures with more than seven bands (based on WT average; Fig. 4J,K), aNPC transplantation restored myelin thickness by nearly half (30.23 ± 0.06% of axons in WT; 1.26 ± 0.01% axons in shi/shi; and 14.05 ± 0.02% of transplanted shi/shi axons possessed >7 myelin bands; N = 3, 2–3 fields/animal; ***p < 0.001 in 1-way ANOVA followed by Tukey's post hoc test).
Figure 4.

Newly formed myelin enwraps axons, creating regions with dense, dark, compact myelin and enabling CAP prediction using computer models. MBP gene product is absent in shiverer mice but produced by transplanted cells. A, Merged image of transplanted eYFP+ stem cells (green) expressing MBP (red). B, Orthogonal section of eYFP/MBP+ cell processes, which interact with NF200+ axons. C, TEM sections of transplanted dysmyelinated shiverer animals, which were preselected for eYFP+ cell presence, showed myelin thickness reaching ∼200 nm. D, G, Normal WT myelin compacts around axons, forming multilamellar structures. E, H, Shiverer myelin congenitally lacks this. F, I, In transplanted shiverer animals, aNPCs enwrapped axons, restoring a multilamellar myelination banding pattern. J, Bands were quantified and a frequency histogram was created for all axons with ≥7 lamellae. K, Transplantation in shi/shi animals restored an intermediate phenotype in the percentage of myelinated cells with at least seven bands. L, Based on TEM lamellar quantification, published literature, and histological measurements (above, Table 1), a computer model of action potential properties was generated to gauge stimulation, recorded by a measuring electrode. M, Unilamellar myelin is predicted to have a CAP speed of 0.62 m/s, with multilamellar myelin producing a signal with approximately double unilamellar CV. N = 3 per group. ***p < 0.001 in one-way ANOVA, followed by Tukey's post hoc test. Data are presented as means ± SEM.
Computer simulation predicts significant action potential restoration after remyelination
To better understand the significance of myelination on functional recovery after aNPC transplantation, we used a published computer model to assess CV of newly myelinated axons based on TEM, immunohistochemistry, and previously published input (McIntyre et al., 2002; Bakiri et al., 2011), summarized in Tables 1 and 2. Action potential CVs were estimated in WT myelinated, shi/shi dysmyelinated, and aNPC-transplanted myelinated axons, using the double-cable axon model (Fig. 4L; Lasiene et al., 2008). Based on these parameters, simulation CV in a bare CC axon is 0.57 m/s; axons with a single myelin layer show CV of 0.62 m/s. In myelinated axons, our TEM measurements show the emergence of 7–40 banded myelin profiles surrounding axons. From these measurements, we can estimate the effects of additional myelin on CV. At n = 10 layers, CV = 1.36 m/s; at n = 20 layers, CV = 1.57 m/s; at n = 30 layers, CV = 1.67 m/s; and finally at n = 40 layers, CV = 1.73 m/s (Fig. 4M). Therefore, even moderate aNPC-induced myelination can functionally double CV in this model.
aNPC transplantation normalizes ion channel profiling
A key role of the myelin sheath is to organize, at the node and paranode, different voltage-dependent ion channels that are required for saltatory conduction; nodal Na+ channels are separated from juxtaparanodal Kv+ channels by contactin-associated protein-positive (Caspr+) paranodal loops. As this spatial distinction is a physiological marker of the potential for cell polarization, nodal structure and ion channel reorganization are highly suggestive indicators of functional remyelination (Rasband et al., 1999; Arroyo et al., 2002). Although these markers are spatially distinct in WT animals (Fig. 5A,E,I), shiverer mice possess disrupted ion channel organization, with Kv, Na, and Caspr immunopositivity arbitrarily interspersed along bare axons (Fig. 5B,F,J). Transplantation of aNPCs resulted in increased levels of compact myelin attachment, as determined by significantly decreased Caspr length (2.18 ± 0.10 μm) in transplanted mice compared with control shi/shi mice (2.60 ± 0.12 μm; Fig. 5D). aNPC transplantation rescued WT phenotype in transplanted shi/shi animals, as WT levels were 1.93 ± 0.065 μm (p < 0.05; Fig. 5D). Next, node length was determined by measuring the distance between Caspr pairs. In WT mice, node-length measurement was 0.84 ± 0.03 μm. Dysmyelinated shi/shi mice possessed a significantly reduced node length (0.59 ± 0.03 μm), which was not altered following aNPC transplantation (0.62 ± 0.03 μm; p > 0.05).
Figure 5.

Transplanted aNPC-derived myelin normalizes ion channel profiling. A, B, Caspr-mediated compact myelin attachment is present in WT (A) but lost in shi/shi (B) mice. C, D, aNPC transplantation results in significantly more compact Caspr, reverting it to WT phenotype. E, F, Voltage-gated Na+ channels (Nav1.6) are normally distributed in nodes of Ranvier (E), but are disturbed in shi/shi mice (F). G, H, aNPC transplantation does not affect Na+ channel distribution and number of nodal structures quantified by Nav1.6-Caspr double-staining. I, J, Distribution of voltage-gated K+ channels (Kv1.2), normally present in juxtaparanodal regions (I), is disturbed in shi/shi mice (J). K, aNPC transplantation reorganizes Kv1.2 to its native juxtaparanodal region. L, Quantification of nodal structures using Kv1.2-Caspr double-staining shows significant increase in number of nodes after aNPC transplantation (N = 3 per group). M, Pharmacological assessment of voltage-dependent K channels in shi/shi CC after aNPC transplantation shows that 4-AP can enhance CAP in the slow N2 component in the WT axons, but not in the fast N1 component. It can enhance the N2 component in the shi/shi animals. N, It can enhance both N1 and N2 components in the aNPC-transplanted animals, described in statistical summary (N = 5 per group). *p < 0.05, **p < 0.01, ***p < 0.001 respectively, in one-way ANOVA, followed by Tukey's post hoc test. Data are presented as means ± SEM.
Nav1.6 is a subunit of the voltage-gated Na+ channel normally found in the nodal region of myelinated axons; it was used in this study to examine the localization of the voltage-gated Na+ channels. There was no significant difference in the distribution of Nav1.6 in WT, shi/shi, and transplanted mice. Nodal aggregates of Nav1.6 were identified in all three groups (Fig. 5H). No detectable changes in Nav1.6 distribution were observed after aNPC transplantation (Fig. 5G). Localization of Nav1.6 was further examined by quantifying Caspr pair number using Nav1.6 and Caspr double staining. Similar to Kv1.2/Caspr double staining, Nav1.6/Caspr double staining identified the highest number of Caspr pairs in WT mice (33.3 ± 3.3) and the lowest number of Caspr pairs in shi/shi mice (13.2 ± 1.0). However, transplant did not restore physiological organization of Nav1.6/Caspr+ nodes (16.3 ± 1.6).
