

Abstract
Motivation: Converting a pyrosequencing signal into a nucleotide sequence appears highly challenging when signal intensities are low (unitary peak heights 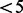 ) or when complex signals are produced by several target amplicons. In these cases, the pyrosequencing software fails to provide correct nucleotide sequences. Accordingly, the objective was to develop the AdvISER-PYRO algorithm, performing an automated, fast and reliable analysis of pyrosequencing signals that circumvents those limitations.
) or when complex signals are produced by several target amplicons. In these cases, the pyrosequencing software fails to provide correct nucleotide sequences. Accordingly, the objective was to develop the AdvISER-PYRO algorithm, performing an automated, fast and reliable analysis of pyrosequencing signals that circumvents those limitations.
Results: In the current mycobacterial amplicon genotyping application, AdvISER-PYRO performed much better than the pyrosequencing software in the following two situations: when converting Single Amplicon Sample (SAS) signals into a correct single sequence (97.2% versus 56.5%), and when translating Multiple Amplicon Sample (MAS) signals into the correct sequence pair (74.5%).
Availability: AdvISER-PYRO is implemented in an R package (http://sites.uclouvain.be/md-ctma/index.php/softwares) and can be used in broad range of clinical applications including multiplex pyrosequencing and oncogene re-sequencing in heterogeneous tumor cell samples.
Contact: jerome.ambroise@uclouvain.be or jean-luc.gala@uclouvain.be
1 INTRODUCTION
Pyrosequencing is a DNA sequencing technology that has many applications including rapid genotyping of a broad spectrum of bacteria. In this type of application, bacterial 16S rRNA gene sequence is a commonly used target for identifying organisms at the species and even strain level (Ronaghi and Elahi, 2002). High throughput sequencing (NGS) is now emerging as a powerful technology able to characterize at the finest scale the diversity in natural microbial and viral populations (Rosen et al., 2012). However, NGS is expensive and requires complex sample preparation and elaborate data analysis. Despite the increased use of NGS for the study of microbial diversity, pyrosequencing therefore remains a cost-effective solution for genotyping a portion of the bacterial genome that allows rapid bacterial or viral genotyping as well as rapid assessment of microbial antibiotic resistance (Amoako et al., 2012; Deccache et al., 2011).
Pyrosequencing is based on pyrophosphate release during nucleotide incorporation (Ronaghi, 2001). The four possible nucleotides are sequentially dispensed in a predetermined order. The first chemi-luminescent signal produced during nucleotide incorporation is detected by a charge-coupled device camera in the pyrosequencer and displayed in a 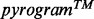 . The
. The 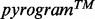 can then be converted automatically into a nucleotide sequence by dedicated software or visually by an experienced operator. The number of incorporated nucleotides at each position is computed from the corresponding peak height. The pyrosequencing data analysis software frequently produces reading errors in homopolymer regions due to the nonlinear light response following incorporation of consecutive identical nucleotides. However, pyrosequencing software interpretation is mostly reliable when a
can then be converted automatically into a nucleotide sequence by dedicated software or visually by an experienced operator. The number of incorporated nucleotides at each position is computed from the corresponding peak height. The pyrosequencing data analysis software frequently produces reading errors in homopolymer regions due to the nonlinear light response following incorporation of consecutive identical nucleotides. However, pyrosequencing software interpretation is mostly reliable when a 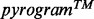 with intermediate (
with intermediate (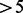 ) unitary peak heights (i.e. the peak heights observed after incorporation of a single nucleotide) is obtained from a Single Amplicon Sample (SAS, i.e. a sample that includes a single target amplicon), as in Figure 1A where unitary peak heights are close to 30.
) unitary peak heights (i.e. the peak heights observed after incorporation of a single nucleotide) is obtained from a Single Amplicon Sample (SAS, i.e. a sample that includes a single target amplicon), as in Figure 1A where unitary peak heights are close to 30.
Fig. 1.
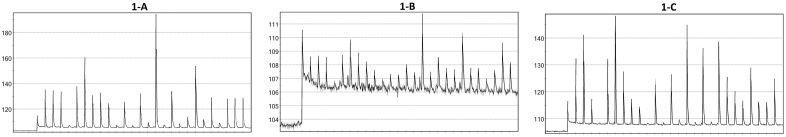
Examples of pyrosequencing signal. (A) Pyrosequencing signal obtained with high DNA concentration in an SAS. The noise intensity is close to 105 while intensities of unitary peaks are close to 135. The unitary peak heights are therefore close to 30. (B) Pyrosequencing signal obtained with low DNA concentration in an SAS. The unitary peak heights are close to 2.5. (C): Pyrosequencing signal obtained with an MAS including two distinct amplicons
Two main situations generate signals preventing automated translation into a correct nucleotide sequence. This happens first when a sample contains a very low DNA concentration, which induces a signal with peak heights close to the noise level (Fig. 1B). It happens also when the 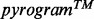 compiles signals from a Multiple Amplicon Sample (MAS, i.e. a sample that includes multiple target amplicons). In this case, the complex signal reflects indeed the integration of signals produced by each amplicon (Fig. 1C). The pyrosequencing data analysis software is not able to distinguish each amplicon-specific signal; hence, it has a limited capacity to produce correct amplicon-specific nucleotide sequences. In such situations, the only option left is a cumbersome, time-consuming and usually very inefficient visual interpretation.
compiles signals from a Multiple Amplicon Sample (MAS, i.e. a sample that includes multiple target amplicons). In this case, the complex signal reflects indeed the integration of signals produced by each amplicon (Fig. 1C). The pyrosequencing data analysis software is not able to distinguish each amplicon-specific signal; hence, it has a limited capacity to produce correct amplicon-specific nucleotide sequences. In such situations, the only option left is a cumbersome, time-consuming and usually very inefficient visual interpretation.
