

Abstract
Objective
To familiarize clinicians with advances in computational disease modeling applied to trauma and sepsis.
Data Sources
PubMed search and review of relevant medical literature.
Summary
Definitions, key methods, and applications of computational modeling to trauma and sepsis are reviewed.
Conclusions
Computational modeling of inflammation and organ dysfunction at the cellular, organ, whole-organism, and population levels has suggested a positive feedback cycle of inflammation → damage → inflammation that manifests via organ-specific inflammatory switching networks. This structure may manifest as multi-compartment “tipping points” that drive multiple organ dysfunction. This process may be amenable to rational inflammation reprogramming.
Keywords: Inflammation, mathematical model, trauma, sepsis
INTRODUCTION
Critical illness comprises a constellation of pathophysiological derangements that ensues in the setting of trauma, hemorrhagic shock, and sepsis. Trauma, often accompanied by hemorrhage, is among the leading causes of morbidity and mortality worldwide, often leading to inflammation-related late complications that include sepsis and multiple organ dysfunction syndrome (MODS) (1–3). Sepsis alone is responsible for more than 215,000 deaths in the United States per year and an annual healthcare cost of over $16 billion (4), while trauma/hemorrhage is the most common cause of death for young people in the U.S., costing over $400 billion annually (5–7).
It is now beyond doubt that the acute immuno-inflammatory response, with its manifold manifestations at the molecular, cellular, tissue, organ, and whole-organism levels, drives outcomes in critical illness. Though properly regulated inflammation allows for timely recognition and effective reaction to injury or infection, both trauma and sepsis are manifestations of disordered and mis-compartmentalized inflammation that in turn impairs physiological functions. This paradox of a robust, evolutionarily conserved network of inflammation whose very structure may lead to disease(8–10) has resulted in a lack of therapeutic options other than supportive care (11, 12). Indeed, the current lack of therapeutic options may result from the failure to fully understand this structure, and thus certain modes of supportive care (e.g. ventilation) may propagate inflammation and organ damage as compared to others (13).
Over a decade ago, there was recognition of the complex interplay between inflammation and physiology in critical illness and of the need to apply complex systems approaches such as computational modeling to unravel this complexity (14, 15). In the context of this review, we use the term “in silico modeling” to refer to the constellation of computational approaches utilized in an attempt to define and, in a sense, defeat the complexity of critical illness (Figure 1; Table 1). The advent of “omics” methodologies, with the theoretical capability of interrogating the complete responses of cells and tissues, spurred the application of these methodologies to critical illness (16–23); the resultant formation of the Inflammation and the Host Response to Injury “glue” consortium (http://www.gluegrant.org/) led to seminal contributions to the understanding of the myriad pathways induced by injury and infection in humans(24, 25). More recently, Translational Systems Biology has been suggested asa rational, systems engineering-oriented, computationally-based frameworkfor integrating data derived from basic biology experiments as well as pre-clinical studies and clinical studies(26–29). This recognition of the need to apply such complex systems approaches critical illness led to the formation of the Society for Complexity in Acute illness (SCAI; www.scai-med.org) in 2003, has been a featured topic of discussion in meetings of various other scientific societies, and has been recognized by funding agencies as an important means by which to grapple with the multidimensional problem of critical illness. The tremendous progress and remaining challenges of computational modeling in critical illnessare reviewed in this article.
Figure 1. The progression from measurement to modeling to modulation in sepsis and trauma.
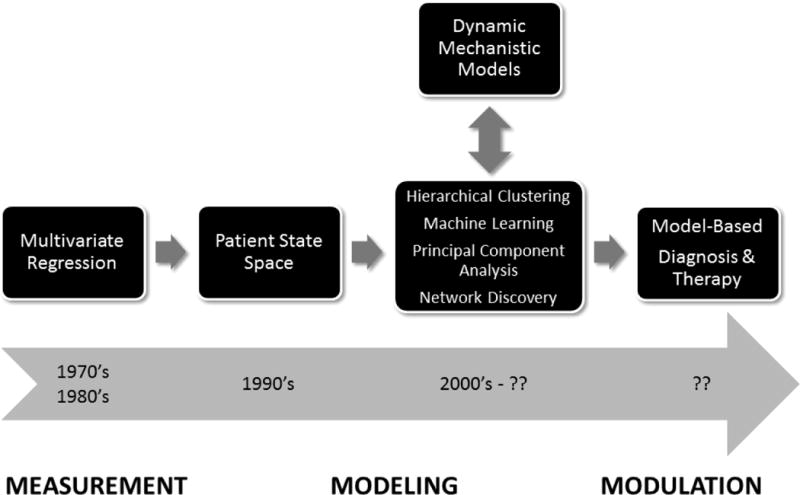
Quantitative (in silico) methods have progressed from purely association-based statistical methods to dynamic mechanistic modeling capable of predicting the responses of patient cohorts and individuals as well as suggesting novel therapies. Data-driven and mechanistic modeling methods are now being integrated. Future possibilities include the design of novel diagnostics and therapies based on in silico modeling.
Table 1. Comparison of modeling methods and applications to sepsis and trauma.
Key primary and review papers are provided to guide the reader.