Finally, we examined voltage-gated K+ channels, which are responsible for postaction potential repolarization. In normal myelinated axons, voltage-gated K+ channels are found in the juxtaparanodal region, flanking Caspr pairs (Fig. 5I); Kv1.2 reveals this regional spatial distribution was absent in shi/shi animals (Fig. 5J). aNPC transplantation resulted in localization of Kv1.2+ channels to the juxtaparanodal region (Fig. 5K), indicating that reorganization of K+ channels, and not Na+ channels, occurs following myelination. This relocalization was further evaluated by the quantification of Caspr pair number using Kv1.2 and Caspr double staining to identify pairs. Shi/shi mice had significantly reduced Caspr pair number (4.4 ± 0.7) compared with WT mice (17.4 ± 1.3; p < 0.001). While a significant increase in Caspr pair number was detected after aNPC transplantation (9.6 ± 0.8; p < 0.01; Fig. 5L), this increase only partially restored nodal phenotype.
In normal, myelinated axons, Kv channels, due to their concealment under the myelin sheath, are not sensitive to potassium channel blocker 4-AP (Nashmi and Fehlings, 2001a). However, in dysmyelinated shiverers, these exposed channels can be blocked by 4-AP (Sinha et al., 2006), thus enhancing axonal conduction (Targ and Kocsis, 1985). Observed restoration of Kv channels to normal submyelin, juxtaparanodal regions (Fig. 5L) gives rise to the possibility of altered pharmacological sensitivity to Kv channel blockers, such as 4-AP.
To investigate physiological sensitivity to 4-AP as a mechanistic assessment of the level of ionic channel reorganization after aNPC-induced myelination, we applied 200 μm 4-AP in the bath while recording CAPs. In WT, with masked K+ channels, the fast N1 component was not affected by 4-AP (Fig. 5M), as measured by CAP area changes before and after 4-AP administration (p > 0.05). In contrast, slow, dysmyelinated axons with exposed K+ channels were affected by 4-AP, as indicated by a prolonged decay phase and increased CAP area (p < 0.01). In shi/shi axons, CAP magnitude was significantly increased by 4-AP (p < 0.01). When 4-AP was applied to aNPC-transplanted, myelinated CC, both N1 and N2 components were enhanced. Results are summarized in Figure 5N. Therefore, voltage-dependent potassium channels were not fully concealed by regenerated myelin, although the moderate concealment achieved partially restored axonal insensitivity to 4-AP after aNPC transplantation.
Creation of bimodal CAP in aNPC-transplanted shi/shi CC
To test the model prediction, as well as the capacity for a minimum amount of myelin to produce functional benefit, we recorded CC CAPs in P21-transplanted mice, which showed the most sparse myelination. The CC is the largest white matter structure in the brain; as such, it would be most likely to show a functional correlate of anatomically incorporated transplanted aNPCs. To record CAPs carried by CC axons, fibers were stimulated by administering electric current and, after electrophysiological identification of myelinated axons, distance between the stimulation electrode and each of two recording electrodes was recorded. CV was then elucidated by measuring time intervals between the artifact and peaks of the N1 and N2 components, using the formula v = d/t (Fig. 6A). Since immunohistochemistry revealed that YFP-positive aNPC-derived OLs distributed nonhomogeneously in the CC, we hypothesized that this would introduce variability in electrophysiological recordings. To ensure successful recordings from myelinated axons, locations of recording electrodes were not constrained, but rather the characterized CAP profile was used to signal myelination. Transplanted cells led to creation of a CAP, representative of a “myelinated” phenotype, which was nearly twofold faster than that recorded across unmyelinated axons (Fig. 6B,C).
Figure 6.
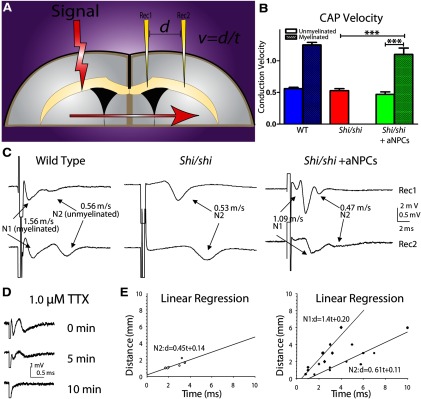
Electrophysiological evidence of myelination and enhancement in axonal conductance after aNPC transplantation into shiverer CC. A, Dual recording of CAPs from the CC in shiverer mouse brain slices. Positions of the stimulation electrode and two recording electrodes are shown. B, Bimodal signal velocity is restored in transplanted shi/shi animals to normal levels. C, Dual-channel recording reveals single-peak CAPs in the shiverer CC, but double peaks in P21 aNPC-transplanted shi/shi (shi-shi + aNPCs) CC, comparable to double peaks visible in WT sections (Crawford et al., 2009a). D, CAPs mediated by both newly myelinated and dysmyelinated axons in the aNPC-transplanted shi/shi CC could be blocked by application of sodium channel blocker TTX (1.0 μm) for ∼10 min. E, Estimated CV using linear regression from multiple shi/shi slices (N = 7) and aNPC-transplanted shi/shi slices (N = 10) reveals restoration of CAPs with accumulated velocities comparable to model predictions. ***p < 0.001 in one-way ANOVA, followed by Tukey's post hoc test. Data are presented as means ± SEM.
Figure 6C shows a typical CAP recorded from a shi/shi mouse, which features a single-peak, monophasic profile mediated by the exclusively unmyelinated axons in the CC. The CV of these axons is 0.53 ± 0.03 m/s, which is comparable to unmyelinated axons in WT CC (0.54 ± 0.02 m/s; Crawford et al., 2009a). In contrast, electrophysiological recordings from aNPC-transplanted shi/shis showed a restored bimodal “double-peak” profile. The maximum size of both components varied from 0.5 to 3 mV, depending on axonal recruitment by the electric currents and location of recording electrodes. In dual recording, CAPs recorded by the distal electrode had better separation of the two components than those recorded by the proximal electrode, since its unique spatial placement allowed further propagation of action potential and temporal separation of the two fiber groups. In aNPC-transplanted animals, the fast peak was associated with a CV of 1.10 ± 0.10 m/s and the slow peak was associated with a CV of 0.47 ± 0.04 m/s. It has been proven in previous literature that the fast components of CAPs are mediated by faster-conducting myelinated fibers and slow peaks are mediated by the unmyelinated contingent (Preston et al., 1983; Baker et al., 2002; Crawford et al., 2009a,b). CVs recorded from myelinated axons were comparable to those of myelinated axons recorded in the WT CC (1.67 ± 0.21 m/s, Crawford et al., 2009a), suggesting a full recovery of axonal conduction after aNPC transplantation. Sodium channel blocker TTX (1.0 μm) was used to confirm that recorded CAPs were sodium-dependent events (Fig. 6D) and linear regression analyses (Fig. 6E) revealed this emergence of a bimodal signal in transplanted but not naive shi/shi animals. In aNPC-transplanted shi/shi mice, we recorded CAPs containing a two-peak profile in 10 of 19 transplanted slices (52.6%) from five transplanted animals, suggesting that a new subpopulation of fast-conducting axons were generated by this intervention.