MAS signals are generated in numerous diagnostic applications. A first one is dedicated to multiplex pyrosequencing. In this case, several primers are used simultaneously, which leads to overlapping of primer-specific pyrosequencing signals. The mPSQed and the MultiPSQ softwares were recently developed to aid researchers in designing and analyzing multiplex pyrosequencing assays (Dabrowski and Nitsche, 2012; Dabrowski et al., 2013). The mPSQed software can be used to avoid situations where competing signals from SNPs in different sequences cancel each other out. The MultiPSQ software enables the analysis of multiplex pyrograms originating from various pyrosequencing primers. A second application is found in clinical molecular diagnostic laboratories testing mutations in KRAS, BRAF, PIK3CA and EGFR genes (Chen et al., 2012; Shen and Qin, 2012; Sundström et al., 2010). Recently, a virtual pyrogram generator (Pyromaker) was developed to resolve complex pyrosequencing results (Chen et al., 2012) and could be used to generate simulated  based on user inputs. The interpretation of MAS-pyrosequencing signals was also addressed by Shen et al. who developed a pyrosequencing data analysis software for EGFR, KRAS and BRAF mutation analysis (Shen and Qin, 2012). The software aimed at identifying the presence of mutated cells as well as their proportions. In a first step, this software compared peak heights with a known wild-type peak pattern. If the signal did not fit with the expected wild-type pattern, the software compared it with the mutant peak patterns. When a mutation was identified, the percentage of the candidate mutant gene in the specimen was computed using a built-in formula specific for each mutation. The main drawback of this software was the need for a built-in formula, defined specifically for each mutation and not based on objective parameter computation exploiting a statistical method. A third application that generates MAS signals is related to samples including a heterogeneous microbial population. In this context, a novel approach based on a single Sanger-sequencing reaction was recently proposed for identifying each microbial population from the original population mixture (Amir and Zuk, 2011). This novel approach was based on the reconstruction of a sparse signal using a small number of measurements.
based on user inputs. The interpretation of MAS-pyrosequencing signals was also addressed by Shen et al. who developed a pyrosequencing data analysis software for EGFR, KRAS and BRAF mutation analysis (Shen and Qin, 2012). The software aimed at identifying the presence of mutated cells as well as their proportions. In a first step, this software compared peak heights with a known wild-type peak pattern. If the signal did not fit with the expected wild-type pattern, the software compared it with the mutant peak patterns. When a mutation was identified, the percentage of the candidate mutant gene in the specimen was computed using a built-in formula specific for each mutation. The main drawback of this software was the need for a built-in formula, defined specifically for each mutation and not based on objective parameter computation exploiting a statistical method. A third application that generates MAS signals is related to samples including a heterogeneous microbial population. In this context, a novel approach based on a single Sanger-sequencing reaction was recently proposed for identifying each microbial population from the original population mixture (Amir and Zuk, 2011). This novel approach was based on the reconstruction of a sparse signal using a small number of measurements.
Sparse representations of signals have received a lot of attention in recent years (Huang and Aviyente, 2007; Zheng et al., 2011). The problem solved by sparse representation is to look for a compact representation of signals in terms of linear combination of atoms in an over-complete dictionary [i.e. a dictionary including a number of atoms (p) that exceeds the dimension of the signal space (n)]. In the present study, each atom of the dictionary corresponds to a pyrosequencing signal generated from a known amplicon. For a y testing signal of length n, the issue for sparse representation is to find a vector 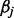 (j = 1 , … , p) such that the following objective function is minimized:
(j = 1 , … , p) such that the following objective function is minimized:
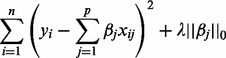 |
(1) |
where 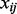 is
is 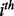 element of the
element of the 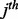 atom, and
atom, and 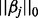 is the
is the  norm of vector
norm of vector  and is equivalent to its number of nonzero components. After having constructed the model, the values of
and is equivalent to its number of nonzero components. After having constructed the model, the values of 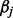 regression coefficients are used for identifying which of the atoms are contributing to the y testing signal. Unfortunately, finding the solution to this problem is NP-hard. However, a solution can be obtained by replacing the
regression coefficients are used for identifying which of the atoms are contributing to the y testing signal. Unfortunately, finding the solution to this problem is NP-hard. However, a solution can be obtained by replacing the 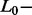 norm by a
norm by a 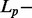 norm penalty on the regression coefficients.
norm penalty on the regression coefficients. 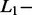 norm penalties are used in lasso regression while
norm penalties are used in lasso regression while 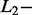 norm penalties are used in ridge regression and a combination of
norm penalties are used in ridge regression and a combination of 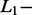 and
and 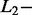 norm penalties are used in Elastic Net (ELNET) (Tibshirani, 1996; Zou and Hastie, 2005).
norm penalties are used in Elastic Net (ELNET) (Tibshirani, 1996; Zou and Hastie, 2005).