| Modeling method | Description | Examples of Applications to sepsis/trauma | Key References |
|---|---|---|---|
| Data-driven Modeling | A compendium of methods that are primarily based on associations among data variables. These methods can be applied to either static or dynamic data | Prediction of likelihood to worsen or improve clinical state | Reviews: (31, 109, 110) |
| Multivariate regression | Methods for exploring the relationships of each of multiple variables to a given outcome | Associating inflammatory mediator levels with clinical outcomes | Primary papers: (50, 111, 112) |
| Hierarchical clustering | A technique for grouping multivariate data based on similarity in vectors of values. | Highlighting the natural variability, as well as any overlap, in gene transcripts, inflammatory mediators, or physiologic/biochemical clinical data | Primary papers:(22, 24, 50) |
| Principal Component Analysis | A non-parametric statistical method of reducing a multidimensional dataset to a few principal components. These are the components that account for the most variability in the dataset. The underlying hypothesis is that a variable that changes during a specific process is important to that process. | Suggesting principal inflammatory drivers or biomarkers of sepsis or trauma, either to guide further study directly or to suggest which variables should be included in a mechanistic model. | Primary papers: (50, 91, 113) |
| Network Discovery Methods | Methods that can suggest relationships among variables as well as key features of connectivity in a multivariate dataset. | Suggesting biomarkers and possible interactions among mediators in sepsis or trauma. | Primary papers: (22, 24, 50) |
|
| |||
| Mechanistic Modeling | A compendium of methods that are primarily based on mechanistic abstractions that simulate key intracellular, cell/cell, tissue/organ, organ system, or whole-organism level, or that can tie across levels of organization (multiscale modeling) | Gaining mechanistic knowledge about sepsis/trauma; simulating clinical trials; prediction of likelihood to worsen or improve clinical state | Reviews: (9, 26, 27, 92, 114, 115) |
| Equation-based models | A compendium of methods based on ordinary or partial differential equations that typically describe the change over time of variables. These models are typically deterministic but can be stochastic, and are are based on the average behavior of components in a well-mixed system. | Qualitatively and quantitatively predictive models of inflammation (at the cellular, tissue/organ, and whole-organism levels) in sepsis and trauma, including in silico clinical trials and individual-specific models. | Primary papers:(63–65, 91, 100, 116) |
| Agent-based models | Cellular automa models in which individual agents interact with each other and with their environment in a given space by following rules that are applied in a probabilistic manner. These models are therefore generally stochastic. | Qualitatively and quantitatively predictive models of inflammation (at the cellular, tissue/organ, and whole-organism levels) in sepsis and trauma, including in silico clinical trials and individual-specific models. | Primary papers:(60–62) |
| Rule-basedmodels | Similar to agent-based models, rules-based models are typically used to model detailed biochemical intreactions among molecules. These models are typically stochastic. | Models of inflammatory signal transduction pathways. | Primary papers:(74, 75, 117) |
CONCEPTS, APPROACHES, AND FRAMEWORKS FOR COMPUTATIONAL MODELING IN CRITICAL ILLNESS
There is a fairly long history of quantitative modelingapproaches to critical illness, as summarized in Figure 1 and detailed below. Initial studies were based on statistical, data-driven methods that have been employed to better define the patient state and predict clinical outcomes (e.g. the APACHEscore and numerous other scoring systems utilized in sepsis and trauma (30)); these approaches are summarized in Table 1, which is an expansion of the excellent summary given in (31). These methods are based on associations among variables, and include time-tested logistic regression techniques as well as more recent tools that enable graphical views of network interconnectivity (31). Such methods were used predominantly in an attempt to develop better prognostic and diagnostic scoring systems for critically ill patients, and incorporated both clinical data and levels of circulating inflammation biomarkers in recognition of the interrelated nature of inflammation and (patho)physiology (32–34). More recently, the advent of multiple high-content (“omic”) technologies has resulted in a deluge of data in all fields of biology(35), including critical illness(24, 25, 36–42). In addition, there has been an explosion in the use of multiple computational techniques that could be classified as “complex systems” approaches, including signal processing techniques, multivariate dynamic clustering, and machine-learningand network discovery algorithms based on physiologic measurements as well as inflammation biomarkers (22, 43–47). Importantly, these systems biology and computational biology studies verified the importance of known biological pathways in critical illness and injury, as well as suggesting potentially novel pathways for further study (22, 36, 48–50).
The increasing use of these techniques has led to a growing recognition that more data leads to more possible explanations for those data; that there are multiple technical, practical, and economic challenges to implementing these purely data-driven systems biology approaches as diagnostic strategies; and thatinvestigators’ intuition is not up to the task of unraveling causal mechanisms from highly-dimensional, data-based associations (3, 13, 51). In contrast to data-driven modeling, mechanistic computational simulations depict biological interactions (e.g. among cells, their products, and the outcomes that result under a given set of conditions). Such computational models simulations may be used as “knowledge stores” that may be queried as to the emergent behavior of the sum total of known or hypothesized reductionist biological interactions (52–56); to suggest novel interactions not yet described by experimental data(57); and to address controversies based on diverseexperimental/clinical conditions or other experimental differences among groups studying any given complex biological system(58). Unlike data-driven models, dynamic mechanistic models offer the possibility of prediction outside of the time range or other specific conditions of the data on which they were trained(3, 13, 29, 51). The primary methods of dynamic mechanistic modeling used to datein acute inflammation and other phenomena related to critical illnessare agent-based modeling(57, 59–62) and equation-based modeling(63–71), though rules-based modeling has also been used for some studies of inflammatory and immune intracellular signaling (26, 72–76). These modeling frameworks have their respective strengths and weaknesses(26, 27, 77, 78), but have all proven valuable in the critical care arena(8, 9, 77, 79–82).
However, as useful as mechanistic computational modeling has been in integrating known interactions gleaned from the literature, this approach is inherently biased given the tremendous volume of information that could, in theory, be incorporated into models and that is deemed irrelevant or unnecessary for the degree of abstraction chosen by the modeler. Bias is also introduced based on the level of interest devoted to a particular pathway by the scientific community and thus ignores potentially important pathways that are less studied or yet to be discovered. In response to this concern,there has been an attempt to couple the less-biased data-driven approach with mechanistic mathematical modeling, in studies focused on the acute inflammatory response(3, 9, 13, 28, 29, 51, 83, 84). In these studies, mechanistic computational simulations were created based on biology abstracted from “omics” data (85–90) or inferred from data-driven analysis of principal drivers (91). This type of combined data-driven and mechanistic modeling allows for a further, intermediate validation step with regards to hypotheses inferred from the original data. Furthermore, these studiesreflect the maturity of computational modeling in acute illness. Importantly, this combined data-driven and mechanistic approach is likely to be the area of study with the most growth in coming years due to the inherent appeal of unifying – and gaining testable mechanistic insights from – the growing repository of “omics” data.
