

Abstract
Neuropsychiatric pathologies, including neurodegenerative diseases and neurodevelopmental syndromes, are frequently associated with dysregulation of various essential cellular mechanisms, such as transcription, mitochondrial respiration and protein degradation. In these complex scenarios, it is difficult to pinpoint the specific molecular dysfunction that initiated the pathology or that led to the fatal cascade of events that ends with the death of the neuron. Among the possible original factors, epigenetic dysregulation has attracted special attention. This review focuses on two highly related epigenetic factors that are directly involved in a number of neurological disorders, the lysine acetyltransferases CREB-binding protein (CBP) and E1A-associated protein p300 (p300). We first comment on the role of chromatin acetylation and the enzymes that control it, particularly CBP and p300, in neuronal plasticity and cognition. Next, we describe the involvement of these proteins in intellectual disability and in different neurodegenerative diseases. Finally, we discuss the potential of ameliorative strategies targeting CBP/p300 for the treatment of these disorders.
Keywords: Histone acetylation, CBP, p300, Rubinstein-Taybi syndrome, Huntington’s disease, Alzheimer’s disease, HDACi, KATe, KATi.
1. CHROMATIN ACETYLATION AND COGNITION
1.1. Epigenetic Regulation of Cognitive Function
Epigenetic mechanisms, such as DNA methylation and post-translational modification of histones, provide a molecular interface between genes and environment that allows the modulation of gene expression by experience. These mechanisms are known to play a key role in the specification of cell lineages during embryonic development. Throughout cell differentiation, cells with the same genome acquire unique properties by switching on and off specific gene programs; their acquired gene expression profiles will then be inherited by daughter cells along successive rounds of division [1]. It has been known for years that this process depends on the epigenetic modification of the chromatin of differentiating cells. In comparison, the idea that epigenetic mechanisms are involved in cognition is relatively new. The relevance of these mechanisms to brain function was long overlooked by neuroscientists because adult neurons do not divide and are thought to be terminally differentiated. However, studies during the last decade have indicated that these mechanisms might allow neurons to customize their gene expression patterns according to experience, widening the computational and storage capabilities of neural networks. Epigenetic mechanisms also represent a suitable basis for the phenotypic variation in behavioral and cognitive traits observed in humans. Thus, in monozygotic twins who share the same genomic sequence, the differences in the contents and distributions of epigenetic marks grow throughout life along with the discordance of psychological traits [2].
Among these epigenetic mechanisms, the acetylation and deacetylation of histone tails play a prominent and particularly well-studied role. The N-terminus of the four nucleosome histones (H2A, H2B, H3 and H4) is unstructured and has high intrinsic flexibility that enables the dynamic interaction of histones with DNA. The addition of an acetyl group to lysine residues located in these tails neutralizes its basic charge and loosens up the contacts between histones and DNA, which causes the relaxation of the chromatin and may facilitate the recruitment of transcription factors and the basal transcriptional machinery to specific DNA sequences. Thus, the degree of chromatin compaction is thought to contribute to both the definition of global patterns of transcriptional competence and to the local regulation of transcription initiation and elongation at specific loci [3]. Histone acetylation is mediated by histone acetyltransferase activities (HATs) and has been generally associated with gene activation, whereas histone deacetylation is mediated by histone deacetylases (HDACs) and has been associated with gene repression. However, this association is more complex than initially expected because both HATs and HDACs bind to gene promoters regardless of their transcriptional activity [4]. The term poised (also referred to as primed or bookmarked) gene has recently been coined to explain the presence of histone modifications associated with transcriptionally active genes at promoters of inactive genes that likely enable their induction in response to particular stimuli [4-6].
Importantly, the balance between histone acetylation and deacetylation is very dynamic. In neurons, chromatin acetylation is modulated in response to neuronal activity and during plasticity processes [7]. Extensive correlative evidence supports a role for histone acetylation in memory storage that seems highly conserved through evolution [8-13]. Although we present below some seminal findings supporting a role for histone acetylation in plasticity and memory, we do not intend to review the extensive literature on this topic here because several excellent review articles have recently been published [14-18].
Both in invertebrates and vertebrates, memory acquisition has been associated with changes in histone acetylation in the neuronal nucleus. In some of these studies, the changes were restricted to specific gene promoters and investigated through chromatin immunoprecipitation (ChIP) assays, in others the changes were so general that they could be detected at the bulk chromatin level by western blot or immunohistochemistry. For example, in Aplysia californica, stimuli that trigger long-term facilitation (LTF) or long-term depression (LTD), two forms of plasticity that are thought to be cellular correlates of memory formation in this organism, bidirectionally regulate the acetylation of H3-lysine (K)14 and H4-K8 in the promoter of the gene encoding the CCAAT-box-enhanced binding protein (C/EBP), a transcription factor involved in the maintenance of LTF [19]. More recently, experiments in another invertebrate, the crab Chasmagnathus, demonstrated that strong context-signal training triggers increases in bulk histone H3 acetylation in the neurons involved in this form of learning [20]. Experiments in rodents indicate that several nucleosome positions are acetylated during learning tasks. These marks are associated with specific memory stages and have been detected in the chromatin of neuronal types known to be engaged in that type of memory. For instance, bulk H3 acetylation increases in the hippocampus shortly after contextual fear conditioning [21], whereas spatial memory acquisition in a water maze task is associated with acetylation of hippocampal H2B and H4 [22]. H3 acetylation increases have also been observed in the lateral amygdala after cued fear conditioning training [23], and a variety of modifications have been recently reported to occur during object recognition memory formation and recall [24]. Like in invertebrates, some studies in rodents have demonstrated that the changes in acetylation can be restricted to the promoters of genes known to be involved in memory formation, such as that encoding the neurotrophin BDNF. Thus, histone H3 was hyperacetylated at the BDNF gene promoter after retrieval of contextual fear memory, and this hyperacetylation was required for reconsolidation of that memory [25], whereas extinction of tone fear memory triggered H4 hyperacetylation at the same promoter in neurons of the prefrontal cortex [26].
Collectively, these data indicate that an array of histone positions can be differentially acetylated during memory formation. However, we should note that all the histone acetylation marks that have been reported to occur during memory formation thus far are transient. These marks may be part of the regulatory mechanisms that control the timing of activity dependent events, but it is unlikely that they directly participate in encoding the memory [9]. More stable epigenetic marks, such as DNA methylation and other posttranslational histone modifications (methylation, ubiquitination, sumoylation or more stable acetylation events) might correlate better with the duration of the memory process [27].
1.2. Histone Acetyltransferases and Deacetylases as Memory Gates
As previously mentioned, histone acetylation is regulated by the opposing enzymatic activities of HATs and HDACs. However, despite the names of these enzymes, histones are not their only substrates. Non-histone substrates include other nuclear proteins like transcription and chromatin remodeling factors and diverse cytoplasmic and mitochondrial proteins. In fact, computational predictions [28] and high throughput mass spectrometry analyses [29] indicate that hundreds of proteins may be susceptible to acetylation. For this reason, it has recently been proposed that the term HAT should be replaced by KAT, wherein the K stands for lysine [30]. We will use this terminology in the following sections. Similarly, HDAC could have been renamed as KDAC, but the scientists proposing the new nomenclature felt that the HDAC families already had a coherent nomenclature and that renaming them would serve to confuse rather than clarify [30].
KAT proteins catalyze the formation of an amide bond between the ε-amino group of a lysine residue and acetyl-Coenzyme A. They have been grouped into two classes (Fig. 1). Type A KATs are thought to acetylate nucleosomal histones during or in close temporal proximity to transcriptional events, whereas type B KATs primarily acetylate newly synthesized histones in the cytoplasm before their assembly into chromatin. Type A KATs are separated into five families based on similarities in their primary sequences [31, 32]. Counteracting KAT activity, HDAC proteins remove acetyl groups from acetyl-lysines and are classified, according to protein function and sequence similarities, into five families that includes HDAC1 to 11 and sirtuins SIRT1 to 7 [33] (Fig. 1).
Fig. (1).

KATs and HDAC classes. Schematic depicting the balance of KAT and HDAC activities and the impact of this balance on chromatin conformation. The presence of acetyl groups (black balls) in the nucleosome histone tails relaxes the chromatin and facilitates the access of transcription factors. The boxes show the general classification of KAT and HDAC proteins and list representative examples for each group. For the KATs, we used the new nomenclature, but the former name or names are also indicated [30]. See the review articles [31, 156] for additional details.
Both KATs and HDACs have been postulated to be key regulators of memory storage. Although most KATs and HDACs are ubiquitously expressed, some are expressed at particularly high levels in brain areas involved in learning and memory, such as the hippocampus and prefrontal cortex [34]. Deficiencies in KAT activity have been associated with intellectual disability (ID) and cognitive impairment; thus, KATs are thought to be positive regulators of cognitive processes. This is the case for the KAT3 proteins CBP and p300, which will be discussed in detail in the next section, and for KAT2B (a.k.a. p300/CBP-associated factor, PCAF) [35, 36]. Based on these studies, PCAF and CBP/p300 seem to have non-overlapping functions in memory formation. Conversely, different HDAC activities have been postulated to be negative regulators of memory formation. The evidence is especially robust for Class I HDACs. Thus, HDAC2 levels seem to regulate memory formation in a bidirectional manner because contextual and cued fear memory are enhanced in HDAC2 knockout mice and impaired in mice overexpressing HDAC2 [37]. Moreover, HDAC2 knockout mice show increased numbers of synapses and facilitated L-LTP, while mice overexpressing HDAC2 show the opposed phenotype [37]. Similar to HDAC2, conditional deletion of HDAC3 in the CA1 region, achieved by injecting adeno-associated virus expressing cre recombinase in HDAC3 floxed mice, leads to a persistent enhancement of LTM [38]. Strikingly, two recent studies have shown that the loss of either HDAC4 [39] or HDAC5 [40] impairs memory function, suggesting that Class IIa enzymes, in contrast to Class I enzymes, positively regulate memory consolidation. The family of NAD+-dependent Class III HDACs includes sirtuin proteins that have also been postulated to be positive regulators of memory formation. Thus, mice bearing a conditional brain deletion of Sirt1 show deficits in contextual and cued fear memory [41].
1.3. KAT3 Proteins: Molecular Structure, Function and Regulation
CBP and p300 are the only members of the CBP/p300 or KAT3 family [30]. Both are large ubiquitously expressed nuclear proteins with an approximate molecular mass of 250 kDa. CBP was named after its initial description as an interacting partner of the transcription factor CREB (cAMP responsive element binding) [42], whereas p300 was initially described as the host factor interacting with the protein E1A from adenovirus type 5 [43, 44]. It was later found that both proteins really interact with hundreds of proteins with different functions [32, 45-47]; thus, it has recently been proposed that CBP and p300 should be renamed KAT3A and KAT3B, respectively, in an attempt to standardize the nomenclature of chromatin-remodeling enzymes [30].
KAT3 proteins have diverse functions related to transcription activation and regulation. Thus, they are usually described as molecular scaffolds that bring different proteins together to the promoters. Their large size (over 2400 aas) and modular organization enable interaction with several proteins at the same time. For example, the interaction of CBP with MAPKs and the E-Cdk2 complex not only promotes the phosphorylation of CBP but also the phosphorylation of several CBP-interacting transcription factors [48, 49]. The following domains can be distinguished in both CBP and p300 (Fig. 2): (i) three cysteine/histidine-rich regions (CH1 to CH3) that bind zinc and are involved in protein-protein interactions; (ii) a lysine acetyltransferase (KAT) domain in the center of the protein; (iii) a bromodomain (BD) that binds acetylated lysines in histones and specific transcription factors [50]; (iv) two transactivation domains located at either end of the protein; and (v) multiple specific interaction domains for different transcription factors, such as the KIX domain that mediates the interaction between CBP/p300 and CREB phosphorylated at Ser133 [51]. An important consequence of this structure is that CBP/p300 can act as a molecular bridge between DNA-binding transcription factors and components of the basal transcription machinery, such as the TATA-box-binding protein (TBP) and the RNApol II complex. In addition, the KAT activity of CBP/p300 can relax the configuration of the chromatin around the bound DNA sequences by acetylation of histones. These are thought to be the molecular mechanisms responsible for the function of KAT3 proteins as transcriptional co-activators.
Fig. (2).

Structure of KAT3 proteins. CBP and p300 share a number of structural domains including three cysteine/histidine rich regions (CH1-CH3) for protein-protein interaction, the KIX domain that mediates the interaction with CREB and other transcription factors, and the KAT domain. The domains of highest homology and the percentage of amino acid identity between the two proteins are indicated. Regions of high homology between the human CBP and p300 proteins expressed as % identity. NRID, nuclear hormone receptor interacting domain; CH1-3, cysteine/histidine-rich regions 1-3; TAZ1-2, transcriptional adaptor Zn-finger domain 1-2; KIX, kinase inducible domain; Br, bromodomain; PHD, plant homeodomain; ZZ, ZZ-type Zn-finger domain; SID, SRC- 1 interacting domain; MDM2, p53 E3-ubiquitin protein ligase homolog; ATF, activation transcription factors; TBP, TATA-binding protein. Figure modified from [52].