To the best of our knowledge, it is the first time that sparse representation of signals is used to analyze pyrosequencing signals. Accordingly, the objective of the present study was to develop a new algorithm for improving the analysis of pyrosequencing signals. This algorithm, called AdvISER-PYRO, deciphers each amplicon-specific signal that contributes to the resulting global signal. In the present study, AdvISER-PYRO was used to identify mycobacterial species by pyrosequencing. Considering the likely existence of heterogenous mycobacterial populations in a clinical specimen, this case study appears particularly relevant. Indeed, the identification of causative mycobacterial agents in infected samples can be affected by the presence of other ubiquitous mycobacterial species (Covert et al., 1999). Moreover, coinfection with Mycobacterium tuberculosis (MTB) and nontuberculous mycobacteria (NTB) in clinical samples, and notably in AIDS patients, can easily be overlooked when using conventional identification methods, and presents therefore a real challenge in diagnosis and treatment. This probably explains at least partially why evidence of dual infection with MTB and NTB is scanty (Gopinath and Singh, 2009). The performance of AdvISER-PYRO in identifying mycobacterial amplicons was assessed using signals generated by SAS (n = 220) and MAS (n = 144), the latter containing two distinct amplicons. For SAS signals, the AdvISER-PYRO performance was compared with the percentage of correct identification obtained with the pyrosequencing data analysis software ( Software V.2.1.1, Biotage AB, Sweden) and reflecting the
Software V.2.1.1, Biotage AB, Sweden) and reflecting the  translation into a correct nucleotide sequence.
translation into a correct nucleotide sequence.
2 METHODS
Signals were generated with a pyrosequencer  (Biotage AB, Sweden), following successive dispensation of 26 nucleotides. The predefined order of dispensation of these nucleotides was determined according to the sequence tag corresponding to a hypervariable region of the Mycobacterium genome. Accordingly, dispensed nucleotides produced distinct
(Biotage AB, Sweden), following successive dispensation of 26 nucleotides. The predefined order of dispensation of these nucleotides was determined according to the sequence tag corresponding to a hypervariable region of the Mycobacterium genome. Accordingly, dispensed nucleotides produced distinct  peaks, each peak height being proportional to the number of identical nucleotides consecutively incorporated. In this study, a signal is defined as the global pattern integrating the 26 successive peak heights.
peaks, each peak height being proportional to the number of identical nucleotides consecutively incorporated. In this study, a signal is defined as the global pattern integrating the 26 successive peak heights.
All amplicons of the current Mycobacterium target sequence started with the same single nucleotide. Accordingly, the first peak height was named ‘First Unitary Peak Height’ (FUPH) and was used as an indicator of the global signal intensity. Pyrosequencing was performed as classically described. In brief, the Mycobacterium target sequence was first amplified by PCR. The PCR amplification was carried out using a couple of forward and biotinylated reverse primers. The biotinylated amplicons were immobilized on streptavidin-coated magnetic beads and denaturated. After denaturation, the biotinylated single-stranded amplicon was isolated and allowed to hybridize with a sequencing primer. Owing to the close relatedness of some mycobacterial species (e.g. M.marinum and M.ulcerans) on one hand, and the genetic heterogeneity within other species (e.g. M.gordonae), a single amplicon can correspond to more than one mycobacterial species and conversely, a mycobacterial species can be associated with more than one specific amplicon (Table 1).
Table 1.
Correspondence between amplicons and mycobacterial species
| Amplicon | Mycobacterium | Amplicon | Mycobacterium | Amplicon | Mycobacterium |
|---|---|---|---|---|---|
| Amplicon1 | M.avium subsp. avium | Amplicon12 | M.interjectum | Amplicon24 | M.paraffinicum |
| M.avium subsp. paratuberculosis | Amplicon13 | M.marseillense | Amplicon25 | M.scrofulaceum | |
| M.avium subsp. silvaticum | Amplicon14 | M.intracellulare | Amplicon26 | M.scrofulaceum | |
| Amplicon2 | M.bohemicum | Amplicon15 | M.kansasii | Amplicon27 | M.scrofulaceum |
| Amplicon3 | M.celatum | Amplicon16 | M.lentiflavum | M.paraffinicum | |
| Amplicon4 | M.celatum | Amplicon17 | M.lentiflavum | Amplicon28 | M.simiae |
| Amplicon5 | M.chelonae | Amplicon18 | M.malmoense | Amplicon29 | M.simiae |
| M.abscessus | Amplicon19 | M.marinum | Amplicon30 | M.szulgai | |
| Amplicon6 | M.gastri | M.ulcerans | Amplicon31 | M.genavense | |
| Amplicon7 | M.gordonae | Amplicon20 | M.non chromogenicum | M.triplex | |
| Amplicon8 | M.gordonae | M.ratisbonense | Amplicon32 | M.tuberculosis | |
| Amplicon9 | M.gordonae | Amplicon21 | M.non chromogenicum | M.bovis | |
| Amplicon10 | M.hiberniae | Amplicon22 | M.non chromogenicum | M.africanum | |
| Amplicon11 | M.interjectum | Amplicon23 | M.paraffinicum | Amplicon33 | M.xenopi |
Pyrosequencing signals were generated from SAS (n = 220) and MAS (n = 144). SAS were generated from single mycobacterial clinical isolates. Three distinct types of MAS were analyzed in the current study. MAS-1 were generated by mixing in various proportion (50/50%; 33/66%) the amplification products generated from two separate PCR performed on two distinct mycobacterial clinical isolates (n = 84). MAS-2 were generated with a single PCR performed on a reconstructed sample where DNA from two distinct mycobacterial clinical isolates were mixed in various proportions (10/90%; 25/75%; 50/50%; 75/25%; 90/10%) (n = 45). MAS-3 were generated with a single PCR performed on natural clinical samples from patients with a mycobacterial co-infection (n = 15). In MAS-2 and MAS-3, the final proportion of both amplicons after PCR amplification was unknown because of the amplicon-specific efficiency of the PCR reaction likely altering the initial DNA proportions. The estimated proportion of the minor amplicon could therefore vary widely between 0.1% and 50.0%.