This progression from multivariate regression models through various quasi-mechanistic associate methods to dynamic mechanistic modeling (and the integration across methods) is depicted in Figure 1. Below, we discuss the insights gleaned from computational modeling in acute illness, and suggest challenges and opportunities for future work.
KEY INSIGHTS FOR CRITICAL ILLNESS FROM COMPUTATIONAL MODELING
Early studies utilizing complex systems approaches in critical illness suggested the concept of “coupled oscillators” that become uncoupled as inflammation becomes dysregulated and organ dysfunction progresses (14). More recent in silico modeling work has posed specific hypotheses with regard to the mechanisms by which inflammation is coupled nonlinearly to physiological (dys)function, namely due to multiple feedback loopsthat manifest at the cellular, tissue/organ, and whole-organism levels(3, 13, 27, 51, 60–65, 67–70, 84–86, 91–93). Positive feedback loops allow rapid ramping up of a response to biological stress, while the negative feedback works to suppress inflammation and restore homeostasis once the threat (infection, damaged tissue, etc.) has been eliminated.
One of the earliest insights to come from computational modeling was the crucial role of “late” mediators in sepsis (now known as Damage-associated Molecular Pattern [DAMP] molecules), intracellular components whose release into the extracellular environment signals stress, damage, or dysfunction (63), in the establishing and perpetuating the positive feedback loop of inflammation → damage → inflammation (9, 10, 13, 27, 51, 84, 94, 95). In silico modeling studies support the notion that this dysfunction occurs via a positive feedback loop in which inflammation induced by pathogen-derived molecular pattern (PAMP) molecules leads to the secondary release of DAMP molecules. In turn, DAMP’s stimulate nearby inflammatory cells to produce more of the classical inflammatory mediators, leading to further release of DAMP’s and therefore to self-maintaining inflammation even after the pathogen has been cleared. The recognition that PAMP’s and DAMP’s signal via common pathways (e.g. the Toll-like receptors) (96, 97)has helped validate this concept at the molecular level.
The concept of inflammatory preconditioningis another key area in which in silico modeling has yielded key insights. Inflammatory preconditioning refers to the spectrum of possible responses to stimulation with two or more pro-inflammatory stimuli in succession, namely responses that are equal to, greater than, or lesser than each stimulus in isolation. For example, repeated treatment with bacterial lipopolysaccharide can lead to desensitization or enhancement of subsequent pro-inflammatory responses that manifest at the cellular, tissue/organ, and whole-organism levels(98, 99). In silico modeling studies using various platforms have suggested that the aforementioned positive and negative feedback loops of the inflammatory response can explain the various phenotypes characteristic of inflammatory preconditioningboth in vitro and in vivo(62, 68, 74, 85, 100–102).
PERSPECTIVES AND CHALLENGES
In silico modeling has yielded both basic insights and translational applicationsin critical illness (3, 9, 13, 27, 29, 51, 82, 84, 92). Indeed, key translational applications such as in silico clinical trials were pioneered in the arena of critical illness (61, 64). One of these studies suggested mechanistic reasons for the failure of neutralizing anti-TNF-α antibodies in sepsis, due to cohort-specific beneficial and detrimental effects that ultimately resulted in the lack of net benefit for this drug (64). Recent studies showing the potential to predict the individual inflammatory and pathophysiologic outcomes of human subjects (103) and large, outbred animals(91) subjected to acute inflammatory stress suggest the possibility of predicting the outcomes of – and possibly tailoring therapy for – individual patients(29, 82). Recently, we constructed a hybrid equation- and agent-based model that simulates blood flow along with skin injury, inflammation, and ulcer formation(104), since pressure ulcers are a complication that con occur in critically ill patients (105). The relationship between pressure and the course of ulcer formation, as well as several other important characteristic patterns of pressure ulcer formation, was demonstrated in this model. The equation-based portion of this model was calibrated to data related to blood flow following experimental pressure responses in non-injured human subjects or to data from people with spinal cord injury (SCI). This hybrid model predicted a higher propensity to form ulcers in response to pressure in people with SCI vs. non-injured control subjects (both as cohorts and individual patients), and thus may serve as novel diagnostic platform for post-SCI ulcer formation(104). Other emerging applications of computational modeling to understand multi-factorial therapies for critical illness that reprogram the inflammatory response, such as hemoadsorption (106, 107) also point to an exciting and valuable application of in silico methods.
Despite this progress, many challenges remain for this rapidly-evolving field. At the most practical level, in silico modelers must prove the translational benefit of this technology through prospective clinical studies and ultimately through the development of computationally-based diagnostics or therapeutics for critical illness. At the grandest scale, the key challenge revolves around the inherently multi-scale, multi-system nature of critical illness. As has likely occurred in many other complex biological systems (108), inflammation may have evolved to be robust in response to a broad range of perturbations at multiple biological scales of organization, but at a cost of fragility in key control nodes. We speculate that the aforementioned positive and negative feedback loops manifest dynamically as cellular, tissue/organ, and whole-organism “tipping points” that drive MODS (13, 51). In silico modeling could therefore rise to the challenge of integrating inflammatory, neuro-endocrine, and physiologic processes in order to unravel the multi-dimensional, multi-compartment, and highly dynamic landscape of critical illness.
Acknowledgments
The authors would like to acknowledge the invaluable intellectual contributions of Gary An, Ioannis Androulakis, Gilles Clermont, Gregory Constantine, Thomas Dick, Bard Ermentrout, James Faeder, Frank Jacono, Qi Mi, and Jonathan Rubin. This work was supported in part by the National Institutes of Health grants R01GM67240, P50GM53789, R33HL089082, R01HL080926, R01AI080799, R01HL76157, R01DC008290, and UO1DK072146; National Institute on Disability and Rehabilitation Research grant H133E070024; National Science Foundation grant 0830-370-V601; a Shared University Research Award from IBM, Inc.; and grants from the Commonwealth of Pennsylvania, the Pittsburgh Lifesciences Greenhouse, and the Pittsburgh Tissue Engineering Initiative / Department of Defense.
