

Abstract
Motivation: Human cells are organized into compartments of different biochemical cellular processes. Having proteins appear at the right time to the correct locations in the cellular compartments is required to conduct their functions in normal cells, whereas mislocalization of proteins can result in pathological diseases, including cancer.
Results: To reveal the cancer-related protein mislocalizations, we developed an image-based multi-label subcellular location predictor, iLocator, which covers seven cellular localizations. The iLocator incorporates both global and local image descriptors and generates predictions by using an ensemble multi-label classifier. The algorithm has the ability to treat both single- and multiple-location proteins. We first trained and tested iLocator on 3240 normal human tissue images that have known subcellular location information from the human protein atlas. The iLocator was then used to generate protein localization predictions for 3696 protein images from seven cancer tissues that have no location annotations in the human protein atlas. By comparing the output data from normal and cancer tissues, we detected eight potential cancer biomarker proteins that have significant localization differences with P-value < 0.01.
Availability: http://www.csbio.sjtu.edu.cn/bioinf/iLocator/
Contact: hbshen@sjtu.edu.cn or zhng@umich.edu
Supplementary information: Supplementary data are available at Bioinformatics online.
1 INTRODUCTION
The knowledge of the subcellular/organelle localization of protein molecules can provide important help for understanding their functions in cells (Chou and Shen, 2008; Emanuelsson et al., 2007). In the past decade, a variety of methods have been developed for predicting the subcellular localizations of proteins (Nair and Rost, 2009). These methods can be grouped into two categories: one-dimensional (1D) amino acid sequence-based and two-dimensional (2D) image-based methods.
The sequence-based methods usually generate protein subcellular location predictions through homology transfer (Nair and Rost, 2009), target signal search (Emanuelsson et al., 2007) or statistical machine learning (Chou and Shen, 2008). These methods have been successfully applied to genome-wide large-scale function annotations, but they are hard to detect protein translocations, which often cause changes in the related biological network functions. One reason is that when the translocation occurs, the primary sequences of the translocated protein are about the same, which cannot be detected by the sequence-based comparisons. Therefore, the intuitive 2D image-based algorithms are explored in this situation (Peng et al., 2012).
Efforts on the 2D image-based approaches are mainly focused on three aspects: (i) studies on new representative image descriptors, with examples including the subcellular location features (SLFs) for protein distributions as proposed by Boland and Murphy (2001); (ii) studies on improving the accuracy of classification algorithms, with examples including the classification based on multi-resolution subspaces as reported by Chebira et al. (2007), and the two discriminative models by Li et al. (2012b), which extended the logistic regression with structure latent variables; and (iii) applications of existing predictors to the data analysis (Li et al., 2012a).
Most of the 2D methods are single-label predictors, i.e. they assumed that each protein corresponds to only one location. However, nearly 20% of human proteins co-exist at ≥2 different subcellular locations (Zhu et al., 2009). These multi-label proteins often contain significant biological information, such as protein complex transcription in cell cycles. Only few studies have attempted to predict distribution from human protein images in multiple-location cases (Peng et al., 2010; Zhao et al., 2005). These studies tried to use statistics of fluorescence objects to differentiate location patterns. In these approaches, protein fluorescence objects in images are detected and clustered into several types based on their shape, size and position in cells; then each single-label pattern is represented by a set of objects in distinct types. Finally, a multi-label image is linearly decomposed into the object frequencies in each type (Zhao et al., 2005). Such methods have several limitations: (i) overlapping objects and filamentous proteins will confuse the object detection algorithm because a continuous region of pixels was defined as an object; and (ii) prediction accuracy is limited by the simple linear model. The former limitation may lead to incorrect object targets, and the latter tends to cause the constructed classifier to be too sensitive to the noise and overfitting.
Instead of the linear statistics, in this study we seek to develop a more flexible and accurate machine learning predictor, named iLocator, which can handle multi- and single-label samples simultaneously. Three factors critical for machine learning algorithm developments, including high-quality benchmark dataset, highly discriminative image descriptors and accurate multi-label learning algorithms, will be carefully designed and systemically examined.
For benchmark training and testing, we will collect the high-resolution immunohistochemistry (IHC) microscopy images from the human protein atlas (HPA, http://www.proteinatlas.org/) (Pontèn et al., 2008). The efficiency of both global and local image descriptors will be examined as input to the classifiers. The global features contain the Haralick texture features and the DNA distribution features, which both belong to the well-defined SLFs (Boland and Murphy, 2001). The local binary patterns (LBPs) are applied for the first time to this multi-label problem to describe the local details. Moreover, we will implement an ensemble classifier in iLocator composed of two multi-label learning modes, i.e., binary relevance (BR) (Boutell et al., 2004) and classifier chains (CC) (Read et al., 2009), because both have been reported performing well in multi-label image pattern recognition.