All SAS and MAS signals were divided into training (SAS, n = 99), validation (SAS, n = 103; MAS, n = 122) and test (SAS, n = 18; MAS, n = 22) datasets. A standardized learning dictionary was constructed based on signals from the training dataset. AdvISER-PYRO hyperparameters were tuned on the validation dataset while performance was assessed on the test dataset. Given the small size of the test dataset, a bootstrap method was also applied to provide a reliable evaluation of AdvISER-PYRO performance.
In parallel, all  from SAS were also analyzed with the pyrosequencing data analysis software (
from SAS were also analyzed with the pyrosequencing data analysis software ( Software V.2.1.1, Biotage AB, Sweden) and translated into nucleotide sequences.
Software V.2.1.1, Biotage AB, Sweden) and translated into nucleotide sequences.
3 ALGORITHM
The first step in developing the AdvISER-PYRO was to create a standardized learning dictionary from the training dataset (SAS, n = 99) that included at least one signal (i.e. the global pattern integrating the 26 successive peak heights) for each amplicon. Standardization of the dictionary was performed by dividing each signal (i.e. the 26 successive peak heights) by its corresponding FUPH. After standardization, all signals in the learning dictionary were therefore characterized by a FUPH equal to 1.
The second step was to build a penalized linear model with the y testing signal as response variable and all signals from the learning dictionary as predictor variables. In this model, the sum of regression coefficients corresponding to each amplicon was computed and recorded as the amplicon contribution to the signal. As the number of observations (i.e. the length of the signal which was n = 26) was smaller than the number of variables (i.e. the total number of atoms in the learning dictionary which was P > 33), 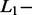 and
and 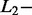 norm penalties were applied for estimating the regression coefficients. These penalties were the first two hyperparameters of AdvISER-PYRO. As the signal contribution from each atom should have a positive value, an additional constraint imposing this prerequisite was implemented. The intercept of the model was also set to 0. The penalized regression models were built using the penalized function of the corresponding R package (Goeman, 2008).
norm penalties were applied for estimating the regression coefficients. These penalties were the first two hyperparameters of AdvISER-PYRO. As the signal contribution from each atom should have a positive value, an additional constraint imposing this prerequisite was implemented. The intercept of the model was also set to 0. The penalized regression models were built using the penalized function of the corresponding R package (Goeman, 2008).
In the third step, amplicons that significantly contributed to the signal were selected. A specific amplicon was considered significant when its contribution to the signal was higher than the Significant Contribution Threshold, which was the third hyperparameter of AdvISER-PYRO.
4 RESULTS
4.1 Hyperparameter optimization on the validation dataset
All signals from the validation dataset (SAS, n = 103; MAS, n = 122) were used to evaluate and optimize AdvISER-PYRO hyperparameters. Accordingly, the percentage of correct identification of SAS and MAS signals were computed with various values of the 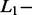 and
and 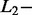 norm penalties and of the Significant Contribution Threshold. For SAS and MAS signals, a right identification was recorded when AdvISER-PYRO correctly identified the unique amplicon (SAS) or the pair thereof (MAS). Any incorrect signal identification included the wrong prediction of an additional (false-positive) amplicon. The percentages of correct SAS and MAS signal identification using the validation dataset are given in Table 2. It was impossible to compute the percentage of correct identification with zero
norm penalties and of the Significant Contribution Threshold. For SAS and MAS signals, a right identification was recorded when AdvISER-PYRO correctly identified the unique amplicon (SAS) or the pair thereof (MAS). Any incorrect signal identification included the wrong prediction of an additional (false-positive) amplicon. The percentages of correct SAS and MAS signal identification using the validation dataset are given in Table 2. It was impossible to compute the percentage of correct identification with zero 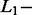 and
and 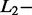 norm penalties, as the number of dimensions (P = 99) exceeded the number of observations (n = 26).
norm penalties, as the number of dimensions (P = 99) exceeded the number of observations (n = 26).
Table 2.
