

Abstract
CD47 is a cell surface glycoprotein that helps mediate neutrophil transmigration across blood vessels. The present study was performed to determine whether absence of the CD47 gene decreases focal ischemic brain damage. Mice were subjected to 90 minutes middle cerebral artery occlusion. CD47 knockout mice were compared against matching wildtype mice. CD47 expression was checked by Western blotting. Infarct volume and ischemic brain swelling were quantified with cresyl violet–stained brain sections at 24 and 72 hours after ischemia. The tight junction protein claudin-5 was detected by imunohistochemistry. Two surrogate markers of neuroinflammation, brain levels of matrix metalloproteinase-9 (MMP-9) and infiltration of neutrophils, were assessed by immunohistochemistry. Western blots confirmed that CD47 was absent in knockout brains. Ischemia did not appear to upregulate total brain levels of CD47 in WT mice. In CD47 knockout mice, infarct volumes were reduced at 24 and 72 hours after ischemia, and hemispheric swelling was decreased at 72 hours. Loss of claudin-5 was observed in ischemic WT brain. This effect was ameliorated in CD47 knockout brains. Extravasation of neutrophils into the brain parenchyma was significantly reduced in CD47 knockout mice compared to wildtype mice. MMP-9 appeared to be upregulated in microvessels within ischemic brain. MMP-9 levels were markedly lower in CD47 knockout brains compared to wildtype brains. We conclude that CD47 is broadly involved in neuroinflammation, and this integrin-associated-protein plays a role in promoting MMP-9 upregulaton, neutrophil extravasation, brain swelling and progression of acute ischemic brain injury.
INTRODUCTION
CD47 or integrin associated protein is an Ig super family transmembrane glycoprotein that is widely expressed in most cells and tissues. Originally, CD47 was identified in association with the αvβ3 integrin. CD47 can interact with thrombopondin-1 (TSP-1) (Sipes, et al., 1999), and has a part in β3 integrin-mediated actions. CD47 plays an important immunoregulatory role in neutrophil transmigration responding to inflammatory stimulation (Parkos, et al., 1996), as demonstrated in CD47-deficient mice that appear to have a defect in phagocyte activation (Lindberg, et al., 1996). In vitro studies indicate that CD47 regulates some adhesion-dependent cell functions, and is involved in neutrophil migration across peripheral endothelial cells and monocyte migration across epithelial cells (Cooper, et al., 1995, Liu, et al., 2001, Rosseau, et al., 2000).
Cerebral ischemic injury is associated with the induction of a series of inflammatory events, including the infiltraction of circulating immune cells (neutrophils and monocytes) and activation of resident cells (Muir, et al., 2007). Of the various types of leukocytes, neutrophils (the major subtype of polymorphonuclear leukocytes) are among the first to infiltrate ischemic brain. Extravasated polymorphonuclear leukocytes release reactive oxygen species and lipid peroxidation products, and promote blood-brain barrier disruption, vascular plugging, edema, and development of infarction (Barone and Feuerstein, 1999, Hernandez, et al., 1987, Zhang, et al., 1994). There may also be positive feedback because in vitro studies suggest that CD47 signaling on endothelium may further amplify the expression of adhesion molecules (Narizhneva, et al., 2005). These experimental observations led us to hypothesize that CD47 might be a potential anti-inflammatory target for stroke therapy. In this study, we examined the effect of CD47 gene knockout on the progression of inflammation and infarction in a mouse model of focal cerebral ischemia.