Lowered threshold of axon activation in aNPC-transplanted shi/shi CC
Axons with different degrees of myelination have different thresholds of excitation to stimulating currents. The myelin sheath provides electrical isolation for associated axons, thus differentially altering membrane potential (Ye et al., 2011) during stimulation. Normal myelinated axons have a much lower threshold of activation than bare axons when generating action potentials (Fehlings and Nashmi, 1997). To examine whether newly myelinated axons had a lower threshold of activation than dysmyelinated axons in the same tissue, we applied electric current with stepwise increases in intensity in aNPC-transplanted shi/shi CC and compared results with WT and shi/shi CC as baseline measurements.
An increase in stimulation intensity did not change overall CAP shape in any of the three groups. Higher-intensity stimulation recruited more axons, whose activation contributed to the overall size of CAPs. CAPs recorded from shi/shi CC maintained a single-peak (N2) profile at various stimulation intensities. CAPs in WT and aNPC-transplanted CC demonstrated a similar two-peak profile at various stimulation intensities (Fig. 7A). These data confirmed that aNPC-transplanted CC likely contained axons from two distinct pools, i.e., myelinated and unmyelinated axons. There was a slight leftward shift of CAP peaks when stimulation intensity increased in all groups, which caused a slight decrease in CAP peak latency. This was probably due to spread of stimulating current to areas closer to the recording site (Velumian et al., 2011).
Figure 7.

Myelin reveals normalized axonal profile: activation threshold, refractoriness, and HFS. A, Myelination lowers the threshold for axon activation. CAPs recorded under different stimulation intensities in the WT, shi/shi, and aNPC-transplanted (shi/shi + aNPCs) CC. Separate traces from A show that the threshold for activation of the fast, myelinated axons (N1) is lower than that for the slow, native dysmyelinated axons (N2) in the aNPC-transplanted CC (n = 6). B, I–O curves for WT (N1 and N2 components), shi/shi (N2 component), and aNPC-transplanted shi-shi animals (N1 and N2 components). The minimal current that can elicit observable CAP is 0.2 mA for the fast N1 component in both the WT and aNPC-transplanted shi/shi animals, but is ∼0.3 mA for the slow N2 component in all three animal groups. There is no statistically significant difference in the activation threshold between the slow component (N2) in the aNPC-transplanted CC and that in shi/shi animals (p > 0.05). In aNPC-transplanted shi/shi, the N1 component shows lower activation threshold than the N2 component. *p < 0.05 at several low stimulation intensities. C, New myelination reduces axonal refractoriness in the shi/shi CC. Superimposed CAPs evoked by paired stimuli of varying intervals from an aNPC-transplanted slice. The fast, newly myelinated axons (N1) have less refractoriness than the slow, dysmyelinated axons (N2), measured by absolute refractory period during pair-pulse stimulation. D, Single traces, as in A, with different pair-pulse intervals. E, Statistical comparison of the ratio of CAP2/CAP1 values in the N1 and N2 components in the CAPs recorded from the aNPC-transplanted axons (N = 6). F, Myelination enhances the response of the axons to HFS. Shi/shi CC under 20 Hz, 5 s stimulation. The CAP peak disappeared after ∼5 s stimulation. The second trace in F shows transplanted CC under 20 Hz, 5 s stimulation. Note the slow peak (N2) disappeared during stimulation while the fast peak (N1) persisted. G, Statistical summary comparing results from shi/shi (N = 6) and aNPC-transplanted shi/shi (shi/shi + aNPC) slices (n = 6). Myelinated axons have much improved capability to propagate action potentials under HFS than native, dysmyelinated axons in aNPC-transplanted CC. No significant difference was observed between the dysmyelinated axons in the shi/shi CC and those in aNPC-transplanted animals. *p < 0.05 and **p < 0.001 respectively, in paired t test or one-way ANOVA, as appropriate. Data are presented as means ± SEM.
We quantitatively compared activation threshold of dysmyelinated and myelinated axons in all groups. We found that threshold for slow (N2) axons in aNPC-transplanted animals was comparable to that in shi/shi animals (p > 0.01); and threshold for fast (N1) axons in aNPC-transplanted animals was comparable to that in WT animals. In aNPC-transplanted animals, threshold for myelinated axons (N1, ∼0.2 mA) was much lower than that of dysmyelinated axons (N2, ∼0.3 mA). This suggests that myelinated axons are more prone to activation than dysmyelinated axons in the same tissue (p < 0.05, paired t test).
To make a direct comparison of input–output (I–O) relations, I–O curves from all groups were normalized to their maximum CAP amplitudes (Fig. 7B). I–O curve for the N2 peak in aNPC-transplanted shi/shi CC was not different from shi/shi CAP and I–O curve for the N1 peaks in aNPC-transplanted shi/shi CC was not different from N1 peaks in WT CAP (p > 0.05, 2-way ANOVA). I–O curve for myelinated axons (N1) was significantly different from that of dysmyelinated (N2) axons in the same slices (p = 0.002, 2-way ANOVA): they responded better to low-intensity stimulation. Thus, myelinated axons have a lower threshold of activation than bare axons, likely reflecting electrical insulation by newly formed myelin.
Reduced axonal refractoriness in aNPC-transplanted shi/shi CC
Previous studies have reported that demyelination leads to an increased axonal refractory period (Nashmi and Fehlings, 2001b). To investigate whether myelination by aNPC transplants could re-establish normal refractoriness profiles in axons, we applied paired-pulse stimulation with varied intervals. Since a CAP represents excitation of a large population of axons, its shape depends on the location of the recording electrode (Fig. 7C). CAPs recorded with the proximal electrode appeared to have narrower CAP and shorter refractory period (data not shown), so refractoriness between myelinated and dysmyelinated axons from the same recording trace were compared.
Varying intervals between the two paired pulses successfully yielded a clear estimation of absolute and relative refractory periods for both myelinated and dysmyelinated axons. Absolute refractory period was 2.0 ± 0.1 ms for myelinated axons and 4.5 ± 1.0 ms for dysmyelinated axons (p = 0.015, paired t test). As illustrated in Figure 7C,D, at 0.5 ms interval, the second stimulus in the pair-pulse protocol could not trigger any CAPs. Excitation of both myelinated and dysmyelinated axons were suppressed by the first stimulus. At 3 ms interval, newly myelinated axons (N1) resumed partial activation, but dysmyelinated axons (N2) were still suppressed. Additional increases of stimuli interval further increased N1 component. At 5 ms interval, many myelinated axons were released from refractoriness but dysmyelinated axons were still suppressed. Dysmyelinated axons were only able to generate and propagate action potentials after 9 ms of suppression. At 16 ms interval, both myelinated and dysmyelinated axons resumed their full activation capability.