Percentage of correct SAS- and MAS-signal identification with AdvISER-PYRO according to 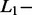 and
and 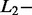 norm penalties and the Significant Contribution Threshold
norm penalties and the Significant Contribution Threshold
| Significant contribution threshold |
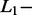 norm norm |
SAS (n = 103) |
MAS (n = 122) |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
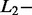 norm norm |
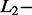 norm norm |
||||||||||
| 0.00 | 0.01 | 0.05 | 0.10 | 0.50 | 0.00 | 0.01 | 0.05 | 0.10 | 0.50 | ||
| 1 | 0.00 | / | 90.3 | 84.5 | 82.5 | 68.9 | / | 62.3 | 58.2 | 52.5 | 29.5 |
| 0.01 | 89.3 | 89.3 | 84.5 | 82.5 | 68.9 | 65.6 | 62.3 | 59.0 | 52.5 | 29.5 | |
| 0.05 | 89.3 | 90.3 | 84.5 | 82.5 | 68.9 | 67.2 | 62.3 | 59.0 | 52.5 | 29.5 | |
| 0.10 | 89.3 | 90.3 | 84.5 | 82.5 | 68.9 | 66.4 | 61.5 | 59.0 | 52.5 | 29.5 | |
| 0.50 | 89.3 | 90.3 | 84.5 | 82.5 | 68.9 | 65.6 | 63.1 | 59.0 | 51.6 | 29.5 | |
| 2 | 0.00 | / | 94.2 | 93.2 | 91.3 | 83.5 | / | 77.0 | 75.4 | 73.0 | 59.0 |
| 0.01 | 94.2 | 94.2 | 93.2 | 91.3 | 83.5 | 77.9 | 77.0 | 74.6 | 73.0 | 59.0 | |
| 0.05 | 94.2 | 94.2 | 93.2 | 91.3 | 83.5 | 77.9 | 77.0 | 73.8 | 73.0 | 59.0 | |
| 0.10 | 94.2 | 94.2 | 93.2 | 91.3 | 83.5 | 77.9 | 77.0 | 74.6 | 73.0 | 59.0 | |
| 0.50 | 94.2 | 94.2 | 93.2 | 91.3 | 83.5 | 77.0 | 77.9 | 75.4 | 71.3 | 59.0 | |
| 3 | 0.00 | / | 95.1 | 95.1 | 94.2 | 90.3 | / | 66.4 | 66.4 | 65.6 | 59.0 |
| 0.01 | 95.1 | 95.1 | 95.1 | 94.2 | 90.3 | 66.4 | 66.4 | 65.6 | 65.6 | 59.0 | |
| 0.05 | 95.1 | 95.1 | 95.1 | 94.2 | 90.3 | 66.4 | 66.4 | 66.4 | 65.6 | 59.0 | |
| 0.10 | 95.1 | 95.1 | 95.1 | 94.2 | 90.3 | 66.4 | 66.4 | 66.4 | 65.6 | 59.0 | |
| 0.50 | 95.1 | 95.1 | 95.1 | 94.2 | 90.3 | 67.2 | 65.6 | 65.6 | 64.8 | 58.2 | |
The effects of 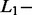 and
and 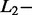 norm penalties were very different, as generally accepted in literature.
norm penalties were very different, as generally accepted in literature. 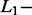 norm penalty tends to produce many regression coefficients shrunk exactly to zero and a few other regression coefficients with comparatively little shrinkage. At the opposite,
norm penalty tends to produce many regression coefficients shrunk exactly to zero and a few other regression coefficients with comparatively little shrinkage. At the opposite, 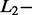 norm penalty tends to result in all small but nonzero regression coefficients (Goeman et al., 2012). In the current application, this second effect induced an important decrease of the percentage of correct identification. The effect of the SCT hyperparameter on the percentage of correct identification was different for SAS and MAS signals. With SAS signals, higher value of SCT improved the results by decreasing the number of false-positive results. With MAS signals, the optimal SCT value resulted from a compromise between the minimisation of false-positive (less frequent with a high SCT value) and false-negative (less frequent with a low SCT value) results.
norm penalty tends to result in all small but nonzero regression coefficients (Goeman et al., 2012). In the current application, this second effect induced an important decrease of the percentage of correct identification. The effect of the SCT hyperparameter on the percentage of correct identification was different for SAS and MAS signals. With SAS signals, higher value of SCT improved the results by decreasing the number of false-positive results. With MAS signals, the optimal SCT value resulted from a compromise between the minimisation of false-positive (less frequent with a high SCT value) and false-negative (less frequent with a low SCT value) results.
4.2 Percentage of correct identification on the test dataset
All SAS (n = 18) and MAS signals (n = 22) of the test dataset were analyzed with AdvISER-PYRO. The algorithm hyperparameters were chosen according to the percentage of correct SAS- and MAS-signal identification using the validation dataset. The Significant Contribution Threshold was therefore set to 2 whereas the 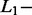 and
and 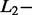 norm penalties were set to 0.05 and 0, respectively. These hyperparameter values produced indeed the best compromise between the percentage of correct identification with SAS (94.2%) and MAS signals (77.9%).
norm penalties were set to 0.05 and 0, respectively. These hyperparameter values produced indeed the best compromise between the percentage of correct identification with SAS (94.2%) and MAS signals (77.9%).