MATERIALS AND METHODS
Mouse Focal Cerebral Ischemia
All experiments were performed following an institutionally approved protocol in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. CD47 gene knock-out (CD47 KO) mice were generated as previously described, and compared against matching C57BL/6 wildtypes (Oldenborg, et al., 2000). Male mice around 12 weeks old were subjected to transient focal cerebral ischemia by intraluminal middle cerebral artery blockade with a 7.0 silicon-coated nylon suture. Anesthesia was induced with 1% isoflurane in a mixture of 70% nitrous oxide and 30% oxygen delivered by face mask. Rectal temperature was maintained at 37 ± 0.5 °C. Systemic parameters including pH and blood gases were all within normal range and there were no differences between wildtype and KO mice. Regional cerebral blood flow was monitored by laser Doppler flowmetry. Probes were placed at the center (from bregma: 3.5 mm lateral, 1.5 mm caudal) and periphery (from bregma: 1.5 mm lateral, 1.5 mm caudal) of the ischemic territory (Shimizu-Sasamata, et al., 1998). After 90 minutes of focal ischemia, CBF was restored by withdrawal of the nylon suture. Blood gases were monitored during the ischemic occlusion.
Calculation of Brain Infarction and Swelling
Brains were removed at 24 or 72 hours after reperfusion. With 24 hours brain samples, 1 mm coronal slices were made from front of brain to brainstem, and 2,3,5-triphenyltetrazolium chloride (TTC) (Sigma Chemical Co., St.Louis, MO) staining was performed to demonstrate the presence of infarction. With 72 hours brain samples, fourteen serial 12 μm-thick coronal sections from each brain were cut at 500 μm intervals beginning at 1 mm from the frontal pole using a cryostat. Each slice was stained with cresyl violet. The infarction areas were measured with the computer-assisted image analysis software ImageJ (NIH). Brain swelling was calculated as = [(ipsilateral hemisphere’s volume/contralateral hemisphere’s volume)-1] x100 (Strbian, et al., 2006). True infarction volumes were corrected by the swelling factor (Takano, et al., 1997).
Immunohistochemistry
Brains were were perfused with PBS (pH 7.4) and quickly frozen in liquid nitrogen. Coronal sections at the caudate level were cut on a cryostat at −20°C to 12μm thickness, and collected on glass slides. Staining was performed for neutrophils (NIMP-R14, Cell Sciences), MMP-9 (antibody generously donated by Robert Senior, Washington University St Louis) (Betsuyaku, et al., 2000) with CD31 (BD Pharmingen) endothelial labeling, and claudin-5 (Zymed) with CD31 labeling. Sections were fixed by 4% paraformaldehyde (PFA) or ethanol, and rinsed three times in PBS (pH 7.4). After blocking with 3% normal goat serum, sections were then incubated at 4°C overnight in a PBS solution containing the primary antibodies in PBS, 0.3% TritonX, 3% bovine serum albumin. The sections were washed and incubated for 1 hour with secondary antibodies with fluorescence conjugations. Subsequently, the slides were covered with VECTASHIELD mounting medium with 4′, 6′-diamidino-2-phenylindole (DAPI) (H-1200; Vector Laboratories). Signals were examined using an Olympus microscope (BX51; Olympus) equipped with FITC and rhodamine filter set. The density of staining was analyzed by using NIH ImageJ software from representative staining sections, and the measurement was repeated 3 times in each brain. All images were collected from microscope image system under the same conditions, and the positive pixel area was calculated after setting threshold range from 50 to 255(Lee, et al., 2006). These imaging processes were automatically completed by batch processing with macro script programming.
Immunoblotting
Whole-cell protein extraction was performed on samples from sham controls and ischemic brain after 6 and 8 hours of focal ischemia. Brain tissues were homogenized in 10X vol of cold protein extraction buffer (complete tablet; Roche Molecular Biochemicals, Mannheim, Germany), centrifuged at 10,000g for 20 mins at 4°C, and the supernatant was used for analysis. After adding the 2X volume of sample buffer (Invitrogen) to the supernatant, equal amounts were loaded per lane. The primary antibody was a 1:1000 dilution of goat polyclonal antibody against CD47 (AF1866, R&D Systems). Western blots were performed with horseradish peroxidase (HRP)-conjugated anti-goat IgG (Sigma) using a chemiluminescence Western blotting detection system kit (ECLTM-RPN 2106, Amersham Pharmacia Biotech, Uppsala, Sweden).
Statistical Analysis
Data were expressed as mean ± s.d.. Statistical analysis was performed using ANOVA and Student’s t-test. Differences with P<0.05 were considered statistically significant.