Because CBP and p300 are highly homologous proteins (with >70% of overall identity) and share most of their known functional domains [52], it is reasonable to assume that both proteins have highly overlapping functions through their interactions with similar partners and substrates. However, these two proteins also play unique roles during development, hematopoiesis, the response to DNA damage, and other processes [46, 53]. Future experiments should determine which protein interactions effectively occur in the nervous system in vivo and distinguish CBP from p300 and both proteins from other KAT activities.
The substrate specificity and preferences of KAT3 proteins also remain unclear. Experiments in CBP-deficient mice [54, 55] and human cell lines [56] with reduced CBP function indicate that the N-terminal tail of histones H2A and H2B, especially the latter, are more sensitive to changes in CBP activity than those of histone H3 and H4. Previous biochemical assays and studies in cultured cells have also identified histone H2B as one substrate for the KAT activity of CBP [3, 57]. In particular, a comprehensive analysis carried out by MacManus and Henzdel in transfected cells indicated that CBP and p300 have both common and specific targets. As such, overexpression of CBP results in increased levels of AcH4K12 but not AcH4K8 while p300 overexpression promotes hyperacetylation of histone H4K8 but not H4K12 [57]. In contrast, experiments in which CBP was specifically knocked-down in the CA1 subfield of the mouse hippocampus produced reduced levels of AcH2BK12, AcH3K14 and AcH4K8 but normal levels of AcH4K12 [58]. Overall, the following histone residues have been identified as substrates of CBP and/or p300 in different studies: H2AK5, H2B(K5, K12, K15, K20), H3(K14, K18, K27) and H4(K5, K8) [55, 58-60]. In addition to histones, other nuclear proteins are also acetylated by KAT3 proteins, such as the tumor suppressors p53 [61, 62] and pRb [63], components of the RNApol II complex (TFIIE and TFIIF) [64] and diverse transcription factors [65-67].
Another important issue that needs further clarification is how the activity of KAT3 proteins is regulated by neuronal activity. These proteins are found at the convergence of various important signaling cascades and are targets of different posttranslational modifications that modulate their activity. Thus, the transactivation potential of CBP is increased by PKA, CaMKIV and p42/44 MAPKs, while the ability to recruit specific transcription factors is selectively increased after PKC phosphorylation [68]. Furthermore, CBP is phosphorylated at Ser301 by CaM kinases [69] and is methylated at Arg residues by the methylase CARM1 [70]. While CBP phosphorylation contributes to CBP-dependent transcription, its methylation appears to block its interaction with CREB and prevent CREB-dependent gene expression. The significance of these regulatory steps in vivo is, however, unclear. Another regulatory mechanism of KAT3-dependent transcription relies on the limited supply of these proteins within the cell. As a consequence, different transcription factors compete for binding to specific sites. For example, because of this competition, CREB activity can be indirectly inhibited by viral proteins that are not functionally or structurally related to CREB, but that sequester CBP from its molecular partners [71]. DNA context can also regulate KAT3 activity. Because CBP and p300 do not bind directly to DNA, their recruitment to gene promoters is highly dependent on transcription factor activities. Thus, a recent study in CBP/p300 depleted cells revealed an inverse correlation between CREB occupancy (determined by the number of CREB binding sites at specific promoters) and CBP/p300 dependence for gene induction in response to cAMP [72]; this result indicates that these proteins likely compete with other cofactors to regulate CREB-dependent expression. Intriguingly, the alternative CREB cofactors do not have KAT activity, which puts the relevance of increased histone acetylation for gene induction into question [73].
2. KAT3 DYSREGULATION IN NEUROLOGICAL DISORDERS
Given the multiple interactions of KAT3 proteins and the role of downstream genetic cascades in neuroprotection, including CREB-dependent gene expression [74, 75], it is not surprising that altered KAT activity had been linked to diverse neurological disorders (Fig. 3). In this section, we will focus exclusively on the consequences of dysregulation of KAT3 proteins in brain function. Their role in other pathologies, like cancer, has been reviewed elsewhere [76].
Fig. (3).

Summary of neuropathologies related to KAT3 proteins and therapeutic approaches targeted to these proteins. The balance between KATs and HDACs define the optimal level of chromatin acetylation required for developmental and cognitive functions. The alteration of this balance, favoring either histone hyperacetylation (right box) or hypoacetylation (left box), may result in cognitive impairment and/or deleterious effects. This is the case of well-known pathological conditions like the Rubinstein-Taybi syndrome (RSTS), Fetal Alcohol Spectrum Disorder (FASD), Huntington’s disease (HD), Alzheimer’s disease (AD), Parkinson’s disease (PD), amyotrophic lateral sclerosis (ALS) and other neurological disorders. Therapeutic approaches aimed at restoring the balance, including HDAC inhibitors (HDACis), KAT enhancers (KATes) and inhibitors (KATis) and genetic overexpression of KAT3 genes, are indicated under the corresponding boxes.
2.1. Neurodevelopmental Disorders
During development, CBP and p300 control the proliferation and differentiation of different cell lineages, including those within the nervous system. In mammals, CBP and p300 are first required for neural tube closure at the three-layer embryonic stage. Homozygous mice bearing cbp or p300 null alleles show exencephalia and die early during development between E9 and E12.5 [77-79]. At that time, the neural plate is folding to generate the neural tube and highly expresses both proteins [80, 81]. Strikingly, the double heterozygous mutation for CBP and p300 is also lethal [79], indicating that these two proteins play redundant roles during early development and can be interchangeable. Afterwards, their expression levels decay [80, 81], but they are still involved in the differentiation of a variety of cellular types, such as motoneurons, cortical progenitors and astrocytes [82]. Given this context, it is not surprising that the deficiency of these proteins is associated with neurodevelopmental disorders.
2.1.1. The Rubinstein-Taybi Syndrome
Rubinstein-Taybi syndrome (RSTS) is a complex autosomal dominant disease affecting 1 out of 100,000 newborns that is characterized by ID among other medical conditions [83, 84]. Most RSTS cases are associated with mutations in the genes encoding CBP (CREBBP, 60% of cases) [85] and p300 (EP300, 3% of cases) [86]. RSTS patients have a characteristic facial appearance and other skeletal abnormalities, such as defects in angulation, positioning or duplication of phalanges and thumb halluces that produce the characteristic broad thumbs and big toes that are used in diagnosis. Neuroanatomical defects, including agenesis of the corpus callosum, Dandy-Walker malformation, cortical clefts and subtler anatomical defects, have sometimes been observed in the brains of RSTS patients. Psychological studies have shown that RSTS patients have low levels of intelligence, short attention spans and poor motor coordination [87]. Although developmental alterations clearly underlie certain clinical manifestations of RSTS such as skeletal defects and growth retardation, experiments in animal models indicate that the lack of KAT3 proteins in the adult brain also contributes to the cognitive impairment in RSTS [88].
Mice bearing genetic disruptions similar to those found in RSTS patients express phenotypes reminiscent of some clinical manifestations of this syndrome. Therefore these mice represent valuable experimental models for the investigation of the molecular etiology of the syndrome and evaluation of therapeutic approaches. As indicated above, studies in mouse mutants have shown that CBP and p300 play roles both during embryonic development and in the adult brain, suggesting that the deficiency at both stages is likely to contribute to ID in humans. CBP and p300 hemizygous mice show some skeletal malformations that resemble those described in RSTS patients, but they have overall normal brain anatomy [54, 77, 89, 90]. The phenotype is more apparent in CBP deficient mice, which show more severe facial dysmorphia and alterations in phalanges and the trunk skeleton including scoliosis and asymmetry, and deficiencies or excesses of ribs and vertebras. These mutants also have reduced body weight and size, consistent with the impaired growth that characterizes RSTS.
In attempts to model the cognitive deficits, the different mouse models of RSTS, including hemizygous mice, conditional knockout (cKO), transgenic mice expressing dominant negative variants and mice with focal depletion of the protein achieved through viral transduction (Table 1), have been examined in a variety of learning and memory tasks. These experiments have revealed memory defects in the step-through passive avoidance, fear-conditioning, object recognition memory and Morris water maze tasks as well as other ID-related phenotypes [91]. Particularly remarkable are the experiments in transgenic mice with regulatable expression of a dominant negative CBP variant with a mutated KAT domain (referred to as CBP{HAT}) suggesting that intact KAT activity in the adult forebrain is required for the consolidation of some forms of explicit memory [88]. Recent experiments in different strains of cKO mice also support this view [55, 58, 92], although the reported memory deficits were in some cases weaker than those observed in hemizygous animals. Most CBP mutant strains showed specific deficits in long-term memory (LTM) but not in short-term memory (STM), suggesting a role for CBP in memory consolidation. However, a recent study on cKO mice in which CBP was completely inactivated in excitatory neurons of the postnatal forebrain revealed deficits in both LTM and STM [92], suggesting that the role of CBP in memory storage may also encompass memory encoding. This effect was however not observed in other similar CBP cKO mice [55, 58]. In addition to memory deficits, several CBP deficient strains, including cbp+/- mice and cbpkix/kix mice bearing a point mutation within the KIX domain that blocked CBP-CREB interaction, exhibited impaired motor skill learning [54, 93], that may resemble the difficulty in planning motor acts and executing locomotor skills that RSTS patients are reported to experience [94].
Table 1.
Neurological Traits in Mouse Strains with Deficient KAT3 Activity
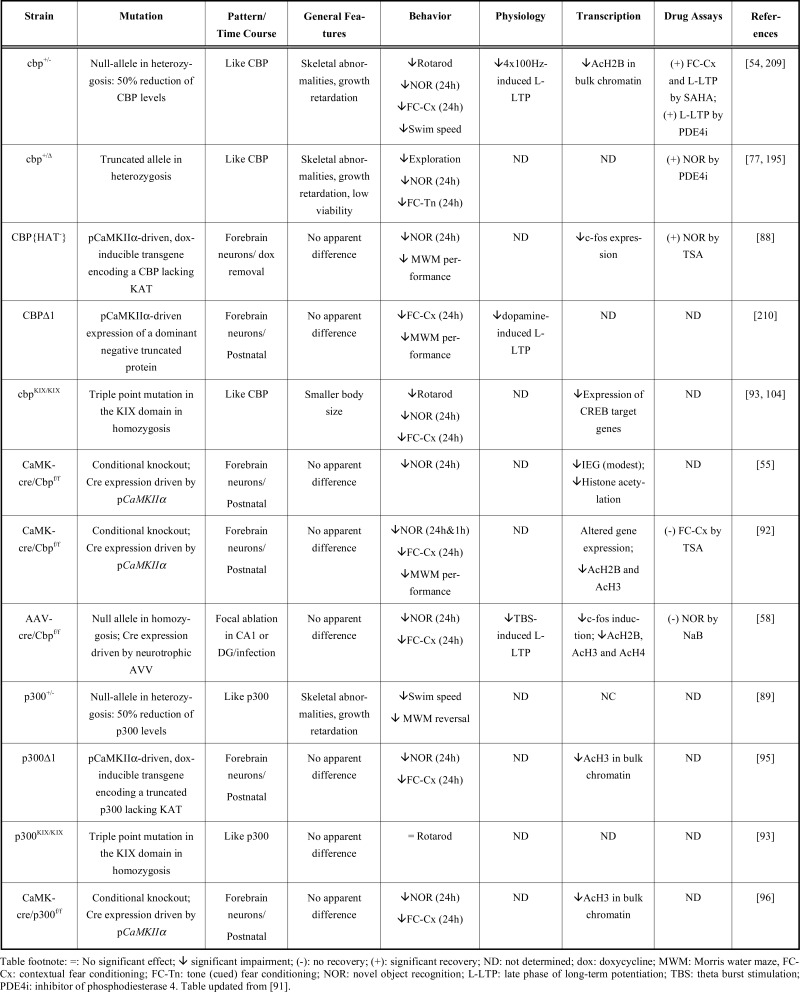 |
In comparison, the analysis of p300 deficient mice revealed weaker defects. Mice expressing a truncated p300 protein (p300∆1) in the hippocampus, amygdala, cortex and cerebellum exhibited memory deficits in contextual fear conditioning and object recognition tasks [95]. Similar memory deficits have been also reported in cKO mice with a p300 deletion restricted to CA1 and the cortex [96]. However, the analyses of p300+/- mice bearing a null allele, a genetic condition closer to RSTS, revealed only mild impairments in the water maze and no impairment in fear conditioning or object recognition memory [89]. P300+/- mice also exhibit mild motor deficits such as abnormal gait and reduced swimming speed. These observations are in agreement with the weaker ID in patients bearing mutations at EP300 compared to CREBBP [97-99] and with the different prevalences of mutations in each of these genes among RSTS patients. In fact, genetic studies indicate that not all EP300 mutations necessarily produce a RSTS condition [99].
Research on RSTS mouse models indicates that impaired neuronal histone acetylation is a critical hallmark of the disease that is likely to play a role in the etiology of the symptoms [100]. Notably, experiments in lymphoblastoid cell lines from RSTS patients have revealed similar histone acetylation deficits [56]. In addition, altered CREB-dependent transcription [101, 102] may also contribute to the cognitive deficits observed in RSTS. Supporting this view, CRE-dependent transcription is impaired in blood cell lines from RSTS patients bearing mutations in the KAT domain of CBP [103] and in neurons of transgenic mice expressing a dominant negative CBP variant [88, 104]. Furthermore, enhancement of CREB activity ameliorates synaptic plasticity deficits in cbp+/- mice [54]. However, it should be noted that experiments in CBP heterozygous and forebrain-specific cKO mice indicate that the reduction or loss of CBP affects differently different activity-driven gene expression programs related to CREB. Thus, whereas the hippocampal induction of immediate early genes in response to novelty is only moderately affected by CBP deficiency [55], the transcriptional program induced in response to environmental enrichment (EE) is clearly impaired [90].