For normal cell function, it is critical to have the proteins appear at the right location at the correct time for forming appropriate interactions with correct molecular partners. Mislocalization will make the proteins inaccessible, and thereby not be integrated into the proper functional biological networks or pathways. Protein function loss caused by the mislocalization will inevitably affect the whole biological system, which has been found to be associated with many human diseases (Hung and Link, 2011). Several potential reasons can result in aberrant protein locations, such as amino acid mutations in targeting signal sequence, changes in the post-translational modifications and the expression level, and trafficking machinery deregulations. To fully understand the underlying mechanisms of the protein mislocalizations, it is important to first identify the mislocalized protein targets. Although studies to look for biomarker proteins have been previously reported (Murphy, 2004), these studies only involved single-label classifiers, which cannot detect changes from multiple locations to a single location, from a single location to multiple locations or from multiple locations to different multiple locations. Considering the fact that there are ∼20% human proteins co-localized in more than one location, these changes can occur frequently. For example, in a healthy cell, protein cylin D1 can shuttle between the nucleus and cytoplasm. In contrast, a reduction of cyclin D1 exported from the nucleus can lead to overexpression of this protein in the nucleus, and the inactivation of retinoblastoma, which is a tumor-suppressing protein (Gladden and Diehl, 2005). Other such multiple-location shuttling proteins associated with cancers include p53, BRCA1, SOX9 and APC (Bratthauer and Vinh, 2009; Fabbro and Henderson, 2003). Detecting these changes requires accurate multi-label predictors. In this study, iLocator has been applied to screen such potential mislocated biomarkers in our protein dataset, which includes multi-label proteins. By comparing the predictions output from iLocator between cancer and normal tissues, several proteins have been detected as potential cancer biomarkers.
2 MATERIALS AND METHODS
The flow chart of the experiment in this study is depicted in Figure 1, which includes two procedures of iLocator development and the application for cancer biomarker detection.
Fig. 1.

The flowchart of the experiment in this study. (A) The procedures of iLocator creation with normal image dataset. (B) The process of biomarker protein detection using iLocator
2.1 Preparing dataset
The basic idea of the statistical machine learning is to learn the discriminative rules from training datasets. Hence, the image data quality is of critical importance for the experiments of this study. For instance, it has been pointed out that the high quality of antibody staining provides a more accurate reflection of subcellular patterns (Uhlen et al., 2010). Here, the benchmark dataset is constructed from the HPA database. We selected high-quality samples based on two indexes: the validation score and the objective score in HPA (Pontèn et al., 2008). Protein labels were obtained from the HPA and UniProt. We collected proteins from both the normal image dataset and the cancer image dataset (Supplementary Table S1).
2.1.1 Normal image dataset
This dataset was used for training and testing classifiers. It contained 3240 images from 28 proteins in normal cells, of which seven proteins with two or more organelle labels (in normal tissues) are considered as multi-label proteins. We considered seven major organelles: cytoplasm, endoplasmic reticulum, Golgi apparatus, lysosome, mitochondria, nucleus and vesicles.
2.1.2 Cancer image dataset
The subcellular locations of cancer images are not annotated in HPA, so the cancer image dataset was to be predicted and compared with the data from normal cells to detect mislocalizations. This dataset contained 3696 cancer images of the same 28 proteins as in the normal dataset. Seven cancers were considered in this study: breast cancer, lung cancer, pancreatic cancer, prostate cancer, renal cancer, thyroid cancer and urothelial cancer.
Each IHC image shows a specific protein in brown and DNA in purple (Pontèn et al., 2008). To avoid artifacts from poorly stained images, such as those with too much black or cyan dye, we removed images that had hue values exceeding a threshold of 13 in the hue, saturation and value color space (Newberg and Murphy, 2008). After this step, 3207 images remained, including 823 multi-label ones. Then, the datasets were partitioned into 2 folds: 1604 images (include 412 multi-label ones) were put into the training set, whereas the other 1603 (include 411 multi-label ones) were put in the testing set.
2.2 Image separation
The original HPA image is the fusion of DNA (purple sections) and protein (brown sections). Because we mainly need the distribution of protein, it is important to separate the protein channel from that of the DNA. We tested two separation techniques, i.e., linear spectral separation (LIN) and blind spectral separation by non-negative matrix factorization (NMF), on the benchmark dataset. Both of them separate purple and brown channels through color transforming (Newberg and Murphy, 2008).
2.3 Feature extraction
After the separation step, we extracted Haralick texture features, DNA distribution features and LBP features from the separated channels. Haralick features describe image texture by some intuitive aspects of image, such as inertia and isotropy (Nanni et al., 2010a). Here, we used 10 Daubechies filters with vanishing moments from 1–10 (db1–db10) when extracting Haralick features. Each of them can generate an independent set of 836 Haralick features. Four DNA distribution features were used because the nucleus is fairly consistent among cells as a common point, and may provide reference for determining the protein localization pattern (Newberg and Murphy, 2008). In this study, LBP is 256-dimensional (See Supplementary Fig. S1 for an example of the LBP descriptor), which characterizes the spatial structure of local image texture and can detect micropatterns in the image, such as flat areas, edges and spots (Nanni et al., 2010a; Tahir et al., 2012).
To reduce computational time and avoid overfitting, feature selection was performed to select the most informative features. We selected features using stepwise discriminant analysis (SDA) (Newberg and Murphy, 2008). The output of SDA is a subset of features ranked in terms of their importance and can be fed into classifiers.