Among the 18 SAS signals, all (100%) were correctly translated into their corresponding single sequence. Among the 22 MAS signals, 16 (72.7%) were translated into their correct sequence pair. The six remaining MAS signals (27.3%) were translated by AdvISER-PYRO into one correct sequence whereas the other expected sequence from the pair was missing (false-negative). Each false-negative sequence resulted from the analysis of a MAS-2 signal where estimated contribution of the corresponding minor amplicon was lower than the Significant Contribution Threshold.
4.3 Bootstrap evaluation of the percentage of correct identification
Given the small size of the test dataset, a 100-fold bootstrap approach was used to obtain a reliable evaluation of the percentage of correct identification. The bootstrap was applied on all SAS (n = 220) and MAS (n = 144) signals. At each iteration, the SAS signals were randomly divided into a training (n = 101) and a test dataset (n = 119). All MAS signals (n = 144) were included in the test dataset. To limit the computation time, the AdvISER-PYRO hyperparameters were not optimised for each iteration (using an internal cross-validation loop) but were kept constant across all iterations (Significant Contribution Threshold = 2; 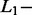 norm = 0.05,
norm = 0.05, 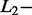 norm = 0).
norm = 0).
A large majority (94.4%) of SAS signals were correctly translated into their corresponding single sequence. Only few (2.5%) SAS signals were falsely translated into two or more distinct sequences, and these always included the correct sequence and another sequence being not present in the sample (i.e. false-positive). The remaining SAS signals (3.1%) were translated into a single wrong sequence.
Most MAS signals (74.5%) were correctly translated into their corresponding sequence pair. However, the percentages of correct identification differed significantly between the three distinct types of MAS signals. For MAS-1, most signals (93.3%) were correctly translated into the correct sequence pair. Few MAS-1 signals (2.6%) were translated by AdvISER-PYRO into one correct sequence whereas the other expected sequence from the pair of amplicons was missing (i.e. false-negative) or wrong. Few MAS-1 signals (4.1%) were predicted with a third additional sequence (i.e. false-positive). The signal contributions of both amplicons were generally well-balanced but not perfectly representative of the amplicon proportion within the sample. The relative signal contribution of the minor amplicon was 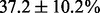 for samples with 50/50% and
for samples with 50/50% and 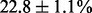 for samples with 33/66% of both amplicons. For MAS-2 and MAS-3, some signals (53.9% for MAS-2 and 30.7% for MAS-3) were correctly translated into the correct sequence pair. Some MAS-2 and MAS-3 signals (46.1% for MAS-2 and 51.5% for MAS3) were translated by AdvISER-PYRO into one correct sequence whereas the other expected sequence from the pair of amplicons was missing (i.e. false-negative) or wrong. Some MAS-3 signals (17.8%) were predicted with a third additional sequence (i.e. false-positive).
for samples with 33/66% of both amplicons. For MAS-2 and MAS-3, some signals (53.9% for MAS-2 and 30.7% for MAS-3) were correctly translated into the correct sequence pair. Some MAS-2 and MAS-3 signals (46.1% for MAS-2 and 51.5% for MAS3) were translated by AdvISER-PYRO into one correct sequence whereas the other expected sequence from the pair of amplicons was missing (i.e. false-negative) or wrong. Some MAS-3 signals (17.8%) were predicted with a third additional sequence (i.e. false-positive).
4.4 Comparison with the 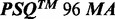 Software V.2.1.1.
Software V.2.1.1.
A leave-one-out cross-validation was applied on AdvISER-PYRO to produce a single and unique answer for each SAS signal. Six amplicons were excluded from the comparison between both methods. These amplicons presented a single pyrosequencing signal that was automatically included within the dictionary and was consequently excluded from the test dataset. The comparison was therefore performed on 114  .
.
Most SAS signals (208/214; 97.2%) were correctly translated into a single correct sequence by AdvISER-PYRO. This percentage of correct identification was much higher than the percentage obtained with the  Software V.2.1.1. that translated 121/214 (56.5%)
Software V.2.1.1. that translated 121/214 (56.5%)  into correct nucleotide sequences. Compared with this software, the percentage of correct identification obtained with AdvISER-PYRO was particularly high at low (FUPH
into correct nucleotide sequences. Compared with this software, the percentage of correct identification obtained with AdvISER-PYRO was particularly high at low (FUPH 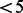 ) signal intensities (Fig. 2).
) signal intensities (Fig. 2).
Fig. 2.
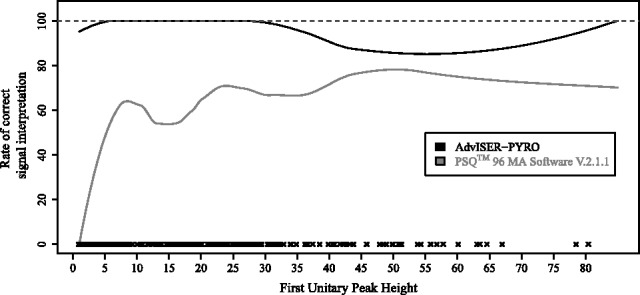
Comparison of the percentage of correct identification as a function of signal intensities (FUPH). The comparison was performed between AdvISER-PYRO and the  Software V.2.1.1, using Local Polynomial Regression Models on identifications obtained with SAS signals. The symbols on the x-axis represent the distribution of the FUPH in the SAS dataset
Software V.2.1.1, using Local Polynomial Regression Models on identifications obtained with SAS signals. The symbols on the x-axis represent the distribution of the FUPH in the SAS dataset
4.5 Illustration of AdvISER-PYRO application
Figure 3 illustrates the results obtained with AdvISER-PYRO when applied on four distinct pyrosequencing signals.