We then quantified the ratio between the second CAPs and the first CAP (CAP2/CAP1) for each axonal group (Fig. 7E). Lower CAP2/CAP1 ratios corresponded to higher axonal refractoriness. Rightward shift of these curves corresponded to increased refractory period of CC axons (Reeves et al., 2005; Crawford et al., 2009a). From these curves, we estimated that the interpulse interval for 50% recovery of the second pulse was 3.0 ± 0.1 ms for myelinated, fast axons and was 6.0 ± 0.1 ms for dysmyelinated, slow axons. Overall, myelinated axons exhibited a significantly enhanced capability to resume activation compared with native, dysmyelinated axons under paired-pulse stimulation (2-way ANOVA, p < 0.001).
Enhanced axonal response to HFS in aNPC-transplanted shi/shi CC
Previous studies have shown that the capability of axons to propagate action potentials at high frequency is impaired where there is a reduced level of myelin sheathing, which normally provides membrane capacitance needed for action potential propagation (Nashmi and Fehlings, 2001b).
To investigate the effect of aNPC-induced myelination on axons' capability to relay high-frequency signals, we stimulated aNPC-transplanted CC with trains of repetitive stimuli composed of frequencies of 20, 50, and 100 Hz (100 pulses in total, Fig. 7F). Similar to previous publications (Nashmi and Fehlings, 2001b; Eftekharpour et al., 2005), quantification analysis of axonal response was performed using the “fatigue ratio” between amplitude of the 100th CAP in the train as a percentage of the first amplitude of the first (Fig. 7G). A higher ratio suggests a better axonal response to HFS.
Axonal responses to HFS were similar in dysmyelinated axons in shi/shi CC and (N2 components) in aNPC-transplanted CC. After 2.5 s 20 Hz stimulation, fatigue ratios were 56.1 ± 5% and 47.5 ± 6.3%, respectively (p > 0.05). A full 5 s, 20 Hz stimulation completely eliminated axonal conduction in shi/shi CC and eliminated those N2 components mediated by native dysmyelinated axons in transplanted CC. In contrast, fast N1 peaks in transplanted CC were maintained during HFS. Fatigue ratio for this group of myelinated axons was 76.3 ± 5.4% after 2.5 s stimulation and 53.9 ± 3.1% after 5 s stimulation; both measures were significantly larger than those of the slow, N1 peaks (p < 0.01) recorded from the same preparation.
Similar results were also obtained from other stimulation frequencies (50, 100 Hz). In all these frequencies, we observed earlier disappearance of N2 peaks than of N1 peaks from aNPC-transplanted animals, leading us to conclude that newly myelinated axons were able to propagate high-frequency signals better than native, dysmyelinated axons.
Increased resilience to ischemia in aNPC-transplanted shi/shi CC
Neural stem cells have been used to treat cerebral ischemic injuries (Daadi et al., 2010). Electrophysiological evidence indicates that unmyelinated axons are more vulnerable than myelinated CC axons to traumatic axonal injury (Reeves et al., 2005). We investigated mechanistically whether aNPC-induced myelination promotes an axon's capability to function in an ischemic environment, with an in vitro OGD protocol. CAP responses in aNPC-transplanted slices were compared with those from WT and shi/shi CC as baselines.
OGD challenge decreased CAP amplitude in both slow (N2) and fast (N1) peaks in aNPC-transplanted shi/shi CC, in WT CC, and in the N2 components in control shi/shi slices (Fig. 8A). To highlight the time dependence of such changes, degree of decline was compared by normalizing the initial CAP amplitude value to 100% in each group (Fig. 8B). After 10 min OGD, the N2 component in aNPC-transplanted CC was reduced to 42.5 ± 2.5% of baseline, which is comparable to that in the control shi/shi group (reduced to 36.9 ± 3.6% of baseline) and to the N2 component in the WT group (36.3 ± 2.4% of baseline). In contrast, a substantial amount of N1 component in the aNPC-transplanted group was maintained after 10 min of OGD challenge (58.0 ± 4.5%, p < 0.05). Degree of N1 decrease in the aNPC-transplanted group is comparable to that in the WT group (60.0 ± 4.1% of baseline).
Figure 8.

Myelination enhances ischemic endurance. A, Representative CAP traces recorded from the WT CC, from shi/shi CC, and from the aNPC-transplanted CC during OGD. Note that after 15 min OGD challenge, CAP mediated by the native, dysmyelinated axons (N2) in the aNPC-transplanted CC disappeared. In contrast, a fairly large fast peak (N1), mediated by newly myelinated axons, persisted, although its amplitude was significantly decreased by OGD challenge. B, Statistical summary of N1 and N2 amplitude changes in OGD for WT CC (n = 6), shi/shi CC (n = 6), and aNPC-transplanted shi-shi (shi/shi + aNPC) CC (n = 6). *p < 0.05, ***p < 0.001 in one-way ANOVA, followed by Tukey's post hoc test. Data are presented as means ± SEM.
A longer OGD challenge further increased the difference between N1 and N2 components in aNPC-transplanted slices. Fifteen minutes of challenge completely abolished the N2 component in all three groups. However, after 15 min OGD, a considerable number (37.9 ± 3.7%) of fast N1 peaks remained in aNPC-transplanted slices. This amount of N1 peaks in the aNPC-transplanted group is comparable to that in the WT group (45.0 ± 2.9%). Together, these data indicate that newly myelinated axons have much better endurance (comparable to WT myelinated axons) to ischemic challenge than native, dysmyelinated axons.
Discussion
Building on our previous successful work using these cells in the spinal cord (Karimi-Abdolrezaee et al., 2006, 2010; Eftekharpour et al., 2007; Hawryluk et al., 2012, 2013), this study is first to compare aNPC transplant and myelination at key clinically relevant developmental time points. Moreover, it provides the first in-depth, mechanistic description of their functional incorporation into the disordered brain and is first to directly compare the physiological properties of aNPC-induced myelinated axons and dysmyelinated axons inherent to shi/shi CC. Cells integrated both anatomically and functionally into local white matter, as well as periventricular areas, and became OLs, which restored ion channel profiling in myelinated regions. This myelination led to significant functional recovery, including faster CV, lower threshold of activation, decreased refractoriness, improved response to HFS and enhanced tolerance to in vitro ischemia. Furthermore, we developed a computer model that can be used to accurately predict axonal properties in newly myelinated axons. This work indicates that many functional properties of axons can be successfully rescued long term by cell-replacement interventions, highlighting their promise for future use in pediatric neurological disease models.
This paper provides a number of key novel insights including (1) the examination of the effect of neural stem cell transplantation at varying developmental time points; (2) the evaluation of neural stem cells in critical cerebral regions, which could have clinical translational relevance for neurodevelopmental disorders, such as CP, and (3) a very detailed, quantitative neurophysiological examination of the functional benefits of remyelination of developmentally dysmyelinated cerebral tracts.