2.4 Multi-label classification
We trained classification models using BR and CC modes, respectively. According to BR, one binary algorithm is trained for predicting the relevance of one class. The method ‘cross-training’ was used in the training section (Tsoumakas et al., 2010). Cross-training means that when creating a label classifier, all the training samples associated with this label are considered as positive samples, whereas all others are considered as negative ones. As in BR, CC also trains seven binary classifiers corresponding to seven labels. However, considering labels that are correlated might indicate multiple possible locations, CC takes the correlation among labels into account. It extends the attribute space of each classifier with the 0/1 label of all previous classifiers, and the seven binary classifiers are linked to form a chain (See Supplementary Fig. S2 for illustrations of BR and CC learning modes) (Read et al., 2009). Because different label orders can give slightly different results, we used a random chain order. We applied support vector machine (SVM) as the classifier, and the LIBSVM-2.91-1 package was used (http://www.csie.ntu.edu.tw/∼cjlin/libsvm/).
Each classifier based on BR or CC can output a seven-dimensional (7D) score vector [s1, s2, … , s7] per testing image, where each score corresponds to a specific class (organelle). The score represents the confidence of belonging to the corresponding class. It is positive if the corresponding binary classifier predicts the image belongs to the class, and negative if not. To decide the label set of a sample from its score vector, we used two criteria, i.e. top criterion and threshold criterion (Boutell et al., 2004), and obtained intersection elements of their results to compose the final label set. The top criterion considers that the label set consists of the labels with positive scores, and if all seven scores are negative, the label with the maximum score is considered as the unique label. The assumption of threshold criterion is that the score values corresponding to the real labels are the largest, and, in the case of multiple labels, have similar scores. Therefore, a threshold T is needed to judge whether the top scores are close enough. When deciding whether the label c should be assigned to the predicted label set, we defined H1 to denote yes and H2 no. And dif was defined as the difference between the biggest score and the cth one:
| (1) |
Then Hb is determined according to magnitude of dif: H1 is true if dif ≥ T, and H2 is true if dif < T. The objective function is:
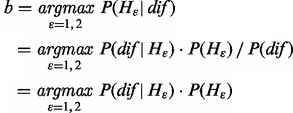 |
(2) |
The probabilities of dif in H1 and H2 (P(dif |H1) and P(dif |H2)), and the probabilities of H1 and H2 (P(H1) and P(H2)), can be calculated from the training set (Fig. 2A–D). The process of determining T uses maximum a posteriori (MAP) principle (Fig. 2E). According to Equation (2), T is at the intersection of P(dif|H1) ċ P(H1) and P(dif|H2) ċ P(H2).
Fig. 2.

The experimental results of determining the threshold T in db8-BR model. (A) The calculation process of dif1 (dif in H1) and dif2 (dif in H2) using 1–3 label images in the training dataset. The predicted score vector [s1, s2, … , s7] can be sorted to be [sr1, sr2, … , sr7], and the gray circles represent real labels. dif1 are the differences between sr1 and scores corresponding to the label not in the real label set but closest to it. dif2 are differences between sr1 and other scores whose label also belongs to the real label set. In theory, dif1 are generally bigger than dif2, which is the key to get T and determine the label set. (B) The histograms of dif1. (C) The histograms of dif2. (D) The Gaussian distribution fitting curves of P(dif|H1) and P(dif|H2). (E) The fitting curves of P(dif|H1)ċP(H1) and P(dif|H2)ċP(H2). In this model, T is 1.084, and the error represented by the gray part is 10.43%
In this study, we evaluated the performance of the classifier by five multi-label classification metrics: subset accuracy, accuracy, recall, precision and average label accuracy (See Supplementary text for details). Among them, subset accuracy is the fraction of samples whose predicted label set is exactly the same as the true label set. We evaluated the performance of classification mainly by it.
2.5 Identifying potential biomarkers
Protein subcellular mislocations are found to have correlations with human diseases (Hung and Link, 2011). To reveal the hidden mechanisms, it is important to know the protein locations in normal and cancer conditions, respectively. Because there are no explicit subcellular location annotation data for proteins in cancer tissues in HPA, we cannot compare these two conditions directly. Therefore, we used the obtained classifiers to give predictions of these 28 proteins in normal and cancer tissues, respectively. The cancer image dataset contains 3696 images, and involves seven cancers, i.e. breast cancer, lung cancer, pancreatic cancer, prostate cancer, renal cancer, thyroid cancer and urothelial cancer (Supplementary Table S1). For each query protein–tissue combination, suppose there are N normal images and M cancer images. Each image has a 7D score vector. With these score vectors, we screened biomarkers by two steps: first, screening by the direct comparison method; second, screening by evaluating the significance of location changes using the t-test. In the first step, we calculate an average vector from these N normal vectors and use it to determine the final label set. The label set corresponding to the cancer state can also be determined with the similar procedures. The direct comparison method selects these protein–tissue combinations satisfying the two conditions: (i) the label set of normal and cancer states are not exactly the same and (ii) sign (+ and −) of the average predicted scores of these changing locations are opposite between normal and cancer states. In the second step, for each selected combination by step 1, an independent sample t-test is conducted on the M and N score vectors. The detailed process on one example protein–tissue combination can be found as Supplementary Figure S3. For each biomarker protein–tissue combination, the t-test will output a P-value vector, where each element represents the confidence of mislocalization in a subcellular location from normal to the cancer condition. These protein candidates are considered as reliable potential biomarkers only if the P-values of their translocations are <0.01.