Fig. 3.

Four examples of signal identification with AdvISER-PYRO. The pyrosequencing signal is represented by vertical black lines. The contribution of each atom is represented with boxes stacked one on top of the other
In Figure 3A, a signal with a low FUPH (2.49) was generated from a SAS. Despite this low signal-to-noise ratio, the signal was correctly converted in the corresponding single nucleotide sequence (amplicon 32). The correlation coefficient (r) between the predicted values of the penalized regression model and the 26 values of the signal was higher than 0.99, confirming the identification reliability obtained with AdvISER-PYRO.
In Figure 3B, the signal was generated from a MAS-1 including PCR product of amplicons 32 and 14 in equivalent proportion (50/50%). Both amplicons were correctly identified by AdvISER-PYRO and the signal contributions of both amplicons were well-balanced but not perfectly equivalent (41/59%). The correlation coefficient (r) between the predicted values of the penalized regression model and the 26 values of the signal was higher than 0.99, confirming the identification reliability obtained with AdvISER-PYRO.
In Figure 3C, the signal was generated from an SAS including a single amplicon, which was excluded from the dictionary. The contributions of atoms corresponding to two distinct amplicons (amplicons 07 and 08) are wrongly identified by AdvISER-PYRO. However, this situation induces a low correlation coefficient (r = 0.759) between the predicted values of the penalized regression model and the 26 values of the signal, pointing out the low reliability of the AdvISER-PYRO identification and allowing the operator to reject this result.
In Figure 3D, the signal was produced from a MAS-2 generated with a single PCR performed on a reconstructed sample where DNA from two distinct mycobacterial clinical isolates (corresponding to amplicon 32 and 14) were mixed in equal proportion (50/50%). The pyrosequencing signal was perfectly (r = 1) modeled as a linear combination of signals corresponding to amplicon 32 showing that initial DNA proportion was strongly altered after PCR amplification.
The computation time for each example was <1 s on an Intel(R) Core(TM) i7-2640M CPU @ 2.80 GHz computer.
5 DISCUSSION
The AdvISER-PYRO algorithm appears as an efficient tool that can reliably be used to identify amplicons in pyrosequencing signals generated by SAS or MAS. The first prerequisite is that pyrosequencing signal analysis by AdvISER-PYRO requires the corresponding amplicon representation in the dictionary. Otherwise, the model produced by AdvISER-PYRO would be wrong. In that case, the fitted values would be weakly correlated with the pyrosequencing signal, which will allow operators to avoid erroneous interpretation.
From this study, it also appears that a quantitative interpretation of signal contributions is not feasible. Indeed, the estimated relative contribution of each amplicon in the MAS-2 pyrosequencing signals did not correspond to the initial ratio of each DNA target. This derives from significant differences in PCR amplification efficiency of these DNA targets, hence to differences in the respective amount of amplicons to be pyrosequenced. Moreover, the estimated relative contribution of each amplicon in the MAS-1 pyrosequencing signals did not correspond to the initial ratio of PCR product, as previously reported in Amoako et al. (2012) who showed that all primer–target association does not perform equally well.
A second prerequisite for using AdvISER-PYRO is that each amplicon produces a specific signal which is different from signals generated by all other amplicons expected to be produced in the genetic identification process. If this is indeed the case, the AdvISER-PYRO algorithm can be applied to a wide spectrum of pyrosequencing-based genotyping applications other than mycobacterial species typing, and is able to analyze genotyping data generated by various types of polymorphisms including single nucleotide polymorphism, single nucleotide repeat sequence, deletion and insertion. A cyclic dispensation order can be used if it satisfies this second prerequisite (i.e. if it produces distinct amplicon-specific signals). However, choosing a selected dispensation order can be advantageous to maximise the signal differences inherent to pyrosequencing signals produced respectively by each type of amplicon according to the genotyping application. Maximising signal differences could also be achieved by increasing the number of dispensed nucleotides with the deleterious consequence that long reads are associated with higher peak height variance. Consequently, the choice of an optimal nucleotide dispensation order is based on a difficult compromise between the quantity and the quality of the acquired information.
In the context of oncogene re-sequencing in heterogeneous tumor cell samples, AdvISER-PYRO could be used as a tool complementary to Pyromaker (Chen et al., 2012). The latter is used to complete the representative learning dictionary by generating a theoretical pyrosequencing signal for each mutation for which no biological sample is yet available; hence, experimental signal is still lacking in the dictionary. If multiplex pyrosequencing needs to be carried out, AdvISER-PYRO could be applied to the analysis of complex signals obtained with multiplex primers designed with the mPSQed software (Dabrowski and Nitsche, 2012). In this study, AdvISER-PYRO showed a high percentage of correct identification with signals generated from samples containing two distinct amplicons. Although this has not been yet tested and needs to be validated, it should be pointed out that AdvISER-PYRO can also be used on samples containing more than two distinct amplicons.