The requirement for p300 in CREB-dependent transcription is also not clear. Although p300 can activate CRE dependent transcription [105], experiments in fibroblasts from p300-/- null mice stimulated with the catalytic subunit of PKA have revealed normal activation of a CRE-luciferase reporter [79]. Gene expression profiling in the hippocampus of p300 heterozygous mice also did not reveal a specific impairment of CREB-dependent transcription [89].
2.1.2. Fetal Alcohol Spectrum Disorder
A second neurodevelopmental disorder, in this case with an environmental origin, has been recently associated with deficits in CBP. Exposure to alcohol during pregnancy causes alterations in the development of the cerebellum that lead to Fetal Alcohol Spectrum Disorder (FASD), which is characterized by deficits in motor coordination, balance and cerebellar-dependent learning tasks [106]. Experiments in rats indicate that CBP protein and histone H3 and H4 acetylation levels are all reduced in the cerebellum of ethanol-treated pups [107]. The involvement of KAT activity in this disorder deserves further examination, including assessment of the efficacy of drugs that increase histone acetylation (see Section 3) in animal models of this disease.
2.2. Neurodegenerative Diseases
Several neurodegenerative conditions have been associated with a reduction in both CBP activity and histone acetylation levels. Moreover, the restoration of acetylation levels, mainly by pharmacological means, as we will discuss in Section 3, concurs with the reversal of neuropathological traits. Both lines of evidence have led to the hypothesis that unbalanced KAT/HDAC activities contribute to the etiology of neurodegenerative disorders [108]. On the other hand, conditional CBP knockout mice exhibit a dramatic reduction in neuronal histone acetylation, but this does not seem to affect neuronal viability, indicating that sustained low levels of histone acetylation do not necessarily cause neuronal death per se [55, 92]. This conclusion is in agreement with the lack of brain atrophy in RSTS patients and models. Importantly, the primary cause of the lethality observed during development of KAT3 homozygous knockout mice is stalled proliferation. Because KAT3 proteins are required for DNA replication checkpoints, their depletion may trigger the death of proliferative cells [109, 110] or at least the loss of their proliferation capability [72] as a consequence of a mitotic failure. The mechanisms by which the loss of CBP and p300 could hamper the viability or survival of postmitotic neurons is less clear. The requirement of normal levels of KAT3 activity for adult neuron survival is, therefore, still under debate. Regardless of this requirement, neuronal malfunction could contribute to the mood changes, memory impairment and other neurological traits observed in patients with neurological conditions associated with KAT3 deficiency.
2.2.1. Huntington’s Disease
Huntington’s disease (HD) is an inheritable fatal neurodegenerative disorder that is caused by expansions of CAG repeats in the Huntingtin gene (HTT) that lead to an aberrant enlargement of a poly-Q track at the protein level [111]. Experiments in animal and cellular models have associated neuropathology in HD with the loss of CBP function and impaired neuronal histone acetylation [112-117]. Thus, it has been proposed that CBP is sequestered in the protein aggregates of mutant Htt (mHtt) observed in the brain tissue of patients and in most experimental models of the disease. This sequestration would cause the depletion of CBP from the cell nucleus resulting in transcriptional dysregulation and cellular toxicity. However, this effect has not been reproduced in all experimental models of HD [118-121]. In addition to the proposed sequestration in aggregates, soluble mHtt can promote the degradation of CBP [122]. In fact, a recent comparison of aggregated and soluble mHtt suggests that the soluble form is more efficient than the aggregates in reducing CBP levels [123]. Interestingly, the turnover of p300 was not affected by mHtt [122]. A third mechanism by which mHtt can interfere with CBP function is the direct inhibition of its KAT activity [124, 125].
Regardless of the specific molecular mechanism underlying the disruption of CBP function in HD, different processes downstream of CBP can contribute to the cognitive deficits observed in patients and mouse models of the disease. First, the reduction in neuronal histone acetylation levels could interfere with cognitive processes, as discussed in section 1.1. Second, the disruption of CREB-dependent gene expression could affect memory consolidation. Interestingly, a number of studies have confirmed that CREB-dependent transcription is altered in HD, but with important divergences depending on the model; CREB-dependent transcription has been shown to be inhibited in some studies [126-129] and enhanced in others [119, 130]. Third, the interference with CBP-dependent acetylation of non-histone substrates, such as p53, which is also involved in HD [114, 131], and Htt itself, could accelerate the progression of the pathology. Because the turnover of Htt depends on the proper balance of CBP and HDAC1 activities, the reduction of CBP could cause an accumulation of mHtt [132]. Fourth, the instability of CAG repeats during germline transmission, a process that depends on HDAC and KAT3 proteins [133, 134], would produce longer poly-Q stretches and could contribute to explaining the phenomenon of anticipation (i.e., worsening of the disease in the next generation as reflected by earlier onset).
In agreement with the role for CBP in HD outlined above, the overexpression of CBP partially rescued the neuronal loss observed in Drosophila, mouse and cellular models of the disease [112, 113, 115], whereas the genetic depletion of CBP further reduced the life span of HD mice bearing a hemizygous CBP mutation [135].
2.2.2. Other poly-Q Disorders
Other poly-Q diseases share molecular mechanisms with HD and have also been linked to impaired KAT activity. This is the case for spinal and bulbar muscular atrophy (SBMA), which is caused by long poly-Q stretches in the androgen receptor, and spinocerebellar ataxia type 3 (SCA3), which is caused by mutations of the ataxin-3 gene. In experimental models of both diseases, CBP has been found sequestered in the protein aggregates [136], although the finding on SCA3 was not confirmed in a later study [122]. Interestingly, other KAT activities have been linked to different forms of spinocerebellar ataxia. Spinocerebellar ataxia type 7 is caused by mutations in the gene ataxin-7, which encode a component of the KAT GCN5-containing coactivator complexes [137]. In vitro experiments indicate that although mutant ataxin-7 does not disrupt such complexes, it interferes with their KAT activity [138, 139]. The scenario in vivo is more complex because KAT activity is apparently not affected in mouse models of the disease, but these mice exhibit a general chromatin de-condensation and aberrant histone acetylation in transcriptionally altered genes [140].
2.2.3. Alzheimer’s disease
Alzheimer’s disease (AD) is a progressive neurodegenerative disorder that causes the most common form of dementia in elderly people. Its most distinctive hallmark is the appearance of intracellular neurofibrillary tangles and extracellular amyloid plaques in cortex, hippocampus and amygdala [141]. Like in HD, several lines of evidence link KAT3 proteins and AD. First, prior to appreciable neurodegeneration, CBP is downregulated in the absence of presenilin 1 (PS1) and 2 (PS2) concomitantly with a reduction in the expression of CREB-dependent target genes and memory impairment [142]. These findings are consistent with the observation that PS1 variants containing mutations associated with familial AD (FAD) fail to positively regulate either CBP or p300 transcriptional activities [143, 144]. Moreover, CRE/CBP-dependent gene expression has been found to be altered in a high-throughput proteomic screen carried out in PS1 + AbetaPP mice [145]. Second and in apparent contradiction with the previous point, in culture experiments indicate that mutations in PS1 associated with FAD enhance CBP activity. Because the production of the N-Cad/CTF2 peptide is inhibited by these mutations and this peptide binds CBP and promotes its proteasomal degradation, N-Cad/CTF2 reduction causes an increase in CBP function and the upregulation of downstream targets like cFos [146]. Third, activation of amyloid precursor protein-dependent signaling reduces CBP levels in primary neuronal cultures [147], and Aβ peptide interferes with CREB signaling upstream of CBP [148, 149]. Fourth, neuronal histone acetylation has been found to be reduced in some mouse models of AD, and treatment with compounds that increase acetylation levels ameliorates or restores some deficits [150, 151]. Fifth, the nuclear traslocation of the p300/CBP inhibitor EP300-interacting inhibitor of differentiation 1 (EID1) is increased in the brains of AD patients, and the overexpression of EID1 results in reduced levels of histone H3 and p53 acetylation, and βIII-tubulin downregulation [152].
Interestingly, the KAT activity of PCAF has also been shown to be involved in AD. PCAF knockout mice develop a resistance to amyloid toxicity. This effect is mediated by PCAF regulation of the expression of proteins involved in Aβ generation and degradation. Therefore, modulating different KAT activities may offer a new route for the development of anti-amyloid therapies [36].
2.2.4. Parkinson’s disease
In contrast to most of the other disorders discussed here, Parkinson’s disease (PD), a degenerative disorder characterized by severe motor dysfunction and the death of dopamine-producing cells in the substantia nigra, has been linked to enhanced KAT activity. PD is triggered by environmental neurotoxicity and mutations or copy number variations in specific genes, such as α-synuclein (α-syn), whose misfolded version is a major constituent of Lewy bodies [153]. Native α-syn confers neuroprotection against neurotoxic insults. One proposed mechanism by which this beneficial effect is accomplished is the selective down-regulation of p300 levels (but not CBP levels) through a still unknown mechanism. This decrease in p300 could cause the hypoacetylation of the transcription factor NF-κB, thus interfering with the transcriptional activation of the proapoptotic PKCδ gene [154]. Indirectly supporting this model, another report found that exposure to the pesticide dieldrin, which is implicated in PD etiology, produces the opposite effect and induces both histone hyperacetylation and CBP accumulation in dopaminergic neurons [155].
2.2.5. Other Neurodegenerative Conditions
CBP reduction, in parallel with histone hypoacetylation, is also observed in models of amyotrophic lateral sclerosis (ALS) [147], a fatal adult-onset neuromuscular disease characterized by the selective degeneration of upper and lower motor neurons, progressive muscle wasting and paralysis. In addition, research on a wide variety of neuropathological conditions, from brain ischemia to aging, indicates that histone deacetylation may be a common feature associated with neuronal malfunction and death in very different pathological contexts. This conclusion is based on the beneficial effects of treatment with inhibitors of HDAC activity but a direct link with KAT3 proteins is still missing.
3. STRATEGIES FOR REINSTATEMENT OF KAT3 ACTIVITY
The previous section discussed how different neuropathological conditions have been associated with dysregulation of histone acetylation and KAT3 activities. Most restoratives strategies aimed at correcting these conditions involve the use of drugs that inhibit HDAC proteins (referred as HDACis), but alternative approaches, such as the genetic or pharmacological enhancement of KAT3 activity could serve a similar aim, possibly with increased specificity [31]. Also, in some cases such as certain animal models of PD and FAD described above, KAT activity should be repressed rather than enhanced. In this section, we will briefly comment on the neuroprotective properties of HDACi drugs (see other articles in this special issue and recent reviews [156, 157] for a detailed review of these compounds) and discuss other therapeutic strategies that directly target KAT3 function (Fig. 3).
3.1. HDACi
Diverse drugs inhibiting HDAC proteins are currently available. Their specificity is limited because they target several HDAC proteins [156]. Among the most used HDACis are sodium butyrate (NaB) and trichostatin A (TSA), which inhibit Class I and Class II HDACs, suberoylanilide hydroxamic acid (SAHA), which also inhibits HDACs from Class IV [8], and valproic acid (VPA), a Class I HDACs inhibitor that is commonly used as an anticonvulsant and mood-stabilizing agent. None of aforementioned HDACis inhibits Class III HDACs. A number of studies have shown that these compounds can potentiate different types of memory. NaB administration favors both the acquisition and the extinction of contextual fear conditioning [21, 26] and increases the duration of object recognition memory [158, 159]. Similar effects have been reported for other HDACis, such as TSA [67, 88, 160, 161], VPA [26], SAHA [8], and novel, more selective, compounds [37, 162, 163]. Interestingly, SAHA, NaB and TSA also facilitate the induction of long-term LTP (L-LTP) in the Schaffer collateral pathways [21, 54, 67], and chronic NaB treatment triggers remodeling of dendritic arbors and enhanced spine density [150].
Apart from their effects in wild type animals, many studies have reported beneficial effects of HDACi administration in animal models of different neuropathological conditions, from Drosophila and C. elegans to rodents. For example, HDAC inhibition prevents cell loss and increases cell survival in HD models [115, 117, 125, 164-166]. Although the most immediate effect is the restoration of the gene expression program [167-169], HDACis might also modulate CAG instability and enhance mHtt clearance [132-134]. Furthermore, HDACi administration recovers memory-related functions and enhances LTP in AD models [150, 151, 170-174]. Although there is little information about which HDAC isoforms are specifically targeted by these inhibitors, Class I HDACis seem to be the most effective in counteracting memory impairments in different AD models. HDACi treatment also ameliorates both motor performance deficits and the survival of motor neurons in an ALS mouse model, although there are some discrepancies between the existing studies (see review in [31]). Paradoxically, two HDACis, VPA and NaB, induce the expression of neuroprotective α-syn in PD models [175, 176], although it is not known whether this is the main mechanism involved in the observed amelioration of the symptoms. Histone deacetylation and reversal or attenuation of its associated deficits by HDACis have also been observed in animal models of other neuropathologies, like SBMA [177], ischemia [178-180], traumatic brain injury [181], intracerebral hemorrhage [182], and aging [183], to mention some prominent examples. These results may indicate either the existence of common pathways underlying all these conditions of very diverse origin or, more likely, the activation of neuroprotective mechanisms by HDACis that improve the chances of neuronal survival regardless of the identity of the insult.