Neural precursors in CNS myelination paradigms
Previous studies have explored the capacity of human fetal precursors (Gao et al., 2006; Tarasenko et al., 2007; Windrem et al., 2008; Sandrock et al., 2010) or expanded cell line fetal-derived or pluripotent-derived neural progenitors (Yandava et al., 1999; Keirstead et al., 2005; Salazar et al., 2010; Obenaus et al., 2011) to myelinate in congenital dysmyelination or injury models (for review, see Ruff and Fehlings, 2010; Ruff et al., 2012). However, these cells were generally more plastic precursors. Increasing evidence suggests that transplantation of more early-lineage cells (for example, fetal-derived or pluripotent-derived, which are generally developmentally embryonic) can result in increased transplant survival (Wang et al., 2013). However, these early cells are less reflective of cell types present in a chronic, mature injury. Regenerative medicine is moving toward noninvasive techniques involving small molecules, growth factors, and drugs to expand and mobilize adult populations of NPCs (Erlandsson et al., 2011; Karimi-Abdolrezaee et al., 2012; Wang et al., 2012) for eventual application in chronic human conditions. Thus, an understanding of how purified populations of adult cells, as well as more primitive cells, behave in neural demyelination and dysmyelination models is a critical research tool for evaluating translational potential.
Furthermore, xenotype is an important consideration in cell-transplant scenarios. Most research using purified neural or oligodendroglial precursor transplant has been completed using human cells. In fact, to date, the only cells that have been able to show phenotypic reversal in shi/shi are human, with some human cells eventually “overtaking” native glia to create human chimeric brains (Wang et al., 2013). There is little understanding surrounding murine cell transplantation, and perhaps the failure of these cells to show equivalent integration and phenotypic reversal results from xenogenic differences. However, particularly because of the need to understand the mechanisms underlying the behavior of transplanted cells within their transplant niches in transgenic mouse systems, it is essential as a research tool to characterize murine systems and to understand the differences in behavior between human and murine neural precursor cells in vivo.
Relationship between transplantation age, differentiation, and survival
We observed migration and distribution of transplanted cells, which expressed three different OL markers, in the subcortical white matter, primarily in CC, but also in hippocampal fimbria and periventricular parenchyma. The observation that 97% of cells differentiated into olig2+ cells and only 3% differentiated into other forms of neural cells contrasts with ∼80% OL differentiation showed previously by our group in adult spinal cord (Eftekharpour et al., 2007; Karimi-Abdolrezaee et al., 2010). This discrepancy is perhaps due to a developmental gliogenic push or differences specific to the brain, but not the spinal cord, microenvironment. This enhanced oligodendroglial differentiation suggests NPC therapy might be useful in models that show specific perinatal depletion of CC and periventricular OLs. To determine whether postnatal age affected cell survival or engraftment, we used three different developmental windows. P0, almost exclusively discussed in the literature, linked with high levels of immune privilege and corresponding to a premature infant, is not a likely time point for clinical cell replacement therapy. P7, representing a neonatal human, is the earliest feasible human transplantation time point, and P21, approximately equating to a toddler, represents the clinical time point when developmental disorders are most often diagnosed (Shevell and Bodensteiner, 2004). We found an inverse linear relationship between area of cell coverage and age at transplant, indicating that, although early interventions might be of most benefit here, transplant at later time points can also produce a functional benefit. As with any therapy, there is an ideal therapeutic time window for treatment efficacy. Sometimes this corresponds with the most common interventional time point and sometimes this does not. It is not surprising that, in this case, the most effective time point corresponded with highest immune privilege and the most plastic potential. However, it is promising that there still remains therapeutic potential at the clinically relevant P21 injection time point. This provides evidence that NPC transplant at diagnosis might still be a viable clinical option and advocates for early diagnosis and intervention in perinatal disorders of myelin.
Model predictions
In most cases of neurological injury, there is concomitant demyelination, which can lead to further secondary loss of neuromotor function. Indeed, in models of SCI, transplantation of shi/shi aNPCs can functionally impede repair by endogenous neural precursors (Hawryluk et al., 2013). With the re-establishment of normal or near-normal myelination, nodal structure, axonal ensheathment, and normal electrical patterning can be straightforwardly examined. Transplant-derived OLs extruded processes that enwrapped axons and normalized lamellae. TEM sections were selected based on eYFP+ cell presence, so exogenous cells were definitively present in the tissue, and were the only cells therein capable of compact myelination (or fluorescence). The presence of multilamellar myelin in otherwise hypomyelinated shiverer tissue is direct evidence of aNPCs' myelination capability, as native OLs are incapable of producing compact myelin.
Furthermore, there are currently no predictive models in place that can use structural cues present in newly myelinated axons to accurately predict axonal behavior after cell transplant, thereby providing the essential link between structure and function. By combining TEM measurements with a published axon model to estimate CV in newly myelinated axons, we have identified the causal relationship between successful myelination and normalized axon function. Our model results indicated that CV could be doubled or tripled by the presence of moderate levels of myelin in aNPC-transplanted axons. Ex vivo CAP data validated this prediction, thereby describing, for the first time, its utility as a reliable indicator of transplant-mediated functional recovery.
Restoration of myelin led to the normalization of nodal architecture, with Kv1.2+ channel relocalization juxtaparanodally postmyelination. Although the precise mechanism driving K+ channel redistribution following myelination remains unclear, it is thought to involve signals from compact myelin and postsynaptic density protein-95 (Baba et al., 1999). Our data does not support the complete concealment of voltage-dependent Kv channels in newly myelinated axons. It is possible that some axons are partially myelinated with incomplete ion channel profiling. Since exposed K+ channels can cause failure in axonal depolarization (via Na+ current shunting) or dysfunctional axon/glia buffering (Nashmi and Fehlings, 2001a), combinatorial strategies, possibly including pharmacological blocking agents, may be required clinically to enhance and preserve axonal signaling in semiremyelinated axons.
Quantifying function
One challenge in functional assessment lies in distinguishing and carrying out direct comparison between successfully and unsuccessfully remyelinated axons in the same tissue. This would enable comparison of the functional profiles of newly myelinated axons to that of their normally myelinated and dysmyelinated counterparts. Previous studies, using technically challenging intra-axonal recordings, succeeded in comparing myelinated and dysmyelinated axon groups with relatively small sample sizes and showed that myelinated axons allowed for faster action potential conduction (Utzschneider et al., 1994; Honmou et al., 1996; Kohama et al., 2001). However, many important physiological aspects of the newly myelinated axons, including refractoriness, ability to carry high-frequency signals and resilience to pathological conditions, such as hypoxia and hypoglycemia, have never been explored previously at a large-population level.
Here, we showed that newly myelinated shi/shi axons had a lowered activation threshold, decreased refractoriness, and improved ability to propagate high-frequency action potentials. Myelinated axons have a lower threshold for activation (Fehlings and Nashmi, 1997) and refractoriness is an indicator of the capacity of Na+ channels to recover from inactivation after action potential (Hodgkin and Huxley, 1952). The novel observation that newly myelinated axons can carry high-frequency signals suggests that this outcome measure can be used in the future to delineate function in groups of axons. This finding supports the idea that increased capability of axons to propagate action potentials at high frequency is associated with increased myelin sheathing (Nashmi and Fehlings, 2001b). Thus, this comprehensive electrophysiological and mechanistic description of newly myelinated axons' physiology is a novel and vital tool for further stem cell myelination studies, providing the most sensitive parameters to detect direct evidence of myelination and functional improvement.