3 RESULTS
3.1 Creating and validating single classifiers
In the initial experience, we tested LIN and NMF, respectively. The experimental results show that LIN approach outperforms NMF by 5–10% on the testing dataset (Supplementary Fig. S4). This is consistent with a previous report (Newberg and Murphy, 2008). Considering LIN can yield better results and its faster speed, it was adopted in the following study of creating iLocator.
To test whether pre-processing of images by enhancing technique is helpful for improving the classification performance in this study, we compared the contrast limited adaptive histogram equalization enhancing, and nothing is performed on the current dataset. The results show that the latter outperforms contrast limited adaptive histogram equalization by 3–5% in the subset accuracy and thus was adopted in the following experiments, which are similar to a previous study (Paci et al., 2013).
Then we calculated various features from the separated DNA and protein channels without pre-processing, selected image features by SDA, and then trained classification models using BR and CC modes, respectively. We tested these classifiers on the testing set. When deciding the label set of a sample from its score vector, we had to determine the difference threshold T before using threshold criterion (Fig. 2). Consequently, the subset accuracies of these single classifiers range from 57.89 to 67.12%. The performance of CC is superior to BR because CC can capture complex correlations, such as proteins co-existing at different locations due to spatial proximity or functional reasons.
3.2 Effects of discriminative features and classification methods
The SLFs, including DNA features and Haralick features, can make good sense in predicting protein localizations (Newberg and Murphy, 2008). In this study, we added the LBP features to SLF vectors and obtained a 1096 (4 + 836 + 256)-dimensional image descriptor. After feature selection, we obtained the most informative features that can be fed into classifiers. From Figure 3A, we can see the overall proportion of LBP components is not small in the top ranked features, where it is also interesting to find that the LBPs contribute to the top 1 selection. This demonstrates that both the LBP features and SLFs have a significant role in distinguishing different protein location patterns.
Fig. 3.

The experimental results when adding LBP into feature space. (A) The top 30 ranked features output from SDA (totally 72 features) when using db8 filter. The red letter L represents the LBP feature, the blue letter H represents the Haralick feature and the green letter D represents the DNA distribution feature. In this feature rank, there are 12 LBP features, 17 Haralick features and 1 DNA feature. (B) The results of subset accuracy from different combinations of BR, CC, SLFs and SLFs + LBP on db1–db10. (C) The results of five multi-label classification metrics from different combinations of BR, CC, SLFs and SLFs + LBP on db8
The classification results also demonstrate LBPs significantly contribute to the performance improvements (Fig. 3B and C). For instance, on the db8 model, the subset accuracy improved from 52.96 to 61.82% by adding LBP in BR, and similarly, a 7.11% improvement was observed in the CC mode (Fig. 3B). Besides, the results of subset accuracy demonstrate the superiority of CC over BR, indicating CC predicts more samples with all the labels correctly identified. It is also interesting that CC is not always better than BR in some metrics (Fig. 3C). The reason is that CC has a disadvantage: it may add some irrelevant attributes about other labels, which can make confusion in classification. The effect of this disadvantage is obvious on single-label samples, which can be confirmed in later experience.
Although the overall performance was improved by LBP, it was not clear how such enhancement happened. To reveal this point, we separated the testing set into two groups: a single-label image set and a multi-label image set. We further represented the two groups with SLFs alone, and with both SLFs and LBP, respectively. The classification results show that the improvements of each evaluating metric in the multi-label testing set are more salient than in the single-label set when incorporating LBP (Fig. 4). For example, the increments of subset accuracy in multi-label set by BR and CC were 11.68 and 14.84%, whereas in the single-label set, the improvements were 7.89 and 4.45%, respectively. The disparity indicates that the multi-label samples benefit more from adding LBP. The reason is that a multi-label pattern is a mixture of several single-label patterns, and LBPs are local features representing statistics of binary micropatterns. Hence, LBP can catch the subtle local features that are important for multi-label classification.
Fig. 4.

The comparison of subset accuracies in the single-label and multi-label testing dataset classified using BR and CC modes with db8 features
Besides, three more phenomena can be observed from Figure 4. The first is that, in general, multi-label images are much more difficult for classification than the single-label ones. This is demonstrated by the better performance of single-label (BR) and single-label (CC) images than multi-label (BR) and multi-label (CC) ones, respectively. Second, the results of multi-label (CC) images are better than multi-label (BR) ones, especially subset accuracy, which shows a difference of 15.57%. This phenomenon indicates that considering correlations among labels is particularly important for multi-label set prediction. Finally, we can see the single-label (BR) samples are higher than single-label (CC) ones, especially when adding LBP. This phenomenon reveals that CC is not always better than BR on single-label samples because it may take irrelevant attributes into account. Considering the latter two phenomena, to get advantages of BR and CC together, we tended to combine their models.