ABBREVIATIONS
- DAMP
damage-associated molecular pattern molecule
- IL
interleukin
- IP-10
interferon-gamma inducible protein of 10 kDa
- MIG
monokine inducible by gamma interferon
- PAMP
pathogen-associated molecular pattern molecule
- SCI
spinal cord injury
- TNF-α
tumor necrosis factor-α
Footnotes
Dr. Vodovotz received grant support from the National Institutes of Health (#P50-GM-53789 & #UO1-DK072146) and from the National Institute on Disability and Rehabilitation Research (#H133E070024).
Dr. Vodovotz has U.S. Patent for “Modeling Wound Healing” (No. 8,165,819) and U.S. Patent applications for “Self-Regulating Device for Modulating Inflammation” (No. 13/121,013) and "Methods for Modeling Hepatic Inflammation" (No. 13/700,244).
Dr. Billiar’s institution receive grant support from NIH.
Drs. Billiar and Vodovotz received article research support from NIH. Dr. Vodovotz received article research support from NIDRR.
Dr. Vodovotz is the co-founder of Immunetrics, Inc. Dr Billiar and Dr. Vodovotz stock options with Immunetrics. Dr. Vodovotz’s laboratory received a high-performance computing platform valued at approximately $500,000 as a Shared University Award from IBM, Inc.
References
- 1.de Montmollin E, Annane D. Year in review 2010: Critical Care - multiple organ dysfunction and sepsis. Crit Care. 2011;15:236. doi: 10.1186/cc10359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gustot T. Multiple organ failure in sepsis: prognosis and role of systemic inflammatory response. Current opinion in critical care. 2011;17:153–159. doi: 10.1097/MCC.0b013e328344b446. [DOI] [PubMed] [Google Scholar]
- 3.An G, Namas R, Vodovotz Y. Sepsis: From pattern to mechanism and back. Crit Rev Biomed Eng. 2012;40:341–351. doi: 10.1615/critrevbiomedeng.v40.i4.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Angus DC, Linde-Zwirble WT, Lidicker J, et al. Epidemiology of severe sepsis in the United States: analysis of incidence, outcome, and associated costs of care. Crit Care Med. 2001;29:1303–1310. doi: 10.1097/00003246-200107000-00002. [DOI] [PubMed] [Google Scholar]
- 5.Namas R, Ghuma A, Hermus L, et al. The acute inflammatory response in trauma / hemorrhage and traumatic brain injury: Current state and emerging prospects. Libyan J Med. 2009;4:97–103. doi: 10.4176/090325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Patton GC, Coffey C, Sawyer SM, et al. Global patterns of mortality in young people: a systematic analysis of population health data. Lancet. 2009;374:881–892. doi: 10.1016/S0140-6736(09)60741-8. [DOI] [PubMed] [Google Scholar]
- 7.World Health Organization report: Young People: Health Risks and Solutions. 2011. [Google Scholar]
- 8.Vodovotz Y, Constantine G, Rubin J, et al. Mechanistic simulations of inflammation: current state and future prospects. Math Biosci. 2009:1–10. doi: 10.1016/j.mbs.2008.07.013. 2008/10/07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vodovotz Y, An G. Systems Biology and Inflammation. In: Yan Q, editor. Systems Biology in Drug Discovery and Development: Methods and Protocols. Totowa, NJ: Springer Science & Business Media; 2009. pp. 181–201. [Google Scholar]
- 10.Vodovotz Y. Translational systems biology of inflammation and healing. Wound Repair Regen. 2010:3–7. doi: 10.1111/j.1524-475X.2009.00566.x. 2010/01/20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dreyfuss D, Saumon G. Ventilator-induced lung injury: lessons from experimental studies. Am J Respir Crit Care Med. 1998;157:294–323. doi: 10.1164/ajrccm.157.1.9604014. [DOI] [PubMed] [Google Scholar]
- 12.Slutsky AS, Tremblay LN. Multiple system organ failure. Is mechanical ventilation a contributing factor? Am J Respir Crit Care Med. 1998;157:1721–1725. doi: 10.1164/ajrccm.157.6.9709092. [DOI] [PubMed] [Google Scholar]
- 13.An G, Nieman G, Vodovotz Y. Computational and systems biology in trauma and sepsis: Current state and future perspectives. Int J Burns Trauma. 2012;2:1–10. [PMC free article] [PubMed] [Google Scholar]
- 14.Godin PJ, Buchman TG. Uncoupling of biological oscillators: a complementary hypothesis concerning the pathogenesis of multiple organ dysfunction syndrome. Crit Care Med. 1996;24:1107–1116. doi: 10.1097/00003246-199607000-00008. [DOI] [PubMed] [Google Scholar]
- 15.Neugebauer EA, Willy C, Sauerland S. Complexity and non-linearity in shock research: reductionism or synthesis? Shock. 2001;16:252–258. doi: 10.1097/00024382-200116040-00003. [DOI] [PubMed] [Google Scholar]
- 16.Chinnaiyan AM, Huber-Lang M, Kumar-Sinha C, et al. Molecular signatures of sepsis: multiorgan gene expression profiles of systemic inflammation. Am J Pathol. 2001;159:1199–1209. doi: 10.1016/S0002-9440(10)62505-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cobb JP, Brownstein BH, Watson MA, et al. Injury in the era of genomics. Shock. 2001:165–170. doi: 10.1097/00024382-200115030-00001. 2001/03/10. [DOI] [PubMed] [Google Scholar]
- 18.Chung TP, Laramie JM, Province M, et al. Functional genomics of critical illness and injury. Crit Care Med. 2002;30:S51–S57. [PubMed] [Google Scholar]
- 19.Cobb JP, O’Keefe GE. Injury research in the genomic era. Lancet. 2004;363:2076–2083. doi: 10.1016/S0140-6736(04)16460-X. [DOI] [PubMed] [Google Scholar]
- 20.Yu SL, Chen HW, Yang PC, et al. Differential gene expression in gram-negative and gram-positive sepsis. Am J Respir Crit Care Med. 2004;169:1135–1143. doi: 10.1164/rccm.200211-1278OC. [DOI] [PubMed] [Google Scholar]