RESULTS
Immunoblotting of brain homogenates revealed CD47 as a band at approximately 50 kDa. CD47 was confirmed to be absent in the CD47 knockout mouse brain samples (Fig. 1). Clear bands were observed in all wildtype mice brain samples, but no significant difference in CD47 expression could be detected between ischemic versus control wildtype mice, even by 24 hours post-ischemia.
Figure 1.
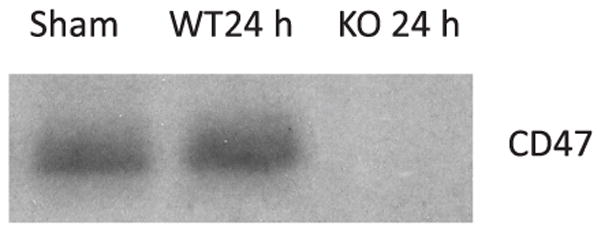
Western blot for CD47 in wildtype (WT) and CD47 knockout (KO) mouse brain tissue homogenates at 24 hours after ischemia. No clear difference in CD47 expression was detected between ischemic animals and control animals. No band was detected in CD47 knockout brains. N=3 per lane.
There were no significant differences in systemic physiologic parameters in the two groups of wildtype and knockout mice (Table 1).After arterial occlusion, regional cerebral perfusion in the center and periphery of the ischemic territory rapidly dropped to approximately 10% and 45% of pre-ischemic baselines, respectively (Fig. 2). Reperfusion induced a return to almost normal blood flow levels in both center and periphery. This occurred uniformly in all mice and there were no differences between wildtype and CD47 knockout blood flow response (Fig. 2).
Table 1.
Physiological Parameters
| pO2 | pCO2 | pH | BP | |
|---|---|---|---|---|
| WT | 114.4 ± 22.2 | 37.6 ± 5.3 | 7.3 ± 0.1 | 92.5 ± 2.9 |
| CD47 KO | 107.8 ± 42.9 | 37.1 ± 4.7 | 7.4 ± 0.1 | 99.2 ± 7.3 |
(mean ± SD)
Figure 2.
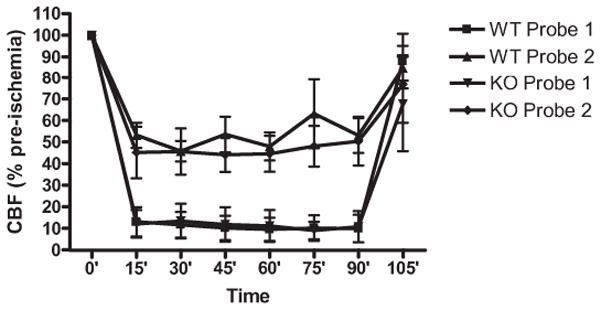
Regional cerebral blood flow (rCBF) in the center of the ischemic focus (probe 1) and peripheral zones (probe 2) of the ischemic distribution. Ischemia led to the expected drops in rCBF. Reperfusion restored rCBF levels close to 100% baselines. There were no significant differences between wildtype (WT) and CD47 knockout (KO) mice.
At 24 hours after arterial occlusion and reperfusion, ischemic brain swelling began to occur in all mice. The degree of swelling was not significantly different in CD47 knockouts (13.6 ± 7.1 %) compared with wildtype mice (17.5 ± 3.2 %, Fig. 3A–B). However, hemispheric swelling continued to develop in the wildtype group, and by 72 hrs, swelling had significantly worsened to 38.6 ± 7.5 %. In contrast, no progression was detected in the CD47 knockouts, and the degree of hemispheric swelling was significantly lower than wildtypes at 72 hrs (13.8 ± 11.7 %, *p<0.01, Fig. 3A–B).
Figure 3.