Within the current literature, no amount of transplant myelination, even with complete glial replacement by transplanted cells (Wang et al., 2013), was able to rescue the shivering phenotype. However, some groups have reported decreased tremor or increased lifespan among shiverers that have been transplanted with various human precursors (Yandava et al., 1999; Windrem et al., 2008; Wang et al., 2013). Decrease in tremor, determined visually, was not present in our aNPC-transplanted group, so we explored remyelination at a more focused level. While we achieved dramatic evidence of focal remyelination, as determined neuroanatomically and electrophysiologically, we recognize that major phenotypic improvements in this developmental mutant would require more widespread application of neural stem cells, which was outside the scope of this experiment. We also recognize that potentially subtle neurobehavioral changes may have occurred, although our main functional assay related to quantitative electrophysiological measurements of the remyelinated cerebral axonal tracts. At the outset of the experiment, we determined that longevity would not be a measured outcome and that a 45 d time point would be the standard survival endpoint, based on a time window that is 1.5× the normal course of myelination. As such, we cannot comment on the lifespan, extended or otherwise, of transplanted animals.
Ischemic performance
Protection from tissue ischemia is an important attribute of myelin. As mentioned in the introduction, the most common subtype of the most frequent neurodevelopmental disorder (CP) presents with OL-specific cell damage due to ischemic injury. It is essential to establish the neuroprotective capacity of new myelin (and its inverse in models of ischemic injury and demyelination) to progress this work to demyelination and dysmyelination paradigms in the future. This experiment represents a critical bridge to future work in this area. OGD provides an ischemic stress to the local tissue environment, which can mimic that observed in perinatal injury. Here, we provide novel evidence that myelinated axons are more resilient to OGD than dysmyelinated axons in the same preparation. This supports reports that myelinated CC axons are more resilient than unmyelinated axons after traumatic injury (Reeves et al., 2005). We propose that aNPC-induced myelination could be a significant neurological mechanism underlying the reported therapeutic effect of stem cell transplantation for cerebral ischemia (Dharmasaroja, 2009; de Vasconcelos Dos Santos et al., 2010).
Given the complexity of demyelinating disorders and their far-reaching implications, it is clear that novel therapeutic interventions are needed to combat these diseases, particularly those involving pediatric populations. The potential of neural stem/progenitor cells to differentiate into myelinating OL-like cells for repairing damaged CNS tissue is undergoing extensive investigation. Indeed, the first NPC transplants are now being used in children with neuronal ceroid lipofuscinosis (Batten's disease; Clinicaltrials.gov ID NCT00337636, NCT01238315). Aiming toward eventual therapeutic translation, our study is first to thoroughly examine murine neurosphere-cultured, allogeneic donor aNPCs' integration into the naive shi/shi CC and differentiation along an oligodendroglial lineage, exploring in depth the electrophysiological mechanisms of resultant functional myelination. Therefore, our work provides the basis for further exploration of the benefits of stem cell therapy for remyelination in pediatric disease models and for potential translation of this therapeutic strategy in clinical practices.
Footnotes
This work was supported by grants from NeuroDevNet, Ontario Brain Institute, Canadian Institutes of Health Research, and Heart and Stroke Foundation of Canada to M.G.F. C.A.R. is a recipient of Ontario Stem Cell Institute and Freedman postdoctoral fellowships. J.M.L. is supported by Natural Sciences and Engineering Research Council of Canada Undergraduate Student Research Award. We thank Giovanni Battista Calvieri and Stephen Doyle for their assistance with TEM and confocal imaging.
The authors declare no competing financial interests.
References
- Arroyo EJ, Xu T, Grinspan J, Lambert S, Levinson SR, Brophy PJ, Peles E, Scherer SS. Genetic dysmyelination alters the molecular architecture of the nodal region. J Neurosci. 2002;22:1726–1737. doi: 10.1523/JNEUROSCI.22-05-01726.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baba H, Akita H, Ishibashi T, Inoue Y, Nakahira K, Ikenaka K. Completion of myelin compaction, but not the attachment of oligodendroglial processes triggers K+ channel clustering. J Neurosci Res. 1999;58:752–764. doi: 10.1002/(SICI)1097-4547(19991215)58:6<752::AID-JNR3>3.0.CO%3B2-D. [DOI] [PubMed] [Google Scholar]
- Baker AJ, Phan N, Moulton RJ, Fehlings MG, Yucel Y, Zhao M, Liu E, Tian GF. Attenuation of the electrophysiological function of the corpus callosum after fluid percussion injury in the rat. J Neurotrauma. 2002;19:587–599. doi: 10.1089/089771502753754064. [DOI] [PubMed] [Google Scholar]
- Bakiri Y, Káradóttir R, Cossell L, Attwell D. Morphological and electrical properties of oligodendrocytes in the white matter of the corpus callosum and cerebellum. J Physiol. 2011;589:559–573. doi: 10.1113/jphysiol.2010.201376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bird TD, Farrell DF, Sumi SM. Brain lipid composition of the shiverer mouse: (genetic defect in myelin development) J Neurochem. 1978;31:387–391. doi: 10.1111/j.1471-4159.1978.tb12479.x. [DOI] [PubMed] [Google Scholar]
- Caillé S, Sauerwein HC, Schiavetto A, Villemure JG, Lassonde M. Sensory and motor interhemispheric integration after section of different portions of the anterior corpus callosum in nonepileptic patients. Neurosurgery. 2005;57:50–59. doi: 10.1227/01.NEU.0000163089.31657.08. [DOI] [PubMed] [Google Scholar]
- Crawford DK, Mangiardi M, Tiwari-Woodruff SK. Assaying the functional effects of demyelination and remyelination: revisiting field potential recordings. J Neurosci Methods. 2009a;182:25–33. doi: 10.1016/j.jneumeth.2009.05.013. [DOI] [PubMed] [Google Scholar]
- Crawford DK, Mangiardi M, Xia X, López-Valdés HE, Tiwari-Woodruff SK. Functional recovery of callosal axons following demyelination: a critical window. Neuroscience. 2009b;164:1407–1421. doi: 10.1016/j.neuroscience.2009.09.069. [DOI] [PubMed] [Google Scholar]