- 21.Wurfel MM. Microarray-based analysis of ventilator-induced lung injury. Proc Am Thorac Soc. 2007;4:77–84. doi: 10.1513/pats.200608-149JG. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Edmonds RD, Vodovotz Y, Lagoa C, et al. Transcriptomic response of murine liver to severe injury and hemorrhagic shock: A dual platform microarray analysis. Physiol Genomics. 2011;43:1170–1183. doi: 10.1152/physiolgenomics.00020.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wong HR. Clinical review: Sepsis and septic shock - the potential of gene arrays. Crit Care. 2012;16:204. doi: 10.1186/cc10537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Calvano SE, Xiao W, Richards DR, et al. A network-based analysis of systemic inflammation in humans. Nature. 2005;437:1032–1037. doi: 10.1038/nature03985. [DOI] [PubMed] [Google Scholar]
- 25.Xiao W, Mindrinos MN, Seok J, et al. A genomic storm in critically injured humans. J Exp Med. 2011;208:2581–2590. doi: 10.1084/jem.20111354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.An G, Faeder J, Vodovotz Y. Translational systems biology: Introduction of an engineering approach to the pathophysiology of the burn patient. J Burn Care Res. 2008;29:277–285. doi: 10.1097/BCR.0b013e31816677c8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vodovotz Y, Csete M, Bartels J, et al. Translational systems biology of inflammation. PLoS Comput Biol. 2008;4:1–6. doi: 10.1371/journal.pcbi.1000014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vodovotz Y, Constantine G, Faeder J, et al. Translational systems approaches to the biology of inflammation and healing. Immunopharmacol Immunotoxicol. 2010;32:181–195. doi: 10.3109/08923970903369867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mi Q, Li NYK, Ziraldo C, et al. Translational systems biology of inflammation: Potential applications to personalized medicine. Personalized Medicine. 2010;7:549–559. doi: 10.2217/pme.10.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vincent JL, Ferreira F, Moreno R. Scoring systems for assessing organ dysfunction and survival. Crit Care Clin. 2000;16:353–366. doi: 10.1016/s0749-0704(05)70114-7. [DOI] [PubMed] [Google Scholar]
- 31.Janes KA, Yaffe MB. Data-driven modelling of signal-transduction networks. Nat Rev Mol Cell Biol. 2006;7:820–828. doi: 10.1038/nrm2041. [DOI] [PubMed] [Google Scholar]
- 32.Rixen D, Siegel JH, Abu-Salih A, et al. Physiologic state severity classification as an indicator of posttrauma cytokine response. Shock. 1995;4:27–38. doi: 10.1097/00024382-199507000-00004. [DOI] [PubMed] [Google Scholar]
- 33.Rixen D, Siegel JH, Friedman HP. "Sepsis/SIRS," physiologic classification, severity stratification, relation to cytokine elaboration and outcome prediction in posttrauma critical illness. J Trauma. 1996;41:581–598. doi: 10.1097/00005373-199610000-00001. [DOI] [PubMed] [Google Scholar]
- 34.Rixen D, Siegel JH, Espina N, et al. Plasma nitric oxide in posttrauma critical illness: A function of sepsis and the physiological state severity classification quantifying the probability of death. Shock. 1997;7:17–28. [PubMed] [Google Scholar]
- 35.Gough NR, Yaffe MB. Focus issue: conquering the data mountain. Sci Signal. 2011;4:eg2. doi: 10.1126/scisignal.2001871. [DOI] [PubMed] [Google Scholar]
- 36.Nguyen A, Yaffe MB. Proteomics and systems biology approaches to signal transduction in sepsis. Crit Care Med. 2003;31:S1–S6. doi: 10.1097/00003246-200301001-00001. [DOI] [PubMed] [Google Scholar]
- 37.McDunn JE, Husain KD, Polpitiya AD, et al. Plasticity of the systemic inflammatory response to acute infection during critical illness: development of the riboleukogram. PLoS ONE. 2008;3:e1564. doi: 10.1371/journal.pone.0001564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wong HR, Cvijanovich N, Lin R, et al. Identification of pediatric septic shock subclasses based on genome-wide expression profiling. BMC Med. 2009;7:34. doi: 10.1186/1741-7015-7-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhou B, Xu W, Herndon D, et al. Analysis of factorial time-course microarrays with application to a clinical study of burn injury. Proc Natl Acad Sci U S A. 2010;107:9923–9928. doi: 10.1073/pnas.1002757107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sutherland A, Thomas M, Brandon RA, et al. Development and validation of a novel molecular biomarker diagnostic test for the early detection of sepsis. Crit Care. 2011;15:R149. doi: 10.1186/cc10274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liu T, Qian WJ, Gritsenko MA, et al. High dynamic range characterization of the trauma patient plasma proteome. Mol Cell Proteomics. 2006;5:1899–1913. doi: 10.1074/mcp.M600068-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Qian WJ, Petritis BO, Kaushal A, et al. Plasma proteome response to severe burn injury revealed by 18O-labeled “universal” reference-based quantitative proteomics. J Proteome Res. 2010;9:4779–4789. doi: 10.1021/pr1005026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gang Y, Malik M. Heart rate variability in critical care medicine. Curr Opin Crit Care. 2002;8:371–375. doi: 10.1097/00075198-200210000-00002. [DOI] [PubMed] [Google Scholar]
- 44.Seely AJ, Macklem PT. Complex systems and the technology of variability analysis. Crit Care. 2004;8:R367–R384. doi: 10.1186/cc2948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cancio LC, Batchinsky AI, Salinas J, et al. Heart-rate complexity for prediction of prehospital lifesaving interventions in trauma patients. J Trauma. 2008;65:813–819. doi: 10.1097/TA.0b013e3181848241. [DOI] [PubMed] [Google Scholar]