We should note that promoting indiscriminate acetylation can also have deleterious effects. Some experiments in cultured neurons have revealed neurotoxic effects associated with HDACi treatment [184-186] especially after chronic administration, whereas pulse treatments have neuroprotective properties [186]. Moreover, excessive NaB concentrations worsen the pathological traits during treatment of SBMA mice models, whereas lower concentrations produce their amelioration [177]. Interestingly, experiments in a cellular model of apoptotic neurodegeneration have demonstrated that the overexpression of either CBP or p300 significantly protects neurons in a pro-apoptotic condition (low K+) but is deleterious for neurons maintained in survival conditions [147]. The inverted U curve defining the relationship between histone acetylation and neuroprotection may explain some apparent contradictions between studies (Fig. 3). For example, whereas a number of studies support a beneficial effect of HDACis in PD [175, 176], others indicate that HDACi administration can exacerbate the pathology [154, 184]. In conclusion, there exists a range of HDACi concentrations that is optimal for treatment, but if that range is exceeded, side effects may appear. Taken together, these results argue that the use of HDACis to treat neurodegenerative diseases will require fine-tuning and targeted delivery to the affected areas.
3.2. KATe
Given the high level of interest in the development of compounds that enhance histone acetylation and the limited specificity and pleiotropic effects of the currently available HDACis, the effort to identify and design molecules that target KAT activation (i.e., a KAT enhancer or KATe) has run in parallel with the development of new, safer and more specific HDACis. However, very few molecules with such activity are known. The best characterized KATe is N-(4-Chloro-3-trifluoromethyl-phenyl)-2-ethoxy-6-pentadecyl-benzamide (CTPB), which was first found to enhance the KAT activity of p300 in cultured cells [187]. Unfortunately, CTPB was found to be cell-impermeable, which limits its therapeutic use. To address this problem, CTPB can be conjugated to carbon spheres, a strategy that has been used successfully to induce hyperacetylation of histone H3 in the mouse brain upon intra-peritoneal injections of the conjugate [188]. More recently, a second activator of p300 acetyltransferase suberoylanilide (a major constituent of floral resins) was identified in a screen of natural compounds. Its ability to penetrate cell membranes and modulate histone acetylation together with its high affinity for the KAT3 proteins in in vitro assays [189] have made this compound an attractive basis for the design of novel drugs aimed at restoring pathological histone deacetylation; however, the efficacy of such drugs in vivo still needs to be evaluated. Notably, targeting a different KAT has been more successful in enhancing memory formation. Thus, the infusion of the KAT2B/ PCAF activator pentadecylidenemalonate 1b (SPV106) enhances memory for fear extinction and prevents fear renewal [190], although we should be aware that this compound, like many HDACis, was initially described as a powerful pro-apoptotic drug in dividing cells [191].
3.3. Combined Strategies
HDACi and KATe drugs are aimed at restoring defective KAT activity. However, due to the highly modular structure of CBP/p300, it may be also desirable to restore other functions diminished by CBP and/or p300 deficiency, including their function as transcriptional co-activators. In this sense, phosphodiesterase 4 inhibitors (PDE4i), such as rolipram, prevent the hydrolysis of cAMP and increase PKA-dependent signaling upstream of CREB and CBP. Rolipram treatment of neuronal cultures and hippocampal slices reverses the effects of Aβ pathogenic peptide at the levels of CREB phosphorylation and LTP deficits [149] and dendritic spine alterations [186]. There is also evidence, including amelioration of learning impairments [186, 192], that this compound is beneficial in AD animal models. Interestingly, this type of inhibitor also improves learning and synaptic plasticity in wild-type animals of different ages [193, 194] and in RSTS models [54, 195]. Moreover, rolipram reduces CBP sequestration and neuronal loss and ameliorates motor impairments in a HD mouse model [196]. Based on this evidence, the combined use of both HDACi and PDE4i to synergistically increase their beneficial actions in the treatment of PS-based AD has been proposed [197]. This strategy may be extendable to other pathologies and to KATe.
3.4. Genetic Approaches
Although its application is currently complicated, gene therapy represents a potential alternative to pharmacological approaches. Genetic approaches can be particularly powerful because, in contrast to HDACis that only address reduced KAT activity, multiple protein functions can be restored at once. As discussed in the previous sections, overexpression of KAT activity in transgenic animals has a beneficial effect in mouse and Drosophila models of HD. A similar approach based on a lentiviral vector, which thus has potential gene therapy applications, was recently tested in a mouse model of AD. That study found that viral delivery of CBP in the brain ameliorates learning and memory deficits. Notably, such improvements occurred without changes in Aβ and tau pathology and were linked to increased levels of brain-derived neurotrophic factor (BDNF) [198]. Based on these results, the authors proposed that increasing CBP expression in adult brains might be a valid therapeutic approach that may not be limited to AD but may also be effective in other brain disorders in which CREB-dependent gene expression is impaired.
3.5. KATi
Therapies aimed at restoring normal KAT3 levels in situations in which KAT3 levels are pathologically enhanced, such as the experimental models of PD and FAD previously discussed, may be even more challenging than those addressing histone hypoacetylation. Few KAT inhibitors (KATi) are known, and it is likely that the indiscriminate or chronic reduction of histone acetylation would have deleterious effects. Among these KATis, we find two naturally occurring compounds: anacardic acid and curcumin that may have beneficial effects in pathological conditions like PD [155, 199, 200] and AD [201, 202]. The former compound inhibits CBP/p300, PCAF and Tip60, whereas the latter is more specific for CBP/p300 [203]. However, neither compound is very specific; in fact, curcumin also interferes with HDAC and DNA methyltransferase activities [204], which reduces its potential as an effective therapeutic drug. More specific KATis might result from the chemical modification of these natural compounds. Two currently available synthetic alternatives are Lys-CoA-Tat and C646, which selectively affect p300 and CBP [205-207]. Both have been reported to influence memory formation, although they do so in an unexpected manner. As with HDACis, pharmacological inhibition of CBP/p300 activity in the infralimbic prefrontal cortex (ILPFC) by C646 enhanced the extinction of fear memory and LTP in this area. These paradoxical observations cast the general role of KAT3 activities as positive regulators of memory formation in question and suggest that, at least in ILPFC, KAT3 proteins may constraint synaptic plasticity and memory [208].
4. PERSPECTIVES
The discovery of the therapeutic potential of activating histone acetylation in neurodegenerative disorders is relatively recent. Fortunately, a wide array of compounds with the potential to activate histone acetylation is already available and new compounds are under development largely thanks to the known efficacy of these compounds in treating different forms of cancer. However, the field still needs to overcome a number of conceptual and technical limitations. Basic questions regarding the function of HDACs and KATs still need to be clarified, and some controversies outlined in this review need to be solved. For instance, we still need to understand the specific roles of the different HDAC and KAT activities in neuronal function, identify the gene sets downstream of each of them, and clarify the requirement of normal histone acetylation levels for neuronal viability. From the technical point of view, prior to assessing the efficacy of HDACis, KATes and KATis in human patients, we need to thoroughly characterize their potential cytotoxicity and pleiotropic effects in animal and cellular models, decipher the gene expression profiles induced by the treatment refine their molecular specificity and improve their targeted delivery to affected brain regions. Many challenges lie ahead, but the prospects are positive and we are confident that new drugs for the treatment of neurological diseases will emerge from the current efforts.
ACKNOWLEDGEMENTS
Research at Barco’s lab is supported by the grants SAF2008-03194-E (part of the coordinated ERA-Net NEURON project Epitherapy), SAF2011-22855 and CSD2007-00023 from the Spanish Ministry of Science and Innovation, the GVA Grant Prometeo/2012/005 and a grant from the Fundació Gent per Gent. LMV is supported by a Ramón y Cajal contract and the grant SAF2011-22506 from the Spanish Ministry of Economy and Competiveness. JLA holds a postdoctoral contract (JAE-doc) from the Program “Junta para la Ampliación de Estudios” co-funded by the Fondo Social Europeo (FSE).
CONFLICT OF INTEREST
The authors confirm that this article content has no conflicts of interest.
REFERENCES
- 1.Hsieh J, Nakashima K, Kuwabara T, Mejia E, Gage FH. Histone deacetylase inhibition-mediated neuronal differentiation of multipotent adult neural progenitor cells. Proc Natl Acad Sci USA. 2004;101:16659–64. doi: 10.1073/pnas.0407643101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fraga MF, Ballestar E, Paz MF, et al. Epigenetic differences arise during the lifetime of monozygotic twins. Proc Natl Acad Sci USA. 2005;102:10604–9. doi: 10.1073/pnas.0500398102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kouzarides T. Chromatin modifications and their function. Cell. 2007;128:693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 4.Wang Z, Zang C, Cui K, et al. Genome-wide mapping of HATs and HDACs reveals distinct functions in active and inactive genes. Cell. 2009;138:1019–31. doi: 10.1016/j.cell.2009.06.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Byun JS, Wong MM, Cui W, et al. Dynamic bookmarking of primary response genes by p300 and RNA polymerase II complexes. Proc Natl Acad Sci USA. 2009;106:19286–91. doi: 10.1073/pnas.0905469106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hazzalin CA, Mahadevan LC. Dynamic acetylation of all lysine 4-methylated histone H3 in the mouse nucleus: analysis at c-fos and c-jun. PLoS Biol. 2005;3:e393. doi: 10.1371/journal.pbio.0030393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Crosio C, Heitz E, Allis CD, Borrelli E, Sassone-Corsi P. Chromatin remodeling and neuronal response: multiple signaling pathways induce specific histone H3 modifications and early gene expression in hippocampal neurons. J Cell Sci. 2003;116:4905–14. doi: 10.1242/jcs.00804. [DOI] [PubMed] [Google Scholar]
- 8.Roth TL, Sweatt JD. Regulation of chromatin structure in memory formation. Curr Opin Neurobiol. 2009;19:336–42. doi: 10.1016/j.conb.2009.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Barrett RM, Wood MA. Beyond transcription factors: the role of chromatin modifying enzymes in regulating transcription required for memory. Learn Mem. 2008;15:460–7. doi: 10.1101/lm.917508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Franklin TB, Russig H, Weiss IC, et al. Epigenetic transmission of the impact of early stress across generations. Biol Psychiatry. 2010;68:408–15. doi: 10.1016/j.biopsych.2010.05.036. [DOI] [PubMed] [Google Scholar]
- 11.Levenson JM, Sweatt JD. Epigenetic mechanisms in memory formation. Nat Rev Neurosci. 2005;6:108–18. doi: 10.1038/nrn1604. [DOI] [PubMed] [Google Scholar]
- 12.Levenson JM, Sweatt JD. Epigenetic mechanisms: a common theme in vertebrate and invertebrate memory formation. Cell Mol Life Sci. 2006;63:1009–16. doi: 10.1007/s00018-006-6026-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sharma SK. Protein acetylation in synaptic plasticity and memory. Neurosci Biobehav Rev. 2010 doi: 10.1016/j.neubiorev.2010.02.009. [DOI] [PubMed] [Google Scholar]