- Daadi MM, Davis AS, Arac A, Li Z, Maag AL, Bhatnagar R, Jiang K, Sun G, Wu JC, Steinberg GK. Human neural stem cell grafts modify microglial response and enhance axonal sprouting in neonatal hypoxic-ischemic brain injury. Stroke. 2010;41:516–523. doi: 10.1161/STROKEAHA.109.573691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Vasconcelos Dos Santos A, da Costa Reis J, Diaz Paredes B, Moraes L, Jasmin, Giraldi-Guimarães A, Mendez-Otero R. Therapeutic window for treatment of cortical ischemia with bone marrow-derived cells in rats. Brain Res. 2010;1306:149–158. doi: 10.1016/j.brainres.2009.09.094. [DOI] [PubMed] [Google Scholar]
- Dharmasaroja P. Bone marrow-derived mesenchymal stem cells for the treatment of ischemic stroke. J Clin Neurosci. 2009;16:12–20. doi: 10.1016/j.jocn.2008.05.006. [DOI] [PubMed] [Google Scholar]
- Eftekharpour E, Karimi-Abdolrezaee S, Sinha K, Velumian AA, Kwiecien JM, Fehlings MG. Structural and functional alterations of spinal cord axons in adult Long Evans Shaker (LES) dysmyelinated rats. Exp Neurol. 2005;193:334–349. doi: 10.1016/j.expneurol.2005.01.019. [DOI] [PubMed] [Google Scholar]
- Eftekharpour E, Karimi-Abdolrezaee S, Wang J, El Beheiry H, Morshead C, Fehlings MG. Myelination of congenitally dysmyelinated spinal cord axons by adult neural precursor cells results in formation of nodes of Ranvier and improved axonal conduction. J Neurosci. 2007;27:3416–3428. doi: 10.1523/JNEUROSCI.0273-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erlandsson A, Lin CH, Yu F, Morshead CM. Immunosuppression promotes endogenous neural stem and progenitor cell migration and tissue regeneration after ischemic injury. Exp Neurol. 2011;230:48–57. doi: 10.1016/j.expneurol.2010.05.018. [DOI] [PubMed] [Google Scholar]
- Fehlings MG, Nashmi R. A new model of acute compressive spinal cord injury in vitro. J Neurosci Methods. 1997;71:215–224. doi: 10.1016/S0165-0270(96)00147-1. [DOI] [PubMed] [Google Scholar]
- Gao J, Prough DS, McAdoo DJ, Grady JJ, Parsley MO, Ma L, Tarensenko YI, Wu P. Transplantation of primed human fetal neural stem cells improves cognitive function in rats after traumatic brain injury. Exp Neurol. 2006;201:281–292. doi: 10.1016/j.expneurol.2006.04.039. [DOI] [PubMed] [Google Scholar]
- Hawryluk GW, Mothe A, Wang J, Wang S, Tator C, Fehlings MG. An in vivo characterization of trophic factor production following neural precursor cell or bone marrow stromal cell transplantation for spinal cord injury. Stem Cells Dev. 2012;21:2222–2238. doi: 10.1089/scd.2011.0596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawryluk GW, Spano S, Chew D, Wang S, Erwin M, Chamankhah M, Forgione N, Fehlings MG. An examination of the mechanisms by which neural precursors augment recovery following spinal cord injury: a key role for remyelination. Cell Transplant. 2013 doi: 10.3727/096368912X662408. doi: 10.3727/096368912X662408. Advance online publication. Retrieved June 16, 2013. [DOI] [PubMed] [Google Scholar]
- Hines ML, Carnevale NT. The NEURON simulation environment. Neural Comput. 1997;9:1179–1209. doi: 10.1162/neco.1997.9.6.1179. [DOI] [PubMed] [Google Scholar]
- Hodgkin AL, Huxley AF. A quantitative description of membrane current and its application to conduction and excitation in nerve. J Physiol. 1952;117:500–544. doi: 10.1113/jphysiol.1952.sp004764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honmou O, Felts PA, Waxman SG, Kocsis JD. Restoration of normal conduction properties in demyelinated spinal cord axons in the adult rat by transplantation of exogenous Schwann cells. J Neurosci. 1996;16:3199–3208. doi: 10.1523/JNEUROSCI.16-10-03199.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karimi-Abdolrezaee S, Eftekharpour E, Wang J, Morshead CM, Fehlings MG. Delayed transplantation of adult neural precursor cells promotes remyelination and functional neurological recovery after spinal cord injury. J Neurosci. 2006;26:3377–3389. doi: 10.1523/JNEUROSCI.4184-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karimi-Abdolrezaee S, Eftekharpour E, Wang J, Schut D, Fehlings MG. Synergistic effects of transplanted adult neural stem/progenitor cells, chondroitinase, and growth factors promote functional repair and plasticity of the chronically injured spinal cord. J Neurosci. 2010;30:1657–1676. doi: 10.1523/JNEUROSCI.3111-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karimi-Abdolrezaee S, Schut D, Wang J, Fehlings MG. Chondroitinase and growth factors enhance activation and oligodendrocyte differentiation of endogenous neural precursor cells after spinal cord injury. PLoS One. 2012;7:e37589. doi: 10.1371/journal.pone.0037589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keirstead HS, Nistor G, Bernal G, Totoiu M, Cloutier F, Sharp K, Steward O. Human embryonic stem cell-derived oligodendrocyte progenitor cell transplants remyelinate and restore locomotion after spinal cord injury. J Neurosci. 2005;25:4694–4705. doi: 10.1523/JNEUROSCI.0311-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohama I, Lankford KL, Preiningerova J, White FA, Vollmer TL, Kocsis JD. Transplantation of cryopreserved adult human Schwann cells enhances axonal conduction in demyelinated spinal cord. J Neurosci. 2001;21:944–950. doi: 10.1523/JNEUROSCI.21-03-00944.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konigsmark BW, Murphy EA. Neuronal populations in the human brain. Nature. 1970;228:1335–1336. doi: 10.1038/2281335a0. [DOI] [PubMed] [Google Scholar]
- Lasiene J, Shupe L, Perlmutter S, Horner P. No evidence for chronic demyelination in spared axons after spinal cord injury in a mouse. J Neurosci. 2008;28:3887–3896. doi: 10.1523/JNEUROSCI.4756-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazzarini RA. Myelin biology and disorders. San Diego: Elsevier Academic; 2004. [Google Scholar]
- McIntyre CC, Richardson AG, Grill WM. Modeling the excitability of mammalian nerve fibers: influence of afterpotentials on the recovery cycle. J Neurophysiol. 2002;87:995–1006. doi: 10.1152/jn.00353.2001. [DOI] [PubMed] [Google Scholar]
- Moyer JR, Jr, Brown TH. Methods for whole-cell recording from visually preselected neurons of perirhinal cortex in brain slices from young and aging rats. J Neurosci Methods. 1998;86:35–54. doi: 10.1016/S0165-0270(98)00143-5. [DOI] [PubMed] [Google Scholar]