- 46.Cohen MJ, Grossman AD, Morabito D, et al. Identification of complex metabolic states in critically injured patients using bioinformatic cluster analysis. Crit Care. 2010;14:R10. doi: 10.1186/cc8864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Convertino VA, Moulton SL, Grudic GZ, et al. Use of advanced machine-learning techniques for noninvasive monitoring of hemorrhage. J Trauma. 2011;71:S25–S32. doi: 10.1097/TA.0b013e3182211601. [DOI] [PubMed] [Google Scholar]
- 48.Cobb JP, Laramie JM, Stormo GD, et al. Sepsis gene expression profiling: murine splenic compared with hepatic responses determined by using complementary DNA microarrays. Crit Care Med. 2002;30:2711–2721. doi: 10.1097/00003246-200212000-00016. [DOI] [PubMed] [Google Scholar]
- 49.Dasu MR, Cobb JP, Laramie JM, et al. Gene expression profiles of livers from thermally injured rats. Gene. 2004;327:51–60. doi: 10.1016/j.gene.2003.11.015. [DOI] [PubMed] [Google Scholar]
- 50.Mi Q, Constantine G, Ziraldo C, et al. A dynamic view of trauma/hemorrhage-induced inflammation in mice: Principal drivers and networks. PLoS ONE. 2011;6:e19424. doi: 10.1371/journal.pone.0019424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.An G, Nieman G, Vodovotz Y. Toward computational identification of multiscale tipping points in multiple organ failure. Ann Biomed Eng. 2012;40:2412–2424. doi: 10.1007/s10439-012-0565-9. [DOI] [PubMed] [Google Scholar]
- 52.An G. Concepts for developing a collaborative in silico model of the acute inflammatory response using agent-based modeling. J Crit Care. 2006;21:105–110. doi: 10.1016/j.jcrc.2005.11.012. [DOI] [PubMed] [Google Scholar]
- 53.An G, Wilensky U. From Artificial Life to In Silico Medicine: NetLogo as a Means of Translational Knowledge Representation in Biomedical Research. In: Adamatsky A, Komosinski M, editors. Artificial Life in Software. London: Springer-Verlag; 2009. pp. 183–209. [Google Scholar]
- 54.An G. Dynamic Knowledge Representation using Agent Based Modeling: Ontology Instantiation and Verification of Conceptual Models. In: Maly I, editor. Systems Biology: Methods in Molecular Biology Series. Totowa, NJ: Humana Press; 2009. pp. 445–468. [DOI] [PubMed] [Google Scholar]
- 55.An GC. Translational systems biology using an agent-based approach for dynamic knowledge representation: An evolutionary paradigm for biomedical research. Wound Rep Reg. 2010;18:8–12. doi: 10.1111/j.1524-475X.2009.00568.x. [DOI] [PubMed] [Google Scholar]
- 56.An G. Closing the scientific loop: Bridging correlation and causality in the petaflop age. Science Translational Med. 2010;2:41ps34. doi: 10.1126/scitranslmed.3000390. [DOI] [PubMed] [Google Scholar]
- 57.Bailey AM, Lawrence MB, Shang H, et al. Agent-based model of therapeutic adipose-derived stromal cell trafficking during ischemia predicts ability to roll on P-selectin. PLoS Comput Biol. 2009;5:e1000294. doi: 10.1371/journal.pcbi.1000294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kitano H. Systems biology: a brief overview. Science. 2002;295:1662–1664. doi: 10.1126/science.1069492. [DOI] [PubMed] [Google Scholar]
- 59.An G, Lee I. Complexity, emergence and pathophysiology: Using agent based computer simulation to characterize the non-adaptive inflammatory response (Manuscript # 344) Inter Journal Complex Systems. 2000 May http://www.interjournal.org.
- 60.An G. Agent-based computer simulation and SIRS: building a bridge between basic science and clinical trials. Shock. 2001;16:266–273. doi: 10.1097/00024382-200116040-00006. [DOI] [PubMed] [Google Scholar]
- 61.An G. In-silico experiments of existing and hypothetical cytokine-directed clinical trials using agent based modeling. Crit Care Med. 2004;32:2050–2060. doi: 10.1097/01.ccm.0000139707.13729.7d. [DOI] [PubMed] [Google Scholar]
- 62.An G. A model of TLR4 signaling and tolerance using a qualitative, particle event-based method: Introduction of Spatially Configured Stochastic Reaction Chambers (SCSRC) Math Biosci. 2009;217:43–52. doi: 10.1016/j.mbs.2008.10.001. [DOI] [PubMed] [Google Scholar]
- 63.Kumar R, Clermont G, Vodovotz Y, et al. The dynamics of acute inflammation. J Theoretical Biol. 2004;230:145–155. doi: 10.1016/j.jtbi.2004.04.044. [DOI] [PubMed] [Google Scholar]
- 64.Clermont G, Bartels J, Kumar R, et al. In silico design of clinical trials: a method coming of age. Crit Care Med. 2004;32:2061–2070. doi: 10.1097/01.ccm.0000142394.28791.c3. [DOI] [PubMed] [Google Scholar]
- 65.Chow CC, Clermont G, Kumar R, et al. The acute inflammatory response in diverse shock states. Shock. 2005;24:74–84. doi: 10.1097/01.shk.0000168526.97716.f3. [DOI] [PubMed] [Google Scholar]
- 66.Vodovotz Y, Chow CC, Bartels J, et al. In silico models of acute inflammation in animals. Shock. 2006;26:235–244. doi: 10.1097/01.shk.0000225413.13866.fo. [DOI] [PubMed] [Google Scholar]
- 67.Reynolds A, Rubin J, Clermont G, et al. A reduced mathematical model of the acute inflammatory response: I. Derivation of model and analysis of anti-inflammation. J Theor Biol. 2006;242:220–236. doi: 10.1016/j.jtbi.2006.02.016. [DOI] [PubMed] [Google Scholar]
- 68.Day J, Rubin J, Vodovotz Y, et al. A reduced mathematical model of the acute inflammatory response: II. Capturing scenarios of repeated endotoxin administration. J Theor Biol. 2006;242:237–256. doi: 10.1016/j.jtbi.2006.02.015. [DOI] [PubMed] [Google Scholar]