- 14.Borrelli E, Nestler EJ, Allis CD, Sassone-Corsi P. Decoding the epigenetic language of neuronal plasticity. Neuron. 2008;60:961–74. doi: 10.1016/j.neuron.2008.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sweatt JD. Experience-dependent epigenetic modifications in the central nervous system. Biol Psychiatry. 2009;65:191–7. doi: 10.1016/j.biopsych.2008.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Riccio A. Dynamic epigenetic regulation in neurons: enzymes, stimuli and signaling pathways. Nat Neurosci. 2010;13:1330–7. doi: 10.1038/nn.2671. [DOI] [PubMed] [Google Scholar]
- 17.Zocchi L, Sassone-Corsi P. Joining the dots: from chromatin remodeling to neuronal plasticity. Curr Opin Neurobiol. 2010;20:432–40. doi: 10.1016/j.conb.2010.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Graff J, Kim D, Dobbin MM, Tsai LH. Epigenetic regulation of gene expression in physiological and pathological brain processes. Physiol Rev. 2011;91:603–49. doi: 10.1152/physrev.00012.2010. [DOI] [PubMed] [Google Scholar]
- 19.Guan Z, Giustetto M, Lomvardas S, et al. Integration of long-term-memory-related synaptic plasticity involves bidirectional regulation of gene expression and chromatin structure. Cell. 2002;111:483–93. doi: 10.1016/s0092-8674(02)01074-7. [DOI] [PubMed] [Google Scholar]
- 20.Federman N, Fustinana MS, Romano A. Histone acetylation is recruited in consolidation as a molecular feature of stronger memories. Learn Mem. 2009;16:600–6. doi: 10.1101/lm.1537009. [DOI] [PubMed] [Google Scholar]
- 21.Levenson JM, O'Riordan KJ, Brown KD, Trinh MA, Molfese DL, Sweatt JD. Regulation of histone acetylation during memory formation in the hippocampus. J Biol Chem. 2004;279:40545–59. doi: 10.1074/jbc.M402229200. [DOI] [PubMed] [Google Scholar]
- 22.Bousiges O, Vasconcelos AP, Neidl R, et al. Spatial memory consolidation is associated with induction of several lysine-acetyltransferase (histone acetyltransferase) expression levels and H2B/H4 acetylation-dependent transcriptional events in the rat hippocampus. Neuropsychopharmacology. 2010;35:2521–37. doi: 10.1038/npp.2010.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Monsey MS, Ota KT, Akingbade IF, Hong ES, Schafe GE. Epigenetic alterations are critical for fear memory consolidation and synaptic plasticity in the lateral amygdala. PLoS One. 2011;6:e19958. doi: 10.1371/journal.pone.0019958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Graff J, Woldemichael BT, Berchtold D, Dewarrat G, Mansuy IM. Dynamic histone marks in the hippocampus and cortex facilitate memory consolidation. Nat Commun. 2012;3:991. doi: 10.1038/ncomms1997. [DOI] [PubMed] [Google Scholar]
- 25.Lubin FD, Sweatt JD. The IkappaB kinase regulates chromatin structure during reconsolidation of conditioned fear memories. Neuron. 2007;55:942–57. doi: 10.1016/j.neuron.2007.07.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bredy TW, Wu H, Crego C, Zellhoefer J, Sun YE, Barad M. Histone modifications around individual BDNF gene promoters in prefrontal cortex are associated with extinction of conditioned fear. Learn Mem. 2007;14:268–76. doi: 10.1101/lm.500907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li B, Carey M, Workman JL. The role of chromatin during transcription. Cell. 2007;128:707–19. doi: 10.1016/j.cell.2007.01.015. [DOI] [PubMed] [Google Scholar]
- 28.Basu A, Rose KL, Zhang J, et al. Proteome-wide prediction of acetylation substrates. Proc Natl Acad Sci USA. 2009;106:13785–90. doi: 10.1073/pnas.0906801106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Choudhary C, Kumar C, Gnad F, et al. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science. 2009;325:834–40. doi: 10.1126/science.1175371. [DOI] [PubMed] [Google Scholar]
- 30.Allis CD, Berger SL, Cote J, et al. New nomenclature for chromatin-modifying enzymes. Cell. 2007;131:633–6. doi: 10.1016/j.cell.2007.10.039. [DOI] [PubMed] [Google Scholar]
- 31.Selvi BR, Cassel JC, Kundu TK, Boutillier AL. Tuning acetylation levels with HAT activators: therapeutic strategy in neurodegenerative diseases. Biochim Biophys Acta. 2010;1799:840–53. doi: 10.1016/j.bbagrm.2010.08.012. [DOI] [PubMed] [Google Scholar]
- 32.Bedford DC, Kasper LH, Fukuyama T, Brindle PK. Target gene context influences the transcriptional requirement for the KAT3 family of CBP and p300 histone acetyltransferases. Epigenetics. 2010;5:9–15. doi: 10.4161/epi.5.1.10449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yang XJ, Seto E. HATs and HDACs: from structure, function and regulation to novel strategies for therapy and prevention. Oncogene. 2007;26:5310–8. doi: 10.1038/sj.onc.1210599. [DOI] [PubMed] [Google Scholar]
- 34.Broide RS, Redwine JM, Aftahi N, Young W, Bloom FE, Winrow CJ. Distribution of histone deacetylases 1-11 in the rat brain. J Mol Neurosci. 2007;31:47–58. doi: 10.1007/BF02686117. [DOI] [PubMed] [Google Scholar]
- 35.Maurice T, Duclot F, Meunier J, et al. Altered memory capacities and response to stress in p300/CBP-associated factor (PCAF) histone acetylase knockout mice. Neuropsychopharmacology. 2008;33:1584–602. doi: 10.1038/sj.npp.1301551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Duclot F, Jacquet C, Gongora C, Maurice T. Alteration of working memory but not in anxiety or stress response in p300/CBP associated factor (PCAF) histone acetylase knockout mice bred on a C57BL/6 background. Neurosci Lett. 2010;475:179–83. doi: 10.1016/j.neulet.2010.03.077. [DOI] [PubMed] [Google Scholar]
- 37.Guan JS, Haggarty SJ, Giacometti E, et al. HDAC2 negatively regulates memory formation and synaptic plasticity. Nature. 2009;459:55–60. doi: 10.1038/nature07925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.McQuown SC, Barrett RM, Matheos DP, et al. HDAC3 is a critical negative regulator of long-term memory formation. J Neurosci. 2011;31:764–74. doi: 10.1523/JNEUROSCI.5052-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kim MS, Akhtar MW, Adachi M, et al. An essential role for histone deacetylase 4 in synaptic plasticity and memory formation. J Neurosci. 2012;32:10879–86. doi: 10.1523/JNEUROSCI.2089-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Agis-Balboa RC, Pavelka Z, Kerimoglu C, Fischer A. Loss of HDAC5 Impairs Memory Function: Implications for Alzheimer's Disease. J Alzheimers Dis. 2012 doi: 10.3233/JAD-2012-121009. [DOI] [PubMed] [Google Scholar]
- 41.Gao J, Wang WY, Mao YW, et al. A novel pathway regulates memory and plasticity via SIRT1 and miR-134. Nature. 2010;466:1105–9. doi: 10.1038/nature09271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chrivia JC, Kwok RP, Lamb N, Hagiwara M, Montminy MR, Goodman RH. Phosphorylated CREB binds specifically to the nuclear protein CBP. Nature. 1993;365:855–9. doi: 10.1038/365855a0. [DOI] [PubMed] [Google Scholar]
- 43.Egan C, Jelsma TN, Howe JA, Bayley ST, Ferguson B, Branton PE. Mapping of cellular protein-binding sites on the products of early-region 1A of human adenovirus type 5. Mol Cell Biol. 1988;8:3955–9. doi: 10.1128/mcb.8.9.3955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Whyte P, Williamson NM, Harlow E. Cellular targets for transformation by the adenovirus E1A proteins. Cell. 1989;56:67–75. doi: 10.1016/0092-8674(89)90984-7. [DOI] [PubMed] [Google Scholar]
- 45.Janknecht R. The versatile functions of the transcriptional coactivators p300 and CBP and their roles in disease. Histol Histopathol. 2002;17:657–68. doi: 10.14670/HH-17.657. [DOI] [PubMed] [Google Scholar]
- 46.Kalkhoven E. CBP and p300: HATs for different occasions. Biochem Pharmacol. 2004;68:1145–55. doi: 10.1016/j.bcp.2004.03.045. [DOI] [PubMed] [Google Scholar]
- 47.Wang L, Tang Y, Cole PA, Marmorstein R. Structure and chemistry of the p300/CBP and Rtt109 histone acetyltransferases: implications for histone acetyltransferase evolution and function. Curr Opin Struct Biol. 2008;18:741–7. doi: 10.1016/j.sbi.2008.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Perkins ND, Felzien LK, Betts JC, Leung K, Beach DH, Nabel GJ. Regulation of NF-kappaB by cyclin-dependent kinases associated with the p300 coactivator. Science. 1997;275:523–7. doi: 10.1126/science.275.5299.523. [DOI] [PubMed] [Google Scholar]
- 49.Liu L, Scolnick DM, Trievel RC, et al. p53 sites acetylated in vitro by PCAF and p300 are acetylated in vivo in response to DNA damage. Mol Cell Biol. 1999;19:1202–9. doi: 10.1128/mcb.19.2.1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Polesskaya A, Harel-Bellan A. Acetylation of MyoD by p300 requires more than its histone acetyltransferase domain. J Biol Chem. 2001;276:44502–3. doi: 10.1074/jbc.M106501200. [DOI] [PubMed] [Google Scholar]
- 51.Parker D, Ferreri K, Nakajima T, et al. Phosphorylation of CREB at Ser-133 induces complex formation with CREB-binding protein via a direct mechanism. Mol Cell Biol. 1996;16:694–703. doi: 10.1128/mcb.16.2.694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chan HM, La Thangue NB. p300/CBP proteins: HATs for transcriptional bridges and scaffolds. J Cell Sci. 2001;114:2363–73. doi: 10.1242/jcs.114.13.2363. [DOI] [PubMed] [Google Scholar]
- 53.Kasper LH, Fukuyama T, Biesen MA, et al. Conditional knockout mice reveal distinct functions for the global transcriptional coactivators CBP and p300 in T-cell development. Mol Cell Biol. 2006;26:789–809. doi: 10.1128/MCB.26.3.789-809.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Alarcon JM, Malleret G, Touzani K, et al. Chromatin acetylation, memory, and LTP are impaired in CBP+/- mice: a model for the cognitive deficit in Rubinstein-Taybi syndrome and its amelioration. Neuron. 2004;42:947–59. doi: 10.1016/j.neuron.2004.05.021. [DOI] [PubMed] [Google Scholar]
- 55.Valor LM, Pulopulos MM, Jimenez-Minchan M, Olivares R, Lutz B, Barco A. Ablation of CBP in forebrain principal neurons causes modest memory and transcriptional defects and a dramatic reduction of histone acetylation, but does not affect cell viability. J. Neurosci. 2011;31:1652–1663. doi: 10.1523/JNEUROSCI.4737-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lopez-Atalaya JP, Gervasini C, Mottadelli F, et al. Histone acetylation deficits in lymphoblastoid cell lines from patients with Rubinstein-Taybi syndrome. J Med Genet. 2012;49:66–74. doi: 10.1136/jmedgenet-2011-100354. [DOI] [PubMed] [Google Scholar]
- 57.McManus KJ, Hendzel MJ. Quantitative analysis of CBP- and P300-induced histone acetylations in vivo using native chromatin. Mol Cell Biol. 2003;23:7611–27. doi: 10.1128/MCB.23.21.7611-7627.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Barrett RM, Malvaez M, Kramar E, et al. Hippocampal Focal Knockout of CBP Affects Specific Histone Modifications, Long-Term Potentiation, and Long-Term Memory. Neuropsychopharmacology. 2011 doi: 10.1038/npp.2011.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tie F, Banerjee R, Stratton CA, et al. CBP-mediated acetylation of histone H3 lysine 27 antagonizes Drosophila Polycomb silencing. Development. 2009;136:3131–41. doi: 10.1242/dev.037127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mullighan CG, Zhang J, Kasper LH, et al. CREBBP mutations in relapsed acute lymphoblastic leukaemia. Nature. 2011;471:235–9. doi: 10.1038/nature09727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gu W, Roeder RG. Activation of p53 sequence-specific DNA binding by acetylation of the p53 C-terminal domain. Cell. 1997;90:595–606. doi: 10.1016/s0092-8674(00)80521-8. [DOI] [PubMed] [Google Scholar]
- 62.Tang Y, Zhao W, Chen Y, Zhao Y, Gu W. Acetylation is indispensable for p53 activation. Cell. 2008;133:612–26. doi: 10.1016/j.cell.2008.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chan HM, Krstic-Demonacos M, Smith L, Demonacos C, La Thangue NB. Acetylation control of the retinoblastoma tumour-suppressor protein. Nat Cell Biol. 2001;3:667–74. doi: 10.1038/35083062. [DOI] [PubMed] [Google Scholar]
- 64.Imhof A, Yang XJ, Ogryzko VV, Nakatani Y, Wolffe AP, Ge H. Acetylation of general transcription factors by histone acetyltransferases. Curr Biol. 1997;7:689–92. doi: 10.1016/s0960-9822(06)00296-x. [DOI] [PubMed] [Google Scholar]