In the present study, the optimisation of AdvISER-PYRO hyperparameters was done on a validation dataset to obtain the higher percentage of correct identification, irrespective of the impact of a false-positive or -negative results. However, such optimisation should ideally be performed for each genotyping application by considering the global clinical context. In oncogene re-sequencing applications, the SCT could indeed be defined in terms of relative contribution by estimating the Limit of Blank (LoB) from a dilution series experiment. This LoB could be modulated to limit the probability of either false-negative or -positive results by considering the clinical impact relative to both types of error.
As illustrated here, AdvISER-PYRO is expected to substantially help improve the reading and translation of the  into a correct sequence or set of sequences in case of SAS and MAS signals, respectively. Validation and optimization of AdvISER-PYRO in clinical applications other than mycobacterial genotyping are already under way.
into a correct sequence or set of sequences in case of SAS and MAS signals, respectively. Validation and optimization of AdvISER-PYRO in clinical applications other than mycobacterial genotyping are already under way.
Acknowledgement
We would like to thank Michel Heusterspreute for his scientific and technical support in the design and development of pyrosequencing assays.
Funding: This work was supported by the Defence Laboratories Department of the Belgian Armed Forces.
Conflict of Interest: none declared.
References
- Amir A, Zuk O. Bacterial community reconstruction using compressed sensing. J. Comput. Biol. 2011;18:1723–1741. doi: 10.1089/cmb.2011.0189. [DOI] [PubMed] [Google Scholar]
- Amoako K, et al. Rapid detection and antimicrobial resistance gene profiling of yersinia pestis using pyrosequencing technology. J. Microbiol. Methods. 2012;90:228–34. doi: 10.1016/j.mimet.2012.05.012. [DOI] [PubMed] [Google Scholar]
- Chen G, et al. A virtual pyrogram generator to resolve complex pyrosequencing results. J. Mol. Diagn. 2012;14:149–159. doi: 10.1016/j.jmoldx.2011.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Covert T, et al. Occurrence of nontuberculous mycobacteria in environmental samples. Appl. Environ. Microbiol. 1999;65:2492–2496. doi: 10.1128/aem.65.6.2492-2496.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dabrowski P, Nitsche A. mPSQed: a software for the design of multiplex pyrosequencing assays. PloS One. 2012;7:e38140. doi: 10.1371/journal.pone.0038140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dabrowski PW, et al. MultiPSQ: a software solution for the analysis of diagnostic n-plexed pyrosequencing reactions. PloS One. 2013;8:e60055. doi: 10.1371/journal.pone.0060055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deccache Y, et al. Development of a pyrosequencing assay for rapid assessment of quinolone resistance in acinetobacter baumannii isolates. J. Microbiol. Methods. 2011;86:115–118. doi: 10.1016/j.mimet.2011.04.007. [DOI] [PubMed] [Google Scholar]
- Goeman J. Penalized: L1 (lasso) and L2 (ridge) penalized estimation in GLMs and in the cox model. R package version 09-21 2008. 2008 http://cran.r-project.org/web/packages/penalized/ (3 July 2013, date last accessed) [Google Scholar]
- Goeman J, et al. L1 and L2 penalized regression models. cran.r-project.or. 2012 [Google Scholar]
- Gopinath K, Singh S. Multiplex PCR assay for simultaneous detection and differentiation of mycobacterium tuberculosis, mycobacterium avium complexes and other mycobacterial species directly from clinical specimens. J. Appl. Microbiol. 2009;107:425–435. doi: 10.1111/j.1365-2672.2009.04218.x. [DOI] [PubMed] [Google Scholar]
- Huang K, Aviyente S. Sparse representation for signal classification. Adv. Neural Inform. Proces. Syst. 2007;19:609. [Google Scholar]
- Ronaghi M. Pyrosequencing sheds light on DNA sequencing. Genome Res. 2001;11:3–11. doi: 10.1101/gr.11.1.3. [DOI] [PubMed] [Google Scholar]
- Ronaghi M, Elahi E. Pyrosequencing for microbial typing. J. Chromatography B. 2002;782:67–72. doi: 10.1016/s1570-0232(02)00693-1. [DOI] [PubMed] [Google Scholar]
- Rosen M, et al. Denoising PCR-amplified metagenome data. BMC Bioinformatics. 2012;13:283. doi: 10.1186/1471-2105-13-283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen S, Qin D. Pyrosequencing data analysis software: a useful tool for EGFR, KRAS and BRAF mutation analysis. Diagnostic Pathol. 2012;7:56. doi: 10.1186/1746-1596-7-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundström M, et al. KRAS analysis in colorectal carcinoma: analytical aspects of pyrosequencing and allele-specific PCR in clinical practice. BMC Cancer. 2010;10:660. doi: 10.1186/1471-2407-10-660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tibshirani R. Regression shrinkage and selection via the lasso. J. R. Stat. Soc. B Methodol. 1996:267–288. [Google Scholar]
- Zheng C, et al. Metasample-based sparse representation for tumor classification. Comput. Biol. Bioinform. 2011;8:1273–1282. doi: 10.1109/TCBB.2011.20. [DOI] [PubMed] [Google Scholar]
- Zou H, Hastie T. Regularization and variable selection via the elastic net. J. R. Stat. Soc. B Stat. Methodol. 2005;67:301–320. [Google Scholar]