- 69.Prince JM, Levy RM, Bartels J, et al. In silico and in vivo approach to elucidate the inflammatory complexity of CD14-deficient mice. Mol Med. 2006;12:88–96. doi: 10.2119/2006-00012.Prince. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lagoa CE, Bartels J, Baratt A, et al. The role of initial trauma in the host’s response to injury and hemorrhage: Insights from a comparison of mathematical simulations and hepatic transcriptomic analysis. Shock. 2006;26:592–600. doi: 10.1097/01.shk.0000232272.03602.0a. [DOI] [PubMed] [Google Scholar]
- 71.Ben David I, Price SE, Bortz DM, et al. Dynamics of intrapulmonary bacterial growth in a murine model of repeated microaspiration. Am J Respir Cell Mol Biol. 2005;33:476–482. doi: 10.1165/rcmb.2005-0053OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Faeder JR, Blinov ML, Goldstein B, et al. Rule-based modeling of biochemical networks. Complexity. 2005;10:22–41. [Google Scholar]
- 73.Lipniacki T, Hat B, Faeder JR, et al. Stochastic effects and bistability in T cell receptor signaling. J Theor Biol. 2008;254:110–122. doi: 10.1016/j.jtbi.2008.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.An G, Faeder JR. Detailed qualitative dynamic knowledge representation using a BioNetGen model of TLR-4 signaling and preconditioning. Math Biosci. 2009;217:53–63. doi: 10.1016/j.mbs.2008.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Gong H, Zuliani P, Komuravelli A, et al. Analysis and verification of the HMGB1 signaling pathway. BMC Bioinformatics. 2010;11(Suppl 7):S10. doi: 10.1186/1471-2105-11-S7-S10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kocieniewski P, Faeder JR, Lipniacki T. The interplay of double phosphorylation and scaffolding in MAPK pathways. J Theor Biol. 2012;295:116–124. doi: 10.1016/j.jtbi.2011.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Vodovotz Y, Clermont G, Chow C, et al. Mathematical models of the acute inflammatory response. Curr Opin Crit Care. 2004;10:383–390. doi: 10.1097/01.ccx.0000139360.30327.69. [DOI] [PubMed] [Google Scholar]
- 78.An G, Hunt CA, Clermont G, et al. Challenges and rewards on the road to translational systems biology in acute illness: Four case reports from interdisciplinary teams. J Crit Care. 2007;22:169–175. doi: 10.1016/j.jcrc.2006.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.An G, Faeder J, Vodovotz Y. Translational systems biology: introduction of an engineering approach to the pathophysiology of the burn patient. J Burn Care Res. 2008:277–285. doi: 10.1097/BCR.0b013e31816677c8. 2008/03/21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Vodovotz Y, Csete M, Bartels J, et al. Translational systems biology of inflammation. PLoS Comput Biol. 2008:e1000014. doi: 10.1371/journal.pcbi.1000014. 2008/04/26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Mi Q, Li NY, Ziraldo C, et al. Translational systems biology of inflammation: potential applications to personalized medicine. Per Med. 2010:549–559. doi: 10.2217/pme.10.45. 2011/02/23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.An G, Bartels J, Vodovotz Y. In silico augmentation of the drug development pipeline: Examples from the study of acute inflammation. Drug Dev Res. 2010;72:1–14. doi: 10.1002/ddr.20415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.An G, Bartels J, Vodovotz Y. In Silico Augmentation of the Drug Development Pipeline: Examples from the study of Acute Inflammation. Drug Dev Res. 2011:187–200. doi: 10.1002/ddr.20415. 2011/05/10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Namas R, Zamora R, Namas R, et al. Sepsis: Something old, something new, and a systems view. J Crit Care. 2012(27):314e1–314e11. doi: 10.1016/j.jcrc.2011.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Foteinou PT, Calvano SE, Lowry SF, et al. Modeling endotoxin-induced systemic inflammation using an indirect response approach. Math Biosci. 2009;217:27–42. doi: 10.1016/j.mbs.2008.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Foteinou PT, Calvano SE, Lowry SF, et al. In silico simulation of corticosteroids effect on an NFkB-dependent physicochemical model of systemic inflammation. PLoS ONE. 2009;4:e4706. doi: 10.1371/journal.pone.0004706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Dong X, Foteinou PT, Calvano SE, et al. Agent-Based Modeling of Endotoxin-Induced Acute Inflammatory Response in Human Blood Leukocytes. PLoS ONE. 2010;5:e9249. doi: 10.1371/journal.pone.0009249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Scheff JD, Calvano SE, Lowry SF, et al. Modeling the influence of circadian rhythms on the acute inflammatory response. J Theor Biol. 2010;264:1068–1076. doi: 10.1016/j.jtbi.2010.03.026. [DOI] [PubMed] [Google Scholar]
- 89.Foteinou PT, Calvano SE, Lowry SF, et al. A physiological model for autonomic heart rate regulation in human endotoxemia. Shock. 2011;35:229–239. doi: 10.1097/SHK.0b013e318200032b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Scheff JD, Mavroudis PD, Calvano SE, et al. Modeling autonomic regulation of cardiac function and heart rate variability in human endotoxemia. Physiol Genomics. 2011;43:951–964. doi: 10.1152/physiolgenomics.00040.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Nieman K, Brown D, Sarkar J, et al. A two-compartment mathematical model of endotoxin-induced inflammatory and physiologic alterations in swine. Crit Care Med. 2012;40:1052–1063. doi: 10.1097/CCM.0b013e31823e986a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Vodovotz Y, Constantine G, Rubin J, et al. Mechanistic simulations of inflammation: Current state and future prospects. Math Biosci. 2009;217:1–10. doi: 10.1016/j.mbs.2008.