3.3 Performance of ensembled classifier
Therefore, considering an ensemble of multiple classifiers generally gives a better performance (Shen and Chou, 2006), we constructed an ensembled classifier by combining the BR and CC classifiers on db1–db10. Each of these 20 classifiers will output a classification vector consisting of seven probabilities of the query image to the seven covered subcellular locations. We then took the average of these 20 output vectors to generate the final predictions. This ensembled model is finally used in iLocator (Fig. 1A). Its five evaluation criteria of subset accuracy, accuracy, recall, precision and average label accuracy are 72.49, 77.83, 75.45, 80.50 and 92.71%, respectively. As expected, we can see big improvements in all the metrics compared with any single simple classifier.
3.4 Identifying cancer biomarkers
In the entire dataset, there are ∼3 normal images and ∼24 cancer images for each protein–tissue combination. We screened biomarkers by the two steps presented before. The first step gives predicted label sets of all the protein–tissue combinations in normal and cancer conditions, respectively (Fig. 5A and B). Subcellular location changes in 12 proteins were detected by direct comparison (Fig. 5C). These 12 proteins constitute an initial set of potential biomarkers. Then we conducted an independent sample t-test on their corresponding score vectors to evaluate the significance levels of the localization changes. There are eight proteins with 18 protein–tissue combinations left after the second step (Fig. 5D and Table 1).
Fig. 5.

The protein mislocalizations detected by the iLocator. (A) The predicted subcellular locations of 28 proteins in seven normal tissues. (B) The predicted subcellular locations of 28 proteins in seven cancer tissues. (C) The locations of these 12 proteins screened by direct comparison method. The green lines represent normal condition, the red represents the cancer condition and the blue lines represent that these locations exist in both conditions. (D) Evaluating the significance of location changes of these 12 selected proteins. The black lines represent changes where the P-value < 0.01, and the gray are P-value > 0.01. The eight proteins involving with black lines were considered as reliable potential biomarkers
Table 1.
Subcellular location changes of the final eight proteins between normal and cancer states
| Protein | Tissue | Subcellular location changesa | Significance of P-values in t-testb |
|---|---|---|---|
| NSDHL | Kidney | (ER and Vesi.)→(Cytopl.) | ER(−): 4.75e-5 |
| Vesi.(−): 1.86e-7 | |||
| Cytopl.(+): 1.5e-12 | |||
| Urinary bladder | (ER and Vesi.)→(Cytopl.) | ER(−): 7.81e-7 | |
| Vesi.(−): 3.02e-8 | |||
| Cytopl.(+): 8.6e-16 | |||
| GOLGA5 | Breast | (Gol.)→(ER) | Gol.(−): 5.42e-7 |
| ER(+): 0.0018 | |||
| Lung | (Gol.)→(ER) | Gol.(−): 5.26e-10 | |
| ER(+): 2.77e-5 | |||
| Pancreas | (Gol.)→(ER) | Gol.(−): 2.90e-9 | |
| ER(+): 4.60e-6 | |||
| Prostate | (Gol.)→(ER) | Gol.(−): 3.33e-10 | |
| ER(+): 1.87e-6 | |||
| Thyroid gland | (Gol.)→(Mito.) | Gol.(−): 4.14e-6 | |
| Mito.(+): 1.94e-6 | |||
| Urinary bladder | (Gol.)→(ER) | Gol.(−): 4.40e-7 | |
| ER(+): 5.91e-4 | |||
| HIP1 | Lung | (Vesi.)→(Cytopl.) | Vesi.(−): 0.0039 |
| Cytopl.(+): 0.0067 | |||
| ACTN4 | Lung | (Cytopl. and Nucl.)→(Cytopl.) | Nucl.(−): 0.0071 |
| Cytopl.(*): 0.3314 | |||
| Prostate | (Cytopl. and Nucl.)→(Cytopl.) | Nucl.(−): 3.80e-4 | |
| Cytopl.(*): 0.0457 | |||
| Kidney | (Cytopl. and Nucl.)→(Cytopl.) | Nucl.(−): 0.0028 | |
| Cytopl.(*): 0.0204 | |||
| FHL2 | Thyroid gland | (Cytopl. and Nucl.)→(Cytopl.) | Nucl.(−): 0.0037 |
| Cytopl.(*): 0.7821 | |||
| DBT | Pancreas | (Cytopl. and Mito.)→(Mito.) | Cytopl.(−): 9.60e-4 |
| Mito.(*): 0.0430 | |||
| AHR | Breast | (Cytopl. and Nucl.)→(Cytopl.) | Nucl.(−): 7.71e-6 |
| Cytopl.(*): 0.0375 | |||
| Pancreas | (Cytopl. and Nucl.)→(Cytopl.) | Nucl.(−): 1.21e-4 | |
| Cytopl.(*): 0.0049 | |||
| Prostate | (Cytopl. and Nucl.)→(Cytopl.) | Nucl.(−): 0.0056 | |
| Cytopl.(*): 0.6544 | |||
| CPT2c | lung | (Mito.)→(Cytopl. and Mito.) | Cytopl.(+): 0.0013 |
| Mito.(*): 0.1741 |
a(Subcellular locations in normal tissue)→(Subcellular locations in cancer tissue).
bThe P-values of involved translocations: ‘−’ means location missed in the cancerous tissue compared with the normal tissue, ‘+’ means new location in the cancerous tissue compared with the normal tissue, and ‘*’ means the same location in both normal and cancerous tissues.
cThis biomarker is applicable to lung adenocarcinoma.