- 65.Boyes J, Byfield P, Nakatani Y, Ogryzko V. Regulation of activity of the transcription factor GATA-1 by acetylation. Nature. 1998;396:594–8. doi: 10.1038/25166. [DOI] [PubMed] [Google Scholar]
- 66.Hung HL, Kim AY, Hong W, Rakowski C, Blobel GA. Stimulation of NF-E2 DNA binding by CREB-binding protein (CBP)-mediated acetylation. J Biol Chem. 2001;276:10715–21. doi: 10.1074/jbc.M007846200. [DOI] [PubMed] [Google Scholar]
- 67.Yeh SH, Lin CH, Gean PW. Acetylation of nuclear factor-kappaB in rat amygdala improves long-term but not short-term retention of fear memory. Mol Pharmacol. 2004;65:1286–92. doi: 10.1124/mol.65.5.1286. [DOI] [PubMed] [Google Scholar]
- 68.Zanger K, Radovick S, Wondisford FE. CREB binding protein recruitment to the transcription complex requires growth factor-dependent phosphorylation of its GF box. Mol Cell. 2001;7:551–8. doi: 10.1016/s1097-2765(01)00202-7. [DOI] [PubMed] [Google Scholar]
- 69.Impey S, Fong AL, Wang Y, et al. Phosphorylation of CBP mediates transcriptional activation by neural activity and CaM kinase IV. Neuron. 2002;34:235–44. doi: 10.1016/s0896-6273(02)00654-2. [DOI] [PubMed] [Google Scholar]
- 70.Xu W, Chen H, Du K, et al. A transcriptional switch mediated by cofactor methylation. Science. 2001;294:2507–11. doi: 10.1126/science.1065961. [DOI] [PubMed] [Google Scholar]
- 71.Hottiger MO, Nabel GJ. Viral replication and the coactivators p300 and CBP. Trends Microbiol. 2000;8:560–5. doi: 10.1016/s0966-842x(00)01874-6. [DOI] [PubMed] [Google Scholar]
- 72.Kasper LH, Lerach S, Wang J, Wu S, Jeevan T, Brindle PK. CBP/p300 double null cells reveal effect of coactivator level and diversity on CREB transactivation. EMBO J. 2010;29:3660–72. doi: 10.1038/emboj.2010.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bedford DC, Brindle PK. Is histone acetylation the most important physiological function for CBP and p300? Aging (Albany NY) 2012;4:247–55. doi: 10.18632/aging.100453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lonze BE, Ginty DD. Function and regulation of CREB family transcription factors in the nervous system. Neuron. 2002;35:605–23. doi: 10.1016/s0896-6273(02)00828-0. [DOI] [PubMed] [Google Scholar]
- 75.Sakamoto K, Karelina K, Obrietan K. CREB: a multifaceted regulator of neuronal plasticity and protection. J Neurochem. 2011;116:1–9. doi: 10.1111/j.1471-4159.2010.07080.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Dekker FJ, Haisma HJ. Histone acetyl transferases as emerging drug targets. Drug Discov Today. 2009;14:942–8. doi: 10.1016/j.drudis.2009.06.008. [DOI] [PubMed] [Google Scholar]
- 77.Oike Y, Hata A, Mamiya T, et al. Truncated CBP protein leads to classical Rubinstein-Taybi syndrome phenotypes in mice: implications for a dominant-negative mechanism. Hum Mol Genet. 1999;8:387–96. doi: 10.1093/hmg/8.3.387. [DOI] [PubMed] [Google Scholar]
- 78.Tanaka Y, Naruse I, Hongo T, et al. Extensive brain hemorrhage and embryonic lethality in a mouse null mutant of CREB-binding protein. Mech Dev. 2000;95:133–45. doi: 10.1016/s0925-4773(00)00360-9. [DOI] [PubMed] [Google Scholar]
- 79.Yao TP, Oh SP, Fuchs M, et al. Gene dosage-dependent embryonic development and proliferation defects in mice lacking the transcriptional integrator p300. Cell. 1998;93:361–72. doi: 10.1016/s0092-8674(00)81165-4. [DOI] [PubMed] [Google Scholar]
- 80.Partanen A, Motoyama J, Hui CC. Developmentally regulated expression of the transcriptional cofactors/histone acetyltransferases CBP and p300 during mouse embryogenesis. Int J Dev Biol. 1999;43:487–94. [PubMed] [Google Scholar]
- 81.Bhattacherjee V, Horn KH, Singh S, Webb CL, Pisano MM, Greene RM. CBP/p300 and associated transcriptional co-activators exhibit distinct expression patterns during murine craniofacial and neural tube development. Int J Dev Biol. 2009;53:1097–104. doi: 10.1387/ijdb.072489vb. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Wang J, Weaver IC, Gauthier-Fisher A, et al. CBP histone acetyltransferase activity regulates embryonic neural differentiation in the normal and Rubinstein-Taybi syndrome brain. Dev Cell. 2010;18:114–25. doi: 10.1016/j.devcel.2009.10.023. [DOI] [PubMed] [Google Scholar]
- 83.Hennekam RC. Rubinstein-Taybi syndrome. Eur J Hum Genet. 2006;14:981–5. doi: 10.1038/sj.ejhg.5201594. [DOI] [PubMed] [Google Scholar]
- 84.Roelfsema JH, Peters DJ. Rubinstein-Taybi syndrome: clinical and molecular overview. Expert Rev Mol Med. 2007;9:1–16. doi: 10.1017/S1462399407000415. [DOI] [PubMed] [Google Scholar]
- 85.Petrij F, Giles RH, Dauwerse HG, et al. Rubinstein-Taybi syndrome caused by mutations in the transcriptional co-activator CBP. Nature. 1995;376:348–51. doi: 10.1038/376348a0. [DOI] [PubMed] [Google Scholar]
- 86.Roelfsema JH, White SJ, Ariyurek Y, et al. Genetic heterogeneity in Rubinstein-Taybi syndrome: mutations in both the CBP and EP300 genes cause disease. Am J Hum Genet. 2005;76:572–80. doi: 10.1086/429130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Wiley S, Swayne S, Rubinstein JH, Lanphear NE, Stevens CA. Rubinstein-Taybi syndrome medical guidelines. Am J Med Genet A. 2003;119:101–10. doi: 10.1002/ajmg.a.10009. [DOI] [PubMed] [Google Scholar]
- 88.Korzus E, Rosenfeld MG, Mayford M. CBP histone acetyltransferase activity is a critical component of memory consolidation. Neuron. 2004;42:961–72. doi: 10.1016/j.neuron.2004.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Viosca J, Lopez-Atalaya JP, Olivares R, Eckner R, Barco A. Syndromic features and mild cognitive impairment in mice with genetic reduction on p300 activity: Differential contribution of p300 and CBP to Rubinstein-Taybi syndrome etiology. Neurobiol Dis. 2010;37:186–94. doi: 10.1016/j.nbd.2009.10.001. [DOI] [PubMed] [Google Scholar]
- 90.Lopez-Atalaya JP, Ciccarelli A, Viosca J, et al. CBP is required for environmental enrichment-induced neurogenesis and cognitive enhancement. Embo J. 2011;30:4287–98. doi: 10.1038/emboj.2011.299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Barco A. The Rubinstein-Taybi syndrome: modeling mental impairment in the mouse. Genes Brain Behav. 2007;6(Suppl 1):32–9. doi: 10.1111/j.1601-183X.2007.00320.x. [DOI] [PubMed] [Google Scholar]
- 92.Chen G, Zou X, Watanabe H, van Deursen JM, Shen J. CREB binding protein is required for both short-term and long-term memory formation. J Neurosci. 2010;30:13066–77. doi: 10.1523/JNEUROSCI.2378-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Oliveira AM, Abel T, Brindle PK, Wood MA. Differential Role for CBP and p300 CREB-Binding Domain in Motor Skill Learning. Behav Neurosci. 2006;120:724–9. doi: 10.1037/0735-7044.120.3.724. [DOI] [PubMed] [Google Scholar]
- 94.Gotts EE, Liemohn WP. Behavioral characteristics of three children with the broad thumb-hallux (Rubinstein-Taybi) syndrome. Biol Psychiatry. 1977;12:413–23. [PubMed] [Google Scholar]
- 95.Oliveira AM, Wood MA, McDonough CB, Abel T. Transgenic mice expressing an inhibitory truncated form of p300 exhibit long-term memory deficits. Learn Mem. 2007;14:564–72. doi: 10.1101/lm.656907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Oliveira AM, Estevez MA, Hawk JD, Grimes S, Brindle PK, Abel T. Subregion-specific p300 conditional knock-out mice exhibit long-term memory impairments. Learn Mem. 2011;18:161–9. doi: 10.1101/lm.1939811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Bartholdi D, Roelfsema JH, Papadia F, et al. Genetic heterogeneity in Rubinstein-Taybi syndrome: delineation of the phenotype of the first patients carrying mutations in EP300. J Med Genet. 2007;44:327–33. doi: 10.1136/jmg.2006.046698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Foley P, Bunyan D, Stratton J, Dillon M, Lynch SA. Further case of Rubinstein-Taybi syndrome due to a deletion in EP300. Am J Med Genet A. 2009 doi: 10.1002/ajmg.a.32771. [DOI] [PubMed] [Google Scholar]
- 99.Zimmermann N, Acosta AM, Kohlhase J, Bartsch O. Confirmation of EP300 gene mutations as a rare cause of Rubinstein-Taybi syndrome. Eur J Hum Genet. 2007;15:837–42. doi: 10.1038/sj.ejhg.5201791. [DOI] [PubMed] [Google Scholar]
- 100.Alarcon JM, Hodgman R, Theis M, Huang YS, Kandel ER, Richter JD. Selective modulation of some forms of schaffer collateral-CA1 synaptic plasticity in mice with a disruption of the CPEB-1 gene. Learn Mem. 2004;11:318–27. doi: 10.1101/lm.72704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Benito E, Barco A. CREB's control of intrinsic and synaptic plasticity: implications for CREB-dependent memory models. Trends Neurosci. 2010;33:230–40. doi: 10.1016/j.tins.2010.02.001. [DOI] [PubMed] [Google Scholar]
- 102.Barco A, Marie H. Genetic approaches to investigate the role of CREB in neuronal plasticity and memory. Mol Neurobiol. 2011;44:330–49. doi: 10.1007/s12035-011-8209-x. [DOI] [PubMed] [Google Scholar]
- 103.Kalkhoven E, Roelfsema JH, Teunissen H, et al. Loss of CBP acetyltransferase activity by PHD finger mutations in Rubinstein-Taybi syndrome. Hum Mol Genet. 2003;12:441–50. doi: 10.1093/hmg/ddg039. [DOI] [PubMed] [Google Scholar]
- 104.Wood MA, Attner MA, Oliveira AM, Brindle PK, Abel T. A transcription factor-binding domain of the coactivator CBP is essential for long-term memory and the expression of specific target genes. Learn Mem. 2006;13:609–17. doi: 10.1101/lm.213906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Lundblad JR, Kwok RP, Laurance ME, Harter ML, Goodman RH. Adenoviral E1A-associated protein p300 as a functional homologue of the transcriptional co-activator CBP. Nature. 1995;374:85–8. doi: 10.1038/374085a0. [DOI] [PubMed] [Google Scholar]
- 106.Green JT. The effects of ethanol on the developing cerebellum and eyeblink classical conditioning. Cerebellum. 2004;3:178–87. doi: 10.1080/14734220410017338. [DOI] [PubMed] [Google Scholar]
- 107.Guo W, Crossey EL, Zhang L, et al. Alcohol exposure decreases CREB binding protein expression and histone acetylation in the developing cerebellum. PLoS One. 2011;6:e19351. doi: 10.1371/journal.pone.0019351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Saha RN, Pahan K. HATs and HDACs in neurodegeneration: a tale of disconcerted acetylation homeostasis. Cell Death Differ. 2006;13:539–50. doi: 10.1038/sj.cdd.4401769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Stauffer D, Chang B, Huang J, Dunn A, Thayer M. p300/CREB-binding protein interacts with ATR and is required for the DNA replication checkpoint. J Biol Chem. 2007;282:9678–87. doi: 10.1074/jbc.M609261200. [DOI] [PubMed] [Google Scholar]
- 110.Smolik S, Jones K. Drosophila dCBP is involved in establishing the DNA replication checkpoint. Mol Cell Biol. 2007;27:135–46. doi: 10.1128/MCB.01283-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Morell V. Huntington's gene finally found. Science. 1993;260:28–30. doi: 10.1126/science.8465196. [DOI] [PubMed] [Google Scholar]
- 112.Jiang H, Poirier MA, Liang Y, et al. Depletion of CBP is directly linked with cellular toxicity caused by mutant huntingtin. Neurobiol Dis. 2006;23:543–51. doi: 10.1016/j.nbd.2006.04.011. [DOI] [PubMed] [Google Scholar]
- 113.Nucifora FC, Jr, Sasaki M, Peters MF, et al. Interference by huntingtin and atrophin-1 with cbp-mediated transcription leading to cellular toxicity. Science. 2001;291:2423–8. doi: 10.1126/science.1056784. [DOI] [PubMed] [Google Scholar]
- 114.Steffan JS, Kazantsev A, Spasic-Boskovic O, et al. The Huntington's disease protein interacts with p53 and CREB-binding protein and represses transcription. Proc Natl Acad Sci USA. 2000;97:6763–8. doi: 10.1073/pnas.100110097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Taylor JP, Taye AA, Campbell C, et al. Aberrant histone acetylation, altered transcription, and retinal degeneration in a Drosophila model of polyglutamine disease are rescued by CREB-binding protein. Genes Dev. 2003;17:1463–8. doi: 10.1101/gad.1087503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Giralt A, Puigdellivol M, Carreton O, et al. Long-term memory deficits in Huntington's disease are associated with reduced CBP histone acetylase activity. Hum Mol Genet. 2012;21:1203–16. doi: 10.1093/hmg/ddr552. [DOI] [PubMed] [Google Scholar]