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Dick TE, Molkov Y, Nieman G, et al. Linking Inflammation and Cardiorespiratory Variability in Sepsis via Computational Modeling. Frontiers in Physiology. 2012;3 doi: 10.3389/fphys.2012.00222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Vodovotz Y, Constantine G, Faeder J, et al. Translational systems approaches to the biology of inflammation and healing. Immunopharmacol Immunotoxicol. 2010;32:181–195. doi: 10.3109/08923970903369867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.An G, Namas R, Vodovotz Y. Sepsis: From pattern to mechanism and back. Crit Rev Biomed Eng. 2012 doi: 10.1615/critrevbiomedeng.v40.i4.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Klune JR, Dhupar R, Cardinal J, et al. HMGB1: endogenous danger signaling. Mol Med. 2008;14:476–484. doi: 10.2119/2008-00034.Klune. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Kaczorowski DJ, Mollen KP, Edmonds R, et al. Early events in the recognition of danger signals after tissue injury. J Leukoc Biol. 2008;83:546–552. doi: 10.1189/jlb.0607374. [DOI] [PubMed] [Google Scholar]
- 98.West MA, Heagy W. Endotoxin tolerance: a review. Crit Care Med. 2002;30:S64–S73. [PubMed] [Google Scholar]
- 99.Cavaillon JM, Adrie C, Fitting C, et al. Endotoxin tolerance: is there a clinical relevance? J Endotoxin Res. 2003;9:101–107. doi: 10.1179/096805103125001487. [DOI] [PubMed] [Google Scholar]
- 100.Rivière B, Epshteyn Y, Swigon D, et al. A simple mathematical model of signaling resulting from the binding of lipopolysaccharide with Toll-like receptor 4 demonstrates inherent preconditioning behavior. Math Biosci. 2009;217:19–26. doi: 10.1016/j.mbs.2008.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Yang Q, Calvano SE, Lowry SF, et al. A dual negative regulation model of Toll-like receptor 4 signaling for endotoxin preconditioning in human endotoxemia. Math Biosci. 2011;232:151–163. doi: 10.1016/j.mbs.2011.05.005. [DOI] [PubMed] [Google Scholar]
- 102.Fu Y, Glaros T, Zhu M, et al. Network topologies and dynamics leading to endotoxin tolerance and priming in innate immune cells. PLoS Comput Biol. 2012;8:e1002526. doi: 10.1371/journal.pcbi.1002526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Li NYK, Verdolini K, Clermont G, et al. A patient-specific in silico model of inflammation and healing tested in acute vocal fold injury. PLoS ONE. 2008;3:e2789. doi: 10.1371/journal.pone.0002789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Solovyev A, Mi Q, Tzen Y-T, et al. Hybrid equation-/agent-based model of ischemia-induced hyperemia and pressure ulcer formation predicts greater propensity to ulcerate in subjects with spinal cord injury. PLoS Comp Biol. 2013 doi: 10.1371/journal.pcbi.1003070. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Montgomerie JZ. Infections in patients with spinal cord injuries. Clin Infect Dis. 1997;25:1285–1290. doi: 10.1086/516144. [DOI] [PubMed] [Google Scholar]
- 106.Song SO, Hogg J, Peng ZY, et al. Ensemble models of neutrophil trafficking in severe sepsis. PLoS Comput Biol. 2012;8:e1002422. doi: 10.1371/journal.pcbi.1002422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Namas R, Namas R, Lagoa C, et al. Hemoadsorption reprograms inflammation in experimental Gram-negative septic fibrin peritonitis: Insights from in vivo and in silico studies. Mol Med. 2012 doi: 10.2119/molmed.2012.00106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Csete ME, Doyle JC. Reverse engineering of biological complexity. Science. 2002;295:1664–1669. doi: 10.1126/science.1069981. [DOI] [PubMed] [Google Scholar]
- 109.Hess KR, Zhang W, Baggerly KA, et al. Microarrays: handling the deluge of data and extracting reliable information. Trends Biotechnol. 2001;19:463–468. doi: 10.1016/s0167-7799(01)01792-9. [DOI] [PubMed] [Google Scholar]
- 110.Zak DE, Aderem A. Systems biology of innate immunity. Immunol Rev. 2009;227:264–282. doi: 10.1111/j.1600-065X.2008.00721.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Rixen D, Siegel JH, Friedman HP. "Sepsis/SIRS," physiologic classification, severity stratification, relation to cytokine elaboration and outcome prediction in posttrauma critical illness. J Trauma. 1996;41:581–598. doi: 10.1097/00005373-199610000-00001. [DOI] [PubMed] [Google Scholar]
- 112.Maier B, Lefering R, Lehnert M, et al. Early versus late onset of multiple organ failure is associated with differing patterns of plasma cytokine biomarker expression and outcome after severe trauma. Shock. 2007;28:668–674. [PubMed] [Google Scholar]
- 113.Janes KA, Albeck JG, Gaudet S, et al. A systems model of signaling identifies a molecular basis set for cytokine-induced apoptosis. Science. 2005;310:1646–1653. doi: 10.1126/science.1116598. [DOI] [PubMed] [Google Scholar]
- 114.An G, Mi Q, Dutta-Moscato J, et al. Agent-based models in translational systems biology. WIRES. 2009;1:159–171. doi: 10.1002/wsbm.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Faeder JR. Toward a comprehensive language for biological systems. BMC Biol. 2011;9:68. doi: 10.1186/1741-7007-9-68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Alt W, Lauffenburger DA. Transient behavior of a chemotaxis system modelling certain types of tissue inflammation. J Math Biol. 1987;24:691–722. doi: 10.1007/BF00275511. [DOI] [PubMed] [Google Scholar]
- 117.Blinov ML, Faeder JR, Goldstein B, et al. A network model of early events in epidermal growth factor receptor signaling that accounts for combinatorial complexity. Biosystems. 2006;83:136–151. doi: 10.1016/j.biosystems.2005.06.014. [DOI] [PubMed] [Google Scholar]