According to HPA, there are multiple subtypes for some cancers like breast cancer, lung cancer and thyroid cancer, so it is necessary to study the effectiveness of screened biomarkers listed in Table 1 for subtype cancers. Thus, we conducted further experiments for the following subtypes: breast cancer of duct carcinoma and lobular carcinoma, lung cancer of adenocarcinoma and squamous cell carcinoma, thyroid cancer of follicular adenoma carcinoma and papillary adenocarcinoma. In Table 1, eight protein–tissue combinations relating to the three cancers need to be further certificated. Using the same method as section 2.5, we compared the images from each subtype of cancers with the normal images separately. The final results show that all the tested proteins, except CPT2, have the same altered subcellular locations among all of the cancer subtypes for a given tissue. However, CPT2 is an exception, where its subcellular location in the lung squamous cell carcinoma subtype is mitochondria, the same as the normal condition. This result indicates that CPT2 is only a suitable biomarker for lung adenocarcinoma (Table 1).
From the listed detailed P-values of the involved translocation changes of the eight proteins (Table 1), we can make five interesting observations:
First, biomarker proteins in cancer tissues tend to move away from other structures and function only in the cytoplasm, except for GOLGA5, DBT and CPT2. This is consistent with previous results that a protein’s cytoplasmic localization can serve as an inactivation mechanism that gives rise to uncontrolled cell proliferation and the onset of disease, which was suggested as a general mechanism for the inactivation of tumor suppressors (Salmena and Pandolfi, 2007).
Second, five screened biomarkers changed from multiple locations to only a single location. Among them, translocations from the cytoplasm and nucleus co-localization to the cytoplasm alone are most frequent. Capable of shuttling between cytoplasm and nucleus and performing functions in nucleus subcellular localization are revealed as important features for some tumor suppressors (Fabbro and Henderson, 2003). Loss of functions in the nucleus of these proteins can change their roles in the cell cycle and can be fatal (Hu et al., 2004).
Third, the results also demonstrate that the developed iLocator can deal with both multi-label and single-label proteins. Of the final eight proteins, six involve multiple locations demonstrating the importance of developing sensitive multi-label classifiers; translocations on protein HIP1 and GOLGA5 also demonstrate that iLocator can still be effectively applied to single-label translocation problems.
Fourth, it also can be seen that if mislocalization occurs, the changes are the same for one protein in different tissues, except the protein GOLGA5. GOLGA5 translocates from the Golgi apparatus to the mitochondrion in thyroid cancer, and from the Golgi apparatus to the ER in the other five cancers. This point, as well as the fact that mislocalization does not occur in all the considered cancers, demonstrates that the metabolic process of protein is various in different tissues. So it is a reasonable way to study the protein subcellular mislocalization in different tissues separately.
Finally, the biomarker protein CPT2 works for lung adenocarcinoma but not lung squamous cell carcinoma. This demonstrates that different subtypes of the same cancer might be driven by different protein translocations. This point is reasonable when considering that cancer subtypes have various pathogenesis.
Some information in the literature can also be found to support our predicted biomarker results. In the annotations in UniProt, ACTN4 locates in both the nucleus and cytoplasm in normal tissues. According to our predicted results, it will locate only in the cytoplasm when cancer occurs in the lung, prostate or kidney. This screened result is consistent with the literature, which found that ACTN4 decreases in the nucleus and locates exclusively to the cytoplasm in cancerous samples and may serve as a biomarker (Honda et al., 1998; Jasavala et al., 2007). Our results also showed that the FHL2 protein is an indicator of thyroid cancer when it only functions in cytoplasm and loses its ability to locate to the nucleus (P-value = 0.0037). The literature supports the finding that cytoplasmic FHL2 is involved in cancer invasion (Kahl et al., 2006; König et al., 2010). Besides, it is also detected that the protein GOLGA5 can translocate from the Golgi apparatus to the mitochondrion in thyroid cancer. This is consistent with the literature specifying that the translocations involving GOLGA5 have been found in thyroid tumors (Corvi et al., 2000; Klugbauer, et al., 1998). All these supporting findings confirm the effectiveness of our system.
4 DISCUSSION
We have built an IHC image-based multi-label human protein subcellular location predictor iLocator and presented a framework to screen cancer biomarkers. The iLocator is an efficient bioimage-based predictor that can handle both single- and multiple-location site proteins simultaneously. By applying iLocator to the datasets from both normal and cancer tissues, eight proteins were identified as cancer biomarkers. We showed that those proteins that can shuttle between multiple subcellular locations are important for normal cell functions, and losing one of the locations may cause disease. These identified mislocated disease-causing protein targets have also opened a new avenue for the therapeutic treatment of some human diseases. For example, by relocating these mislocated proteins to the correct subcellular localizations, they can restore their correct functions, which are helpful to cure the diseases. Although these techniques are still in the infancy stage, they have a promising and bright future.