- 117.Bates EA, Victor M, Jones AK, Shi Y, Hart AC. Differential contributions of Caenorhabditis elegans histone deacetylases to huntingtin polyglutamine toxicity. J Neurosci. 2006;26:2830–8. doi: 10.1523/JNEUROSCI.3344-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Yu ZX, Li SH, Nguyen HP, Li XJ. Huntingtin inclusions do not deplete polyglutamine-containing transcription factors in HD mice. Hum Mol Genet. 2002;11:905–14. doi: 10.1093/hmg/11.8.905. [DOI] [PubMed] [Google Scholar]
- 119.Obrietan K, Hoyt KR. CRE-mediated transcription is increased in Huntington's disease transgenic mice. J Neurosci. 2004;24:791–6. doi: 10.1523/JNEUROSCI.3493-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Higgins DS, Hoyt KR, Baic C, Vensel J, Sulka M. Metabolic and glutamatergic disturbances in the Huntington's disease transgenic mouse. Ann N Y Acad Sci. 1999;893:298–300. doi: 10.1111/j.1749-6632.1999.tb07841.x. [DOI] [PubMed] [Google Scholar]
- 121.Tallaksen-Greene SJ, Crouse AB, Hunter JM, Detloff PJ, Albin RL. Neuronal intranuclear inclusions and neuropil aggregates in HdhCAG(150) knockin mice. Neuroscience. 2005;131:843–52. doi: 10.1016/j.neuroscience.2004.10.037. [DOI] [PubMed] [Google Scholar]
- 122.Cong SY, Pepers BA, Evert BO, et al. Mutant huntingtin represses CBP, but not p300, by binding and protein degradation. Mol Cell Neurosci. 2005;30:560–71. [PubMed] [Google Scholar]
- 123.Choi YJ, Kim SI, Lee JW, et al. Suppression of aggregate formation of mutant huntingtin potentiates CREB-binding protein sequestration and apoptotic cell death. Mol Cell Neurosci. 2012;49:127–37. doi: 10.1016/j.mcn.2011.11.003. [DOI] [PubMed] [Google Scholar]
- 124.Igarashi S, Morita H, Bennett KM, et al. Inducible PC12 cell model of Huntington's disease shows toxicity and decreased histone acetylation. Neuroreport. 2003;14:565–8. doi: 10.1097/00001756-200303240-00007. [DOI] [PubMed] [Google Scholar]
- 125.Steffan JS, Bodai L, Pallos J, et al. Histone deacetylase inhibitors arrest polyglutamine-dependent neurodegeneration in Drosophila. Nature. 2001;413:739–43. doi: 10.1038/35099568. [DOI] [PubMed] [Google Scholar]
- 126.Choi YS, Lee B, Cho HY, et al. CREB is a key regulator of striatal vulnerability in chemical and genetic models of Huntington's Disease. Neurobiol Dis. 2009;36:256–68. doi: 10.1016/j.nbd.2009.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Giampa C, DeMarch Z, D'Angelo V, et al. Striatal modulation of cAMP-response-element-binding protein (CREB) after excitotoxic lesions: implications with neuronal vulnerability in Huntington's disease. Eur J Neurosci. 2006;23:11–20. doi: 10.1111/j.1460-9568.2005.04545.x. [DOI] [PubMed] [Google Scholar]
- 128.Wyttenbach A, Swartz J, Kita H, et al. Polyglutamine expansions cause decreased CRE-mediated transcription and early gene expression changes prior to cell death in an inducible cell model of Huntington's disease. Hum Mol Genet. 2001;10:1829–45. doi: 10.1093/hmg/10.17.1829. [DOI] [PubMed] [Google Scholar]
- 129.Sugars KL, Brown R, Cook LJ, Swartz J, Rubinsztein DC. Decreased cAMP response element-mediated transcription: an early event in exon 1 and full-length cell models of Huntington's disease that contributes to polyglutamine pathogenesis. J Biol Chem. 2004;279:4988–99. doi: 10.1074/jbc.M310226200. [DOI] [PubMed] [Google Scholar]
- 130.Benn CL, Sun T, Sadri-Vakili G, et al. Huntingtin modulates transcription, occupies gene promoters in vivo, and binds directly to DNA in a polyglutamine-dependent manner. J Neurosci. 2008;28:10720–33. doi: 10.1523/JNEUROSCI.2126-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Bae BI, Xu H, Igarashi S, et al. p53 mediates cellular dysfunction and behavioral abnormalities in Huntington's disease. Neuron. 2005;47:29–41. doi: 10.1016/j.neuron.2005.06.005. [DOI] [PubMed] [Google Scholar]
- 132.Jeong H, Then F, Melia TJ, Jr, et al. Acetylation targets mutant huntingtin to autophagosomes for degradation. Cell. 2009;137:60–72. doi: 10.1016/j.cell.2009.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Debacker K, Frizzell A, Gleeson O, Kirkham-McCarthy L, Mertz T, Lahue RS. Histone deacetylase complexes promote trinucleotide repeat expansions. PLoS Biol. 2012;10:e1001257. doi: 10.1371/journal.pbio.1001257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Jung J, Bonini N. CREB-binding protein modulates repeat instability in a Drosophila model for polyQ disease. Science. 2007;315:1857–9. doi: 10.1126/science.1139517. [DOI] [PubMed] [Google Scholar]
- 135.Klevytska AM, Tebbenkamp AT, Savonenko AV, Borchelt DR. Partial depletion of CREB-binding protein reduces life expectancy in a mouse model of Huntington disease. J Neuropathol Exp Neurol. 2010;69:396–404. doi: 10.1097/NEN.0b013e3181d6c436. [DOI] [PubMed] [Google Scholar]
- 136.McCampbell A, Taylor JP, Taye AA, et al. CREB-binding protein sequestration by expanded polyglutamine. Hum Mol Genet. 2000;9:2197–202. doi: 10.1093/hmg/9.14.2197. [DOI] [PubMed] [Google Scholar]
- 137.Helmlinger D, Hardy S, Sasorith S, et al. Ataxin-7 is a subunit of GCN5 histone acetyltransferase-containing complexes. Hum Mol Genet. 2004;13:1257–65. doi: 10.1093/hmg/ddh139. [DOI] [PubMed] [Google Scholar]
- 138.McMahon SJ, Pray-Grant MG, Schieltz D, Yates JR, 3rd, Grant PA. Polyglutamine-expanded spinocerebellar ataxia-7 protein disrupts normal SAGA and SLIK histone acetyltransferase activity. Proc Natl Acad Sci USA. 2005;102:8478–82. doi: 10.1073/pnas.0503493102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Palhan VB, Chen S, Peng GH, et al. Polyglutamine-expanded ataxin-7 inhibits STAGA histone acetyltransferase activity to produce retinal degeneration. Proc Natl Acad Sci USA. 2005;102:8472–7. doi: 10.1073/pnas.0503505102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Helmlinger D, Hardy S, Abou-Sleymane G, et al. Glutamine-expanded ataxin-7 alters TFTC/STAGA recruitment and chromatin structure leading to photoreceptor dysfunction. PLoS Biol. 2006;4:e67. doi: 10.1371/journal.pbio.0040067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Blennow K, de Leon MJ, Zetterberg H. Alzheimer's disease. Lancet. 2006;368:387–403. doi: 10.1016/S0140-6736(06)69113-7. [DOI] [PubMed] [Google Scholar]
- 142.Saura CA, Choi SY, Beglopoulos V, et al. Loss of presenilin function causes impairments of memory and synaptic plasticity followed by age-dependent neurodegeneration. Neuron. 2004;42:23–36. doi: 10.1016/s0896-6273(04)00182-5. [DOI] [PubMed] [Google Scholar]
- 143.Francis YI, Stephanou A, Latchman DS. CREB-binding protein activation by presenilin 1 but not by its M146L mutant. Neuroreport. 2006;17:917–21. doi: 10.1097/01.wnr.0000220137.06542.a0. [DOI] [PubMed] [Google Scholar]
- 144.Francis YI, Diss JK, Kariti M, Stephanou A, Latchman DS. p300 activation by Presenilin 1 but not by its M146L mutant. Neurosci Lett. 2007;413:137–40. doi: 10.1016/j.neulet.2006.11.036. [DOI] [PubMed] [Google Scholar]
- 145.Soreghan BA, Lu BW, Thomas SN, et al. Using proteomics and network analysis to elucidate the consequences of synaptic protein oxidation in a PS1 + AbetaPP mouse model of Alzheimer's disease. J Alzheimers Dis. 2005;8:227–41. doi: 10.3233/jad-2005-8302. [DOI] [PubMed] [Google Scholar]
- 146.Marambaud P, Wen PH, Dutt A, et al. A CBP binding transcriptional repressor produced by the PS1/epsilon-cleavage of N-cadherin is inhibited by PS1 FAD mutations. Cell. 2003;114:635–45. doi: 10.1016/j.cell.2003.08.008. [DOI] [PubMed] [Google Scholar]
- 147.Rouaux C, Jokic N, Mbebi C, Boutillier S, Loeffler JP, Boutillier AL. Critical loss of CBP/p300 histone acetylase activity by caspase-6 during neurodegeneration. EMBO J. 2003;22:6537–49. doi: 10.1093/emboj/cdg615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Tong L, Thornton PL, Balazs R, Cotman CW. Beta -amyloid-(1-42) impairs activity-dependent cAMP-response element-binding protein signaling in neurons at concentrations in which cell survival Is not compromised. J Biol Chem. 2001;276:17301–6. doi: 10.1074/jbc.M010450200. [DOI] [PubMed] [Google Scholar]
- 149.Vitolo OV, Sant'Angelo A, Costanzo V, Battaglia F, Arancio O, Shelanski M. Amyloid beta -peptide inhibition of the PKA/CREB pathway and long-term potentiation: reversibility by drugs that enhance cAMP signaling. Proc Natl Acad Sci USA. 2002;99:13217–21. doi: 10.1073/pnas.172504199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Fischer A, Sananbenesi F, Wang X, Dobbin M, Tsai LH. Recovery of learning and memory is associated with chromatin remodelling. Nature. 2007;447:178–82. doi: 10.1038/nature05772. [DOI] [PubMed] [Google Scholar]
- 151.Ricobaraza A, Cuadrado-Tejedor M, Perez-Mediavilla A, Frechilla D, Del Rio J, Garcia-Osta A. Phenylbutyrate ameliorates cognitive deficit and reduces tau pathology in an Alzheimer's disease mouse model. Neuropsychopharmacology. 2009;34:1721–32. doi: 10.1038/npp.2008.229. [DOI] [PubMed] [Google Scholar]
- 152.Liu R, Lei JX, Luo C, et al. Increased EID1 nuclear translocation impairs synaptic plasticity and memory function associated with pathogenesis of Alzheimer's disease. Neurobiol Dis. 2012;45:902–12. doi: 10.1016/j.nbd.2011.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Norris EH, Giasson BI, Lee VM. Alpha-synuclein: normal function and role in neurodegenerative diseases. Curr Top Dev Biol. 2004;60:17–54. doi: 10.1016/S0070-2153(04)60002-0. [DOI] [PubMed] [Google Scholar]
- 154.Jin H, Kanthasamy A, Ghosh A, Yang Y, Anantharam V, Kanthasamy AG. alpha-Synuclein negatively regulates protein kinase Cdelta expression to suppress apoptosis in dopaminergic neurons by reducing p300 histone acetyltransferase activity. J Neurosci. 2011;31:2035–51. doi: 10.1523/JNEUROSCI.5634-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Song C, Kanthasamy A, Anantharam V, Sun F, Kanthasamy AG. Environmental neurotoxic pesticide increases histone acetylation to promote apoptosis in dopaminergic neuronal cells: relevance to epigenetic mechanisms of neurodegeneration. Mol Pharmacol. 2010;77:621–32. doi: 10.1124/mol.109.062174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Fischer A, Sananbenesi F, Mungenast A, Tsai LH. Targeting the correct HDAC(s) to treat cognitive disorders. Trends Pharmacol Sci. 2010;31:605–17. doi: 10.1016/j.tips.2010.09.003. [DOI] [PubMed] [Google Scholar]
- 157.Graff J, Tsai LH. The Potential of HDAC Inhibitors as Cognitive Enhancers. Annu Rev Pharmacol Toxicol. 2013;53:311–30. doi: 10.1146/annurev-pharmtox-011112-140216. [DOI] [PubMed] [Google Scholar]
- 158.Stefanko DP, Barrett RM, Ly AR, Reolon GK, Wood MA. Modulation of long-term memory for object recognition via HDAC inhibition. Proc Natl Acad Sci USA. 2009;106:9447–52. doi: 10.1073/pnas.0903964106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Roozendaal B, Hernandez A, Cabrera SM, et al. Membrane-associated glucocorticoid activity is necessary for modulation of long-term memory via chromatin modification. J Neurosci. 2010;30:5037–46. doi: 10.1523/JNEUROSCI.5717-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Vecsey CG, Hawk JD, Lattal KM, et al. Histone deacetylase inhibitors enhance memory and synaptic plasticity via CREB:CBP-dependent transcriptional activation. J Neurosci. 2007;27:6128–40. doi: 10.1523/JNEUROSCI.0296-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Lattal KM, Barrett RM, Wood MA. Systemic or intrahippocampal delivery of histone deacetylase inhibitors facilitates fear extinction. Behav Neurosci. 2007;121:1125–31. doi: 10.1037/0735-7044.121.5.1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Hawk JD, Florian C, Abel T. Post-training intrahippocampal inhibition of class I histone deacetylases enhances long-term object-location memory. Learn Mem. 2011;18:367–70. doi: 10.1101/lm.2097411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Fass DM, Reis SA, Ghosh B, et al. Crebinostat: A novel cognitive enhancer that inhibits histone deacetylase activity and modulates chromatin-mediated neuroplasticity. Neuropharmacology. 2013;64:81–96. doi: 10.1016/j.neuropharm.2012.06.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Hockly E, Richon VM, Woodman B, et al. Suberoylanilide hydroxamic acid, a histone deacetylase inhibitor, ameliorates motor deficits in a mouse model of Huntington's disease. Proc Natl Acad Sci USA. 2003;100:2041–6. doi: 10.1073/pnas.0437870100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Gardian G, Browne SE, Choi DK, et al. Neuroprotective effects of phenylbutyrate in the N171-82Q transgenic mouse model of Huntington's disease. J Biol Chem. 2005;280:556–63. doi: 10.1074/jbc.M410210200. [DOI] [PubMed] [Google Scholar]