- Nashmi R, Fehlings MG. Mechanisms of axonal dysfunction after spinal cord injury: with an emphasis on the role of voltage-gated potassium channels. Brain Res Brain Res Rev. 2001a;38:165–191. doi: 10.1016/s0165-0173(01)00134-5. [DOI] [PubMed] [Google Scholar]
- Nashmi R, Fehlings MG. Changes in axonal physiology and morphology after chronic compressive injury of the rat thoracic spinal cord. Neuroscience. 2001b;104:235–251. doi: 10.1016/S0306-4522(01)00009-4. [DOI] [PubMed] [Google Scholar]
- Obenaus A, Dilmac N, Tone B, Tian HR, Hartman R, Digicaylioglu M, Snyder EY, Ashwal S. Long-term magnetic resonance imaging of stem cells in neonatal ischemic injury. Ann Neurol. 2011;69:282–291. doi: 10.1002/ana.22168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paxinos G, Halliday GM, Watson C, Koutcherov Y, Wang H. Atlas of the developing mouse brain at E17.5, P0 and P6. Ed 1. San Diego: Academic; 2006. [Google Scholar]
- Preston RJ, Waxman SG, Kocsis JD. Effects of 4-aminopyridine on rapidly and slowly conducting axons of rat corpus callosum. Exp Neurol. 1983;79:808–820. doi: 10.1016/0014-4886(83)90044-4. [DOI] [PubMed] [Google Scholar]
- Rasband MN, Peles E, Trimmer JS, Levinson SR, Lux SE, Shrager P. Dependence of nodal sodium channel clustering on paranodal axoglial contact in the developing CNS. J Neurosci. 1999;19:7516–7528. doi: 10.1523/JNEUROSCI.19-17-07516.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reeves TM, Phillips LL, Povlishock JT. Myelinated and unmyelinated axons of the corpus callosum differ in vulnerability and functional recovery following traumatic brain injury. Exp Neurol. 2005;196:126–137. doi: 10.1016/j.expneurol.2005.07.014. [DOI] [PubMed] [Google Scholar]
- Roach A, Takahashi N, Pravtcheva D, Ruddle F, Hood L. Chromosomal mapping of mouse myelin basic protein gene and structure and transcription of the partially deleted gene in shiverer mutant mice. Cell. 1985;42:149–155. doi: 10.1016/S0092-8674(85)80110-0. [DOI] [PubMed] [Google Scholar]
- Ruff CA, Fehlings MG. Neural stem cells in regenerative medicine: bridging the gap. Panminerva Med. 2010;52:125–147. [PubMed] [Google Scholar]
- Ruff CA, Wilcox JT, Fehlings MG. Cell-based transplantation strategies to promote plasticity following spinal cord injury. Exp Neurol. 2012;235:78–90. doi: 10.1016/j.expneurol.2011.02.010. [DOI] [PubMed] [Google Scholar]
- Salazar DL, Uchida N, Hamers FP, Cummings BJ, Anderson AJ. Human neural stem cells differentiate and promote locomotor recovery in an early chronic spinal cord injury NOD-scid mouse model. PLoS One. 2010;5:e12272. doi: 10.1371/journal.pone.0012272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandrock RW, Wheatley W, Levinthal C, Lawson J, Hashimoto B, Rao M, Campanelli JT. Isolation, characterization and preclinical development of human glial-restricted progenitor cells for treatment of neurological disorders. Regen Med. 2010;5:381–394. doi: 10.2217/rme.10.24. [DOI] [PubMed] [Google Scholar]
- Shevell MI, Bodensteiner JB. Cerebral palsy: defining the problem. Semin Pediatr Neurol. 2004;11:2–4. doi: 10.1016/j.spen.2004.01.001. [DOI] [PubMed] [Google Scholar]
- Sinha K, Karimi-Abdolrezaee S, Velumian AA, Fehlings MG. Functional changes in genetically dysmyelinated spinal cord axons of shiverer mice: role of juxtaparanodal Kv1 family K+ channels. J Neurophysiol. 2006;95:1683–1695. doi: 10.1152/jn.00899.2005. [DOI] [PubMed] [Google Scholar]
- Tarasenko YI, Gao J, Nie L, Johnson KM, Grady JJ, Hulsebosch CE, McAdoo DJ, Wu P. Human fetal neural stem cells grafted into contusion-injured rat spinal cords improve behavior. J Neurosci Res. 2007;85:47–57. doi: 10.1002/jnr.21098. [DOI] [PubMed] [Google Scholar]
- Targ EF, Kocsis JD. 4-Aminopyridine leads to restoration of conduction in demyelinated rat sciatic nerve. Brain Res. 1985;328:358–361. doi: 10.1016/0006-8993(85)91049-2. [DOI] [PubMed] [Google Scholar]
- Tropepe V, Sibilia M, Ciruna BG, Rossant J, Wagner EF, van der Kooy D. Distinct neural stem cells proliferate in response to EGF and FGF in the developing mouse telencephalon. Dev Biol. 1999;208:166–188. doi: 10.1006/dbio.1998.9192. [DOI] [PubMed] [Google Scholar]
- Utzschneider DA, Archer DR, Kocsis JD, Waxman SG, Duncan ID. Transplantation of glial cells enhances action potential conduction of amyelinated spinal cord axons in the myelin-deficient rat. Proc Natl Acad Sci U S A. 1994;91:53–57. doi: 10.1073/pnas.91.1.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velumian AA, Wan Y, Samoilova M, Fehlings MG. Contribution of fast and slow conducting myelinated axons to single-peak compound action potentials in rat spinal cord white matter preparations. J Neurophysiol. 2011;105:929–941. doi: 10.1152/jn.00435.2010. [DOI] [PubMed] [Google Scholar]
- Wang J, Gallagher D, DeVito LM, Cancino GI, Tsui D, He L, Keller GM, Frankland PW, Kaplan DR, Miller FD. Metformin activates an atypical PKC-CBP pathway to promote neurogenesis and enhance spatial memory formation. Cell Stem Cell. 2012;11:23–35. doi: 10.1016/j.stem.2012.03.016. [DOI] [PubMed] [Google Scholar]
- Wang S, Bates J, Li X, Schanz S, Chandler-Militello D, Levine C, Maherali N, Studer L, Hochedlinger K, Windrem M, Goldman SA. Human iPSC-derived oligodendrocyte progenitor cells can myelinate and rescue a mouse model of congenital hypomyelination. Cell Stem Cell. 2013;12:252–264. doi: 10.1016/j.stem.2012.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Windrem MS, Schanz SJ, Guo M, Tian GF, Washco V, Stanwood N, Rasband M, Roy NS, Nedergaard M, Havton LA, Wang S, Goldman SA. Neonatal chimerization with human glial progenitor cells can both remyelinate and rescue the otherwise lethally hypomyelinated shiverer mouse. Cell Stem Cell. 2008;2:553–565. doi: 10.1016/j.stem.2008.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yandava BD, Billinghurst LL, Snyder EY. “Global” cell replacement is feasible via neural stem cell transplantation: evidence from the dysmyelinated shiverer mouse brain. Proc Natl Acad Sci U S A. 1999;96:7029–7034. doi: 10.1073/pnas.96.12.7029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye H, Cotic M, Fehlings MG, Carlen PL. Transmembrane potential generated by a magnetically induced transverse electric field in a cylindrical axonal model. Med Biol Eng Comput. 2011;49:107–119. doi: 10.1007/s11517-010-0704-0. [DOI] [PubMed] [Google Scholar]