Based on the current study, the following efforts will be made in future studies toward improving the performance of iLocator.
(1) In this article, we used both the global and local features as the input to the prediction engine and found that the local features are sensitive to detect the distribution differences among multiple locations. Here, we used LBP as the local feature, which is a baseline texture descriptor. Actually, some LBP variants, such as local ternary patterns and local quinary patterns, have been proposed and proven to be powerful in image descriptions (Nanni et al., 2010b; Paci et al., 2013). Therefore, we will try to use these LBP variants as a future development.
(2) To handle high-dimensional image feature vectors, the feature reduction is demonstrated necessary in our experiments and other studies. The SDA method was used in this article to select representative features and performed well. As an alternative, we also tested the random subspace ensemble of SVM, for it saves the feature selection step, and performs well in other studies (Paci et al., 2013). According to our experiments, the random subspace ensemble of the SVM approach is not as good as the ensemble of BR and CC algorithms used in this study (data not shown). The reason could be that the random selected features are not informative to reflect the multi-label samples, and the output of the follow-up single classifiers can cause the fluctuation of the voting results, which are sensitive to noise. These results demonstrate the importance of the development of an effective control mechanism that is able to make the subspace approach suitable to handle multi-label samples.
(3) Fusion of BR and CC algorithms in iLocator is demonstrated useful for enhancing the classification accuracy of multi-label samples. The performance of CC mode confirms that passing association information among labels is able to reflect the different propensities of cellular component co-localizations of proteins. Thus, for the bioimage-based multi-label protein classification problem, it is also important to dig into the existing large databases to model the associations among labels.
(4) The sequence-based and image-based protein subcellular location prediction approaches differ in many ways, including the feature descriptors and classification algorithms. Considering that they both deal with protein samples, the combination of the two strategies should help enhance the classification accuracy because of the complementarities, which can also provide a better understanding of the protein translocations both intuitively and at the amino acid level. In the next step, we plan to explore the idea and combine our 1D sequence-based protein subcellular location predictor Cell-PLoc (Chou and Shen, 2008) and the 2D image-based iLocator as developed in this work.
Supplementary Material
Acknowledgements
We thank Dr. Richard Jang for reading through the manuscript, and the anonymous reviewers for suggestions and comments, which helped improve the quality of this article.
Funding: This work was supported in part by the National Natural Science Foundation of China (61222306, 91130033, 61175024); Shanghai Science and Technology Commission (11JC1404800); a Foundation for the Author of National Excellent Doctoral Dissertation of PR China (201048); and the National Institute of General Medical Sciences (Grants GM083107 and GM084222).
Conflict of interest: none declared.
References
- Boland MV, Murphy RF. A neural network classifier capable of recognizing the patterns of all major subcellular structures in fluorescence microscope images of HeLa cells. Bioinformatics. 2001;17:1213–1223. doi: 10.1093/bioinformatics/17.12.1213. [DOI] [PubMed] [Google Scholar]
- Boutell MR, et al. Learning multi-label scene classification. Pattern Recognit. 2004;37:1757–1771. [Google Scholar]
- Bratthauer GL, Vinh TN. Intracellular location of the SOX9 protein in breast disease. Open Pathol. J. 2009;3:118–123. [Google Scholar]
- Chebira A, et al. A multiresolution approach to automated classification of protein subcellular location images. BMC Bioinformatics. 2007;8:210. doi: 10.1186/1471-2105-8-210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou KC, Shen HB. Cell-PLoc: a package of web servers for predicting subcellular localization of proteins in various organisms. Nat. Protoc. 2008;3:153–162. doi: 10.1038/nprot.2007.494. [DOI] [PubMed] [Google Scholar]
- Corvi R, et al. RET/PCM-1: a novel fusion gene in papillary thyroid carcinoma. Oncogene. 2000;19:4236–4242. doi: 10.1038/sj.onc.1203772. [DOI] [PubMed] [Google Scholar]
- Emanuelsson O, et al. Locating proteins in the cell using TargetP, SignalP and related tools. Nat. Protoc. 2007;2:953–971. doi: 10.1038/nprot.2007.131. [DOI] [PubMed] [Google Scholar]
- Fabbro M, Henderson BR. Regulation of tumor suppressors by nuclear-cytoplasmic shuttling. Exp. Cell Res. 2003;282:59–69. doi: 10.1016/s0014-4827(02)00019-8. [DOI] [PubMed] [Google Scholar]
- Gladden AB, Diehl JA. Location, location, location: the role of cyclin D1 nuclear localization in cancer. J. Cell Biochem. 2005;96:906–913. doi: 10.1002/jcb.20613. [DOI] [PubMed] [Google Scholar]