- 166.Ferrante RJ, Kubilus JK, Lee J, et al. Histone deacetylase inhibition by sodium butyrate chemotherapy ameliorates the neurodegenerative phenotype in Huntington's disease mice. J Neurosci. 2003;23:9418–27. doi: 10.1523/JNEUROSCI.23-28-09418.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Thomas EA, Coppola G, Desplats PA, et al. The HDAC inhibitor 4b ameliorates the disease phenotype and transcriptional abnormalities in Huntington's disease transgenic mice. Proc Natl Acad Sci USA. 2008;105:15564–9. doi: 10.1073/pnas.0804249105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Jia H, Pallos J, Jacques V, et al. Histone deacetylase (HDAC) inhibitors targeting HDAC3 and HDAC1 ameliorate polyglutamine-elicited phenotypes in model systems of Huntington's disease. Neurobiol Dis. 2012;46:351–61. doi: 10.1016/j.nbd.2012.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Jia H, Kast RJ, Steffan JS, Thomas EA. Selective histone deacetylase (HDAC) inhibition imparts beneficial effects in Huntington's disease mice: implications for the ubiquitin-proteasomal and autophagy systems. Hum Mol Genet. 2012 doi: 10.1093/hmg/dds379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Kilgore M, Miller CA, Fass DM, et al. Inhibitors of class 1 histone deacetylases reverse contextual memory deficits in a mouse model of Alzheimer's disease. Neuropsychopharmacology. 2010;35:870–80. doi: 10.1038/npp.2009.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Ricobaraza A, Cuadrado-Tejedor M, Marco S, Perez-Otano I, Garcia-Osta A. Phenylbutyrate rescues dendritic spine loss associated with memory deficits in a mouse model of Alzheimer disease. Hippocampus. 2012;22:1040–50. doi: 10.1002/hipo.20883. [DOI] [PubMed] [Google Scholar]
- 172.Ricobaraza A, Cuadrado-Tejedor M, Garcia-Osta A. Long-term phenylbutyrate administration prevents memory deficits in Tg2576 mice by decreasing Abeta. Front Biosci (Elite Ed) 2011;3:1375–84. doi: 10.2741/e340. [DOI] [PubMed] [Google Scholar]
- 173.Francis YI, Fa M, Ashraf H, et al. Dysregulation of histone acetylation in the APP/PS1 mouse model of Alzheimer's disease. J Alzheimers Dis. 2009;18:131–9. doi: 10.3233/JAD-2009-1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Green KN, Steffan JS, Martinez-Coria H, et al. Nicotinamide restores cognition in Alzheimer's disease transgenic mice via a mechanism involving sirtuin inhibition and selective reduction of Thr231-phosphotau. J Neurosci. 2008;28:11500–10. doi: 10.1523/JNEUROSCI.3203-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Kidd SK, Schneider JS. Protective effects of valproic acid on the nigrostriatal dopamine system in a 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine mouse model of Parkinson's disease. Neuroscience. 2011;194:189–94. doi: 10.1016/j.neuroscience.2011.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Rane P, Shields J, Heffernan M, Guo Y, Akbarian S, King JA. The histone deacetylase inhibitor, sodium butyrate, alleviates cognitive deficits in pre-motor stage PD. Neuropharmacology. 2012;62:2409–12. doi: 10.1016/j.neuropharm.2012.01.026. [DOI] [PubMed] [Google Scholar]
- 177.Minamiyama M, Katsuno M, Adachi H, et al. Sodium butyrate ameliorates phenotypic expression in a transgenic mouse model of spinal and bulbar muscular atrophy. Hum Mol Genet. 2004;13:1183–92. doi: 10.1093/hmg/ddh131. [DOI] [PubMed] [Google Scholar]
- 178.Yildirim F, Gertz K, Kronenberg G, et al. Inhibition of histone deacetylation protects wildtype but not gelsolin-deficient mice from ischemic brain injury. Exp Neurol. 2008;210:531–42. doi: 10.1016/j.expneurol.2007.11.031. [DOI] [PubMed] [Google Scholar]
- 179.Meisel A, Harms C, Yildirim F, et al. Inhibition of histone deacetylation protects wild-type but not gelsolin-deficient neurons from oxygen/glucose deprivation. J Neurochem. 2006;98:1019–31. doi: 10.1111/j.1471-4159.2006.04016.x. [DOI] [PubMed] [Google Scholar]
- 180.Ren M, Leng Y, Jeong M, Leeds PR, Chuang DM. Valproic acid reduces brain damage induced by transient focal cerebral ischemia in rats: potential roles of histone deacetylase inhibition and heat shock protein induction. J Neurochem. 2004;89:1358–67. doi: 10.1111/j.1471-4159.2004.02406.x. [DOI] [PubMed] [Google Scholar]
- 181.Dash PK, Orsi SA, Moore AN. Histone deactylase inhibition combined with behavioral therapy enhances learning and memory following traumatic brain injury. Neuroscience. 2009;163:1–8. doi: 10.1016/j.neuroscience.2009.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Sinn DI, Kim SJ, Chu K, et al. Valproic acid-mediated neuroprotection in intracerebral hemorrhage via histone deacetylase inhibition and transcriptional activation. Neurobiol Dis. 2007;26:464–72. doi: 10.1016/j.nbd.2007.02.006. [DOI] [PubMed] [Google Scholar]
- 183.Peleg S, Sananbenesi F, Zovoilis A, et al. Altered histone acetylation is associated with age-dependent memory impairment in mice. Science. 2010;328:753–6. doi: 10.1126/science.1186088. [DOI] [PubMed] [Google Scholar]
- 184.Wang Y, Wang X, Liu L, Wang X. HDAC inhibitor trichostatin A-inhibited survival of dopaminergic neuronal cells. Neurosci Lett. 2009;467:212–6. doi: 10.1016/j.neulet.2009.10.037. [DOI] [PubMed] [Google Scholar]
- 185.Salminen A, Tapiola T, Korhonen P, Suuronen T. Neuronal apoptosis induced by histone deacetylase inhibitors. Brain Res Mol Brain Res. 1998;61:203–6. doi: 10.1016/s0169-328x(98)00210-1. [DOI] [PubMed] [Google Scholar]
- 186.Smith DL, Pozueta J, Gong B, Arancio O, Shelanski M. Reversal of long-term dendritic spine alterations in Alzheimer disease models. Proc Natl Acad Sci USA. 2009;106:16877–82. doi: 10.1073/pnas.0908706106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Balasubramanyam K, Swaminathan V, Ranganathan A, Kundu TK. Small molecule modulators of histone acetyltransferase p300. J Biol Chem. 2003;278:19134–40. doi: 10.1074/jbc.M301580200. [DOI] [PubMed] [Google Scholar]
- 188.Selvi BR, Jagadeesan D, Suma BS, et al. Intrinsically fluorescent carbon nanospheres as a nuclear targeting vector: delivery of membrane-impermeable molecule to modulate gene expression in vivo. Nano Lett. 2008;8:3182–8. doi: 10.1021/nl801503m. [DOI] [PubMed] [Google Scholar]
- 189.Dal Piaz F, Tosco A, Eletto D, et al. The identification of a novel natural activator of p300 histone acetyltranferase provides new insights into the modulation mechanism of this enzyme. Chembiochem. 2010;11:818–27. doi: 10.1002/cbic.200900721. [DOI] [PubMed] [Google Scholar]
- 190.Wei W, Coelho CM, Li X, et al. p300/CBP-Associated Factor Selectively Regulates the Extinction of Conditioned Fear. J Neurosci. 2012;32:11930–11941. doi: 10.1523/JNEUROSCI.0178-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Sbardella G, Castellano S, Vicidomini C, et al. Identification of long chain alkylidenemalonates as novel small molecule modulators of histone acetyltransferases. Bioorg Med Chem Lett. 2008;18:2788–92. doi: 10.1016/j.bmcl.2008.04.017. [DOI] [PubMed] [Google Scholar]
- 192.Gong B, Vitolo OV, Trinchese F, Liu S, Shelanski M, Arancio O. Persistent improvement in synaptic and cognitive functions in an Alzheimer mouse model after rolipram treatment. J Clin Invest. 2004;114:1624–34. doi: 10.1172/JCI22831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Barad M, Bourtchouladze R, Winder DG, Golan H, Kandel E. Rolipram, a type IV-specific phosphodiesterase inhibitor, facilitates the establishment of long-lasting long-term potentiation and improves memory. Proc Natl Acad Sci USA. 1998;95:15020–5. doi: 10.1073/pnas.95.25.15020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Bach ME, Barad M, Son H, et al. Age-related defects in spatial memory are correlated with defects in the late phase of hippocampal long-term potentiation in vitro and are attenuated by drugs that enhance the cAMP signaling pathway. Proc Natl Acad Sci USA. 1999;96:5280–5. doi: 10.1073/pnas.96.9.5280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Bourtchouladze R, Lidge R, Catapano R, et al. A mouse model of Rubinstein-Taybi syndrome: Defective long-term memory is ameliorated by inhibitors of phosphodiesterase 4. Proc Natl Acad Sci USA. 2003;100:10518–22. doi: 10.1073/pnas.1834280100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Giampa C, Middei S, Patassini S, et al. Phosphodiesterase type IV inhibition prevents sequestration of CREB binding protein, protects striatal parvalbumin interneurons and rescues motor deficits in the R6/2 mouse model of Huntington's disease. Eur J Neurosci. 2009;29:902–10. doi: 10.1111/j.1460-9568.2009.06649.x. [DOI] [PubMed] [Google Scholar]
- 197.Beglopoulos V, Shen J. Regulation of CRE-dependent transcription by presenilins: prospects for therapy of Alzheimer's disease. Trends Pharmacol Sci. 2006;27:33–40. doi: 10.1016/j.tips.2005.11.010. [DOI] [PubMed] [Google Scholar]
- 198.Caccamo A, Maldonado MA, Bokov AF, Majumder S, Oddo S. CBP gene transfer increases BDNF levels and ameliorates learning and memory deficits in a mouse model of Alzheimer's disease. Proc Natl Acad Sci USA. 2010;107:22687–92. doi: 10.1073/pnas.1012851108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Mythri RB, Bharath MM. Curcumin: a potential neuroprotective agent in Parkinson's disease. Curr Pharm Des. 2012;18:91–9. doi: 10.2174/138161212798918995. [DOI] [PubMed] [Google Scholar]
- 200.Song C, Kanthasamy A, Jin H, Anantharam V, Kanthasamy AG. Paraquat induces epigenetic changes by promoting histone acetylation in cell culture models of dopaminergic degeneration. Neurotoxicology. 2011;32:586–95. doi: 10.1016/j.neuro.2011.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Davinelli S, Sapere N, Zella D, Bracale R, Intrieri M, Scapagnini G. Pleiotropic protective effects of phytochemicals in Alzheimer's disease. Oxid Med Cell Longev. 2012;2012:386527. doi: 10.1155/2012/386527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Belkacemi A, Doggui S, Dao L, Ramassamy C. Challenges associated with curcumin therapy in Alzheimer disease. Expert Rev Mol Med. 2011;13:e34. doi: 10.1017/S1462399411002055. [DOI] [PubMed] [Google Scholar]
- 203.Piaz FD, Vassallo A, Rubio OC, Castellano S, Sbardella G, De Tommasi N. Chemical biology of histone acetyltransferase natural compounds modulators. Mol Divers. 2011;15:401–16. doi: 10.1007/s11030-010-9299-5. [DOI] [PubMed] [Google Scholar]
- 204.Reuter S, Gupta SC, Park B, Goel A, Aggarwal BB. Epigenetic changes induced by curcumin and other natural compounds. Genes Nutr. 2011;6:93–108. doi: 10.1007/s12263-011-0222-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Crump NT, Hazzalin CA, Bowers EM, Alani RM, Cole PA, Mahadevan LC. Dynamic acetylation of all lysine-4 trimethylated histone H3 is evolutionarily conserved and mediated by p300/CBP. Proc Natl Acad Sci USA. 2011;108:7814–9. doi: 10.1073/pnas.1100099108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Bowers EM, Yan G, Mukherjee C, et al. Virtual ligand screening of the p300/CBP histone acetyltransferase: identification of a selective small molecule inhibitor. Chem Biol. 2010;17:471–82. doi: 10.1016/j.chembiol.2010.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Lau OD, Kundu TK, Soccio RE, et al. HATs off: selective synthetic inhibitors of the histone acetyltransferases p300 and PCAF. Mol Cell. 2000;5:589–95. doi: 10.1016/s1097-2765(00)80452-9. [DOI] [PubMed] [Google Scholar]
- 208.Marek R, Coelho CM, Sullivan RK, et al. Paradoxical enhancement of fear extinction memory and synaptic plasticity by inhibition of the histone acetyltransferase p300. J Neurosci. 2011;31:7486–91. doi: 10.1523/JNEUROSCI.0133-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Levine AA, Guan Z, Barco A, Xu S, Kandel ER, Schwartz JH. CREB-binding protein controls response to cocaine by acetylating histones at the fosB promoter in the mouse striatum. Proc Natl Acad Sci USA. 2005;102:19186–91. doi: 10.1073/pnas.0509735102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Wood MA, Kaplan MP, Park A, et al. Transgenic mice expressing a truncated form of CREB-binding protein (CBP) exhibit deficits in hippocampal synaptic plasticity and memory storage. Learn Mem. 2005;12:111–9. doi: 10.1101/lm.86605. [DOI] [PMC free article] [PubMed] [Google Scholar]