Brain infarction and swelling after 90 minutes of transient focal cerebral ischemia. (A) Representative sections of wildtype (WT) and CD47 knockout (KO) brains measured at 24 and 72 hours. (B) Progression of hemispheric swelling occurred from 24 to 72 hrs in WT brains, but not CD47 KO brains. (C) Ischemic infarction volumes were significantly decreased in CD47 KO compared to WT brains. N=8 per group and N=5 per group for 24 hr and 72 hr measurements respectively, *p<0.05.
Because of the profound hemispheric swelling, infarct sizes were recalculated to normalize for ipsilateral versus contralateral volume ratios. With this normalization procedure, infarct volumes were found to be significantly reduced by CD47 gene knockout at both 24 hrs and 72 hrs (108.2 ± 14.5 mm3 in wildtype vs. 77.7 ± 29.7 mm3 in knockouts at 24 hrs, P<0.05; 110.7 ± 14.5 mm3 in wildtype vs. 135.4 ± 9.8 mm3 in knockouts, *p<0.05; Fig. 3C).
Brain swelling is typically associated with blood-brain barrier disruption and edema. So claudin-5 was assessed as a representative tight junction protein. In normal wildtype and knockout contralateral brain tissue, claudin-5 signals appeared to co-localize with CD31-positive microvessels, as expected (Fig. 4). At 72 hrs after ischemia, claudin-5 levels in peri-infarct cortical regions were decreased, but the degree of claudin-5 reduction appeared larger in wildtype brains compared to CD47 knockout brains (Fig 4).
Figure 4.

Double immunostaining for CD31 (red) and claudin-5 (green) at 72 hrs after ischemia. Claudin-5 staining was colocalized with the endothelial maker CD31. Claudin-5 signals were decreased in ischemic (peri-infarct) cortex compared to matching areas in contralateral brain. Loss of claudin-5 signals appeared to be ameliorated in CD47 knockout mice compared to wildtype mice, in spite of the fact that cell densities (DAPI, blue) and microvessel numbers (CD31, red) appeared similar. Con: contralateral, ipsi: ipsilateral. Scale bar: 50 μm.
A great number of neutrophils were observed in the ischemic territory in wildtype brains at 72 hours post-ischemia (Fig. 5A). Neutrophil infiltration was significantly decreased in knockout mice compared to wildtype mice (Fig. 5A–B). Corresponding to areas of neutrophil infiltration, MMP-9 appeared to be upregulated as well. Co-localization with CD31 suggested that MMP-9 was mainly elevated in microvessels of ischemic brain areas (Fig. 6A). Quantitative densitometry demonstrated that MMP-9 levels were significantly reduced in CD47 knockout versus wildtype brains at 72 hrs post-ischemia (Fig. 6B).
Figure 5.

Neutrophil infiltration at 72 hrs after ischemia. (A) Representative sections showing immunoreactivity for neutrophils within the ischemic area in WT and CD47 KO brains. Scale bar: 100 μm. (B) Quantitative data showing a significant reduction of neutrophils in CD47 KO compared to WT brains, *p<0.05.
Figure 6.

MMP-9 expression at 72 hrs after ischemia. (A) Representative photomicrographs showing immunoreactivity for MMP-9 within the ischemic area in WT and CD47 KO brains. MMP-9 appeared co-localized with the endothelial marker CD31. Scale bar: 50 μm. (B) Quantitative data showing a significant reduction of MMP-9 immunostaining image density in CD47 KO compared to WT brains, *p<0.05.
DISCUSSION
The current study suggests that CD47 may play an important role in the pathophysiology of focal cerebral ischemia. We observed that genetic knockout of CD47 (i) reduced infarction at both 24 and 72 hrs post-stroke, (ii) prevented the progression of hemispheric swelling and edema from 24 to 72 hrs, (iii) ameliorated the loss of the tight junction protein claudin-5, (iv) decreased the infiltration of neutrophils into ischemic brain, and (v) downregulated the overall inflammatory MMP-9 response in damaged parenchyma.
CD47 is a widely expressed molecule in brain tissue, with cellular localization including endothelial cells, macrophages and neurons (de Vries, et al., 2002, Reinhold, et al., 1995). Systemically, CD47 is known to contribute to the transmigration of various blood cells across endothelial and epithelial cells during inflammation (Cooper, et al., 1995, Liu, et al., 2001, Rosseau, et al., 2000). More recently, it has been suggested that similar phenomena may also take place in brain, with CD47 mediating monocyte movements across cerebral endothelial layers (de Vries, et al., 2002). In the context of stroke, infiltration of inflammatory blood cells is expected to worsen brain injury. Our finding that the deletion of CD47 reduced ischemic brain injury was consistent with this general principle. What was a bit surprising however, was that we were unable to detect any upregulation of CD47 after ischemic onset. Previous studies have similarly reported that CD47 expression on brain endothelial cells was not significantly increased upon treatment of the cells with proinflammatory cytokines (Cooper, et al., 1995, de Vries, et al., 2002). Taken together, these observations indicate that constitutive expression of CD47 in brain in vivo may act in concert with other factors after ischemic stimulation to mediate injury in stroke.
Emigration of neutrophils, monocytes and other cells from the blood into the CNS is a key event in the development of neuroinflammatory lesions. Experimental studies in animal models of focal ischemic stroke showed that inhibiting the accumulation of neutrophils decreased brain injury (Jean, et al., 1998, Kochanek and Hallenbeck, 1992, Matsuo, et al., 1994, Mori, et al., 1992). Consistent with a previous report (Maier, et al., 2004), neutrophil infiltration in our model appeared to be easily detected by 72 hours after ischemia. In our CD47 knockouts, reduction of neutrophils appeared to coincide with the downregulation of MMP-9 signals in neurovascular locations. Neutrophils, as a source of both reactive oxygen species and MMP-9, infiltrate damaged tissue after ischemia and contribute to delayed secondary tissue damage (Maier, et al., 2004). MMP-9, as a key inflammatory mediator in stroke, further breaks down the blood-brain barrier and exacerbates inflammatory processes (Kochanek and Hallenbeck, 1992, Matsuo, et al., 1994). Matrix metalloproteinases (MMPs) disrupt the blood-brain barrier during ischemia-reperfusion by degrading tight junction proteins (Yang, et al., 2007), and the neurovascular protease activity is increased by neutrophil-derived matrix metalloproteinase-9 (McColl, et al., 2008). In the present study, enhanced preservation of the tight junction protein claudin-5 in CD47 knockout mice may also contribute to the prevention of brain edema (Jin, et al., 2008) and neutrophil influx (Gurney, et al., 2006). These integrated cellular events may explain the amelioration of brain swelling and infarction. Taken together, these results suggest that blocking CD47 signaling may provide a potential target for broadly attenuating inflammation and the progression of ischemic injury after stroke.
Nevertheless, there are several caveats that warrant further study. First, we were unable to detect cell types involved in CD47 signaling. All the antibodies we tested failed in yielding reliable immunostaining (data not shown). Therefore, we cannot be sure how CD47 might differentially function in endothelial cells, glia and neurons during stroke. We have recently shown that CD47 can trigger neuronal death via caspase-dependent and caspase-independent oxidative stress mechanisms (Xing, et al., 2008). Hence it is conceivable that in addition to the neurovascular inflammation discussed thus far, blocking CD47 can also prevent direct neuronal death. A second issue relates to the ability of CD47 stimulate platelet aggregation (Dorahy, et al., 1997, Isenberg, et al., 2008, Kato, et al., 2005). Is it possible that CD47 activation worsens blood flow in penumbral areas? Although we did not detect any perfusion changes in our model, it must be noted that the laser doppler method used here provide only single point measurements. More rigorous autoradiographic or in vivo imaging data will be needed to determine whether blocking CD47 can also rescue brain by improving flow. A third concern relates to quantitative relationships between our various neurovascular endpoints. We observed amelioration of claudin-5 degradation, reduction of neutrophils and downregulation of MMP-9. But we were unable to quantify the spatial correlation between these events at the microvascular scale. Future studies using in vivo optical imaging may help in this regard. Finally, our study, though suggestive, leads to more questions of causality and translational potential for stroke. Although we showed that CD47 gene knockout reduced various parameters of ischemic brain injury, we cannot be sure what mechanisms are truly involved. It is likely that multiple actions may occur, i.e. blocking CD47 may improve blood flow, decrease neurovascular inflammation, decrease neuronal death, ameliorate edema and brain swelling, and reduce tissue infarction. But how can we design compounds to block CD47? What about the time-course? And what is the corresponding therapeutic window for tackling CD47 as a target? Our current study only provides in vivo proof of concept.
In conclusion, we showed that genetic deletion of CD47 reduces extravasation of neutrophils and MMP-9 expression, thereby decreasing infarct volume and ischemic swelling in a mouse model of focal cerebral ischemia. Interference with CD47 pathway may offer a novel approach to reduce inflammatory injury in stroke. Further studies are warranted to explore CD47 as a potential target for stroke therapy.
Acknowledgments
Supported in part by NIH grants R01-NS37074, R01-NS48422, R01-NS53560, P01-NS55104
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Barone FC, Feuerstein GZ. Inflammatory mediators and stroke: new opportunities for novel therapeutics. J Cereb Blood Flow Metab. 1999;19:819–834. doi: 10.1097/00004647-199908000-00001. [DOI] [PubMed] [Google Scholar]
- 2.Betsuyaku T, Fukuda Y, Parks WC, Shipley JM, Senior RM. Gelatinase B is required for alveolar bronchiolization after intratracheal bleomycin. Am J Pathol. 2000;157:525–535. doi: 10.1016/S0002-9440(10)64563-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cooper D, Lindberg FP, Gamble JR, Brown EJ, Vadas MA. Transendothelial migration of neutrophils involves integrin-associated protein (CD47) Proc Natl Acad Sci U S A. 1995;92:3978–3982. doi: 10.1073/pnas.92.9.3978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.de Vries HE, Hendriks JJ, Honing H, De Lavalette CR, van der Pol SM, Hooijberg E, Dijkstra CD, van den Berg TK. Signal-regulatory protein alpha-CD47 interactions are required for the transmigration of monocytes across cerebral endothelium. J Immunol. 2002;168:5832–5839. doi: 10.4049/jimmunol.168.11.5832. [DOI] [PubMed] [Google Scholar]
- 5.Dorahy DJ, Thorne RF, Fecondo JV, Burns GF. Stimulation of platelet activation and aggregation by a carboxyl-terminal peptide from thrombospondin binding to the integrin-associated protein receptor. J Biol Chem. 1997;272:1323–1330. doi: 10.1074/jbc.272.2.1323. [DOI] [PubMed] [Google Scholar]
- 6.Gurney KJ, Estrada EY, Rosenberg GA. Blood-brain barrier disruption by stromelysin-1 facilitates neutrophil infiltration in neuroinflammation. Neurobiol Dis. 2006;23:87–96. doi: 10.1016/j.nbd.2006.02.006. [DOI] [PubMed] [Google Scholar]
- 7.Hernandez LA, Grisham MB, Twohig B, Arfors KE, Harlan JM, Granger DN. Role of neutrophils in ischemia-reperfusion-induced microvascular injury. Am J Physiol. 1987;253:H699–703. doi: 10.1152/ajpheart.1987.253.3.H699. [DOI] [PubMed] [Google Scholar]
- 8.Isenberg JS, Frazier WA, Roberts DD. Thrombospondins: from structure to therapeutics: Thrombospondin-1: a physiological regulator of nitric oxide signaling. Cell Mol Life Sci. 2008;65:728–742. doi: 10.1007/s00018-007-7488-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jean WC, Spellman SR, Nussbaum ES, Low WC. Reperfusion injury after focal cerebral ischemia: the role of inflammation and the therapeutic horizon. Neurosurgery. 1998;43:1382–1396. doi: 10.1097/00006123-199812000-00076. discussion 1396–1387. [DOI] [PubMed] [Google Scholar]
- 10.Jin G, Arai K, Murata Y, Wang S, Stins MF, Lo EH, van Leyen K. Protecting against cerebrovascular injury: contributions of 12/15-lipoxygenase to edema formation after transient focal ischemia. Stroke. 2008;39:2538–2543. doi: 10.1161/STROKEAHA.108.514927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kato H, Honda S, Yoshida H, Kashiwagi H, Shiraga M, Honma N, Kurata Y, Tomiyama Y. SHPS-1 negatively regulates integrin alphaIIbbeta3 function through CD47 without disturbing FAK phosphorylation. J Thromb Haemost. 2005;3:763–774. doi: 10.1111/j.1538-7836.2005.01235.x. [DOI] [PubMed] [Google Scholar]
- 12.Kochanek PM, Hallenbeck JM. Polymorphonuclear leukocytes and monocytes/macrophages in the pathogenesis of cerebral ischemia and stroke. Stroke. 1992;23:1367–1379. doi: 10.1161/01.str.23.9.1367. [DOI] [PubMed] [Google Scholar]
- 13.Lee SR, Kim HY, Rogowska J, Zhao BQ, Bhide P, Parent JM, Lo EH. Involvement of matrix metalloproteinase in neuroblast cell migration from the subventricular zone after stroke. J Neurosci. 2006;26:3491–3495. doi: 10.1523/JNEUROSCI.4085-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lindberg FP, Bullard DC, Caver TE, Gresham HD, Beaudet AL, Brown EJ. Decreased resistance to bacterial infection and granulocyte defects in IAP-deficient mice. Science. 1996;274:795–798. doi: 10.1126/science.274.5288.795. [DOI] [PubMed] [Google Scholar]
- 15.Liu Y, Merlin D, Burst SL, Pochet M, Madara JL, Parkos CA. The role of CD47 in neutrophil transmigration. Increased rate of migration correlates with increased cell surface expression of CD47. J Biol Chem. 2001;276:40156–40166. doi: 10.1074/jbc.M104138200. [DOI] [PubMed] [Google Scholar]
- 16.Maier CM, Hsieh L, Yu F, Bracci P, Chan PH. Matrix metalloproteinase-9 and myeloperoxidase expression: quantitative analysis by antigen immunohistochemistry in a model of transient focal cerebral ischemia. Stroke. 2004;35:1169–1174. doi: 10.1161/01.STR.0000125861.55804.f2. [DOI] [PubMed] [Google Scholar]
- 17.Matsuo Y, Onodera H, Shiga Y, Nakamura M, Ninomiya M, Kihara T, Kogure K. Correlation between myeloperoxidase-quantified neutrophil accumulation and ischemic brain injury in the rat. Effects of neutrophil depletion. Stroke. 1994;25:1469–1475. doi: 10.1161/01.str.25.7.1469. [DOI] [PubMed] [Google Scholar]
- 18.McColl BW, Rothwell NJ, Allan SM. Systemic inflammation alters the kinetics of cerebrovascular tight junction disruption after experimental stroke in mice. J Neurosci. 2008;28:9451–9462. doi: 10.1523/JNEUROSCI.2674-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mori E, del Zoppo GJ, Chambers JD, Copeland BR, Arfors KE. Inhibition of polymorphonuclear leukocyte adherence suppresses no-reflow after focal cerebral ischemia in baboons. Stroke. 1992;23:712–718. doi: 10.1161/01.str.23.5.712. [DOI] [PubMed] [Google Scholar]
- 20.Muir KW, Tyrrell P, Sattar N, Warburton E. Inflammation and ischaemic stroke. Curr Opin Neurol. 2007;20:334–342. doi: 10.1097/WCO.0b013e32813ba151. [DOI] [PubMed] [Google Scholar]
- 21.Narizhneva NV, Razorenova OV, Podrez EA, Chen J, Chandrasekharan UM, DiCorleto PE, Plow EF, Topol EJ, Byzova TV. Thrombospondin-1 up-regulates expression of cell adhesion molecules and promotes monocyte binding to endothelium. FASEB J. 2005;19:1158–1160. doi: 10.1096/fj.04-3310fje. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Oldenborg PA, Zheleznyak A, Fang YF, Lagenaur CF, Gresham HD, Lindberg FP. Role of CD47 as a marker of self on red blood cells. Science. 2000;288:2051–2054. doi: 10.1126/science.288.5473.2051. [DOI] [PubMed] [Google Scholar]
- 23.Parkos CA, Colgan SP, Liang TW, Nusrat A, Bacarra AE, Carnes DK, Madara JL. CD47 mediates post-adhesive events required for neutrophil migration across polarized intestinal epithelia. J Cell Biol. 1996;132:437–450. doi: 10.1083/jcb.132.3.437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Reinhold MI, Lindberg FP, Plas D, Reynolds S, Peters MG, Brown EJ. In vivo expression of alternatively spliced forms of integrin-associated protein (CD47) J Cell Sci. 1995;108 (Pt 11):3419–3425. doi: 10.1242/jcs.108.11.3419. [DOI] [PubMed] [Google Scholar]
- 25.Rosseau S, Selhorst J, Wiechmann K, Leissner K, Maus U, Mayer K, Grimminger F, Seeger W, Lohmeyer J. Monocyte migration through the alveolar epithelial barrier: adhesion molecule mechanisms and impact of chemokines. J Immunol. 2000;164:427–435. doi: 10.4049/jimmunol.164.1.427. [DOI] [PubMed] [Google Scholar]
- 26.Shimizu-Sasamata M, Bosque-Hamilton P, Huang PL, Moskowitz MA, Lo EH. Attenuated neurotransmitter release and spreading depression-like depolarizations after focal ischemia in mutant mice with disrupted type I nitric oxide synthase gene. J Neurosci. 1998;18:9564–9571. doi: 10.1523/JNEUROSCI.18-22-09564.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sipes JM, Krutzsch HC, Lawler J, Roberts DD. Cooperation between thrombospondin-1 type 1 repeat peptides and alpha(v)beta(3) integrin ligands to promote melanoma cell spreading and focal adhesion kinase phosphorylation. J Biol Chem. 1999;274:22755–22762. doi: 10.1074/jbc.274.32.22755. [DOI] [PubMed] [Google Scholar]
- 28.Strbian D, Karjalainen-Lindsberg ML, Tatlisumak T, Lindsberg PJ. Cerebral mast cells regulate early ischemic brain swelling and neutrophil accumulation. J Cereb Blood Flow Metab. 2006;26:605–612. doi: 10.1038/sj.jcbfm.9600228. [DOI] [PubMed] [Google Scholar]
- 29.Takano K, Tatlisumak T, Formato JE, Carano RA, Bergmann AG, Pullan LM, Bare TM, Sotak CH, Fisher M. Glycine site antagonist attenuates infarct size in experimental focal ischemia. Postmortem and diffusion mapping studies. Stroke. 1997;28:1255–1262. doi: 10.1161/01.str.28.6.1255. discussion 1263. [DOI] [PubMed] [Google Scholar]
- 30.Xing C, Lee S, Kim W, Jin G, Yang Y, Ji X, Wang X, Lo EH. Role of oxidative stress and caspase-3 in CD47-mediated neuronal cell death. J Neurochem. 2009;108:430–436. doi: 10.1111/j.1471-4159.2008.05777.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yang Y, Estrada EY, Thompson JF, Liu W, Rosenberg GA. Matrix metalloproteinase-mediated disruption of tight junction proteins in cerebral vessels is reversed by synthetic matrix metalloproteinase inhibitor in focal ischemia in rat. J Cereb Blood Flow Metab. 2007;27:697–709. doi: 10.1038/sj.jcbfm.9600375. [DOI] [PubMed] [Google Scholar]
- 32.Zhang RL, Chopp M, Chen H, Garcia JH. Temporal profile of ischemic tissue damage, neutrophil response, and vascular plugging following permanent and transient (2H) middle cerebral