- Honda K, et al. Actinin-4, a novel actin-bundling protein associated with cell motility and cancer invasion. J. Cell Biol. 1998;140:1383–1393. doi: 10.1083/jcb.140.6.1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu MC, et al. IκB kinase promotes tumorigenesis through inhibition of forkhead FOXO3a. Cell. 2004;117:225–237. doi: 10.1016/s0092-8674(04)00302-2. [DOI] [PubMed] [Google Scholar]
- Hung MC, Link W. Protein localization in disease and therapy. J. Cell Sci. 2011;124:3381–3392. doi: 10.1242/jcs.089110. [DOI] [PubMed] [Google Scholar]
- Jasavala R, et al. Identification of putative androgen receptor interaction protein modules cytoskeleton and endosomes modulate androgen receptor signaling in prostate cancer cells. Mol. Cell. Proteomics. 2007;6:252–271. doi: 10.1074/mcp.M600169-MCP200. [DOI] [PubMed] [Google Scholar]
- Kahl P, et al. Androgen receptor coactivators lysine-specific histone demethylase 1 and four and a half LIM domain protein 2 predict risk of prostate cancer recurrence. Cancer Res. 2006;66:11341–11347. doi: 10.1158/0008-5472.CAN-06-1570. [DOI] [PubMed] [Google Scholar]
- Klugbauer S, et al. Detection of a novel type of RET rearrangement (PTC5) in thyroid carcinomas after Chernobyl and analysis of the involved RET-fused gene RFG5. Cancer Res. 1998;58:198–203. [PubMed] [Google Scholar]
- König K, et al. Four-and-a-half LIM domain protein 2 is a novel regulator of sphingosine 1-phosphate receptor 1 in CCL19-induced dendritic cell migration. J. Immunol. 2010;185:1466–1475. doi: 10.4049/jimmunol.0903449. [DOI] [PubMed] [Google Scholar]
- Li J, et al. Automated analysis and reannotation of subcellular locations in confocal images from the human protein atlas. PLoS One. 2012a;7:e50514. doi: 10.1371/journal.pone.0050514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, et al. Protein subcellular location pattern classification in cellular images using latent discriminative models. Bioinformatics. 2012b;28:i32–i39. doi: 10.1093/bioinformatics/bts230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy RF. Automated interpretation of protein subcellular location patterns: implications for early cancer detection and assessment. Ann. N. Y. Acad. Sci. 2004;1020:124–131. doi: 10.1196/annals.1310.013. [DOI] [PubMed] [Google Scholar]
- Nair R, Rost B. Sequence conserved for subcellular localization. Protein Sci. 2009;11:2836–2847. doi: 10.1110/ps.0207402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nanni L, et al. Advances in Computational Biology. New York: Springer; 2010a. Novel features for automated cell phenotype image classification; pp. 207–213. [DOI] [PubMed] [Google Scholar]
- Nanni L, et al. Local binary patterns variants as texture descriptors for medical image analysis. Artif. Intell. Med. 2010b;49:117–125. doi: 10.1016/j.artmed.2010.02.006. [DOI] [PubMed] [Google Scholar]
- Newberg J, Murphy RF. A framework for the automated analysis of subcellular patterns in human protein atlas images. J. Proteome Res. 2008;7:2300–2308. doi: 10.1021/pr7007626. [DOI] [PubMed] [Google Scholar]
- Paci M, et al. Non-binary coding for texture descriptors in sub-cellular and stem cell image classification. Curr. Bioinform. 2013;8:208–219. [Google Scholar]
- Peng H, et al. Bioimage informatics: a new category in bioinformatics. Bioinformatics. 2012;28:1057. doi: 10.1093/bioinformatics/bts111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng T, et al. Determining the distribution of probes between different subcellular locations through automated unmixing of subcellular patterns. Proc. Natl Acad. Sci. USA. 2010;107:2944–2949. doi: 10.1073/pnas.0912090107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pontèn F, et al. The human protein atlas—a tool for pathology. J. Pathol. 2008;216:387–393. doi: 10.1002/path.2440. [DOI] [PubMed] [Google Scholar]
- Read J, et al. Classifier chains for multi-label classification. Lect. Notes Artif. Int. 2009;5782:254–269. [Google Scholar]
- Salmena L, Pandolfi PP. Changing venues for tumour suppression: balancing destruction and localization by monoubiquitylation. Nat. Rev. Cancer. 2007;7:409–413. doi: 10.1038/nrc2145. [DOI] [PubMed] [Google Scholar]
- Shen HB, Chou KC. Ensemble classifier for protein fold pattern recognition. Bioinformatics. 2006;22:1717–1722. doi: 10.1093/bioinformatics/btl170. [DOI] [PubMed] [Google Scholar]
- Tahir M, et al. Protein subcellular localization of fluorescence imagery using spatial and transform domain features. Bioinformatics. 2012;28:91–97. doi: 10.1093/bioinformatics/btr624. [DOI] [PubMed] [Google Scholar]
- Tsoumakas G, et al. Data Mining and Knowledge Discovery Handbook. USA: Springer; 2010. Mining multi-label data; pp. 667–685. [Google Scholar]
- Uhlen M, et al. Towards a knowledge-based human protein atlas. Nat. Biotechnol. 2010;28:1248–1250. doi: 10.1038/nbt1210-1248. [DOI] [PubMed] [Google Scholar]
- Zhao T, et al. Object type recognition for automated analysis of protein subcellular location. IEEE Trans. Image Process. 2005;14:1351–1359. doi: 10.1109/tip.2005.852456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu L, et al. Multi label learning for prediction of human protein subcellular localizations. Protein J. 2009;28:384–390. doi: 10.1007/s10930-009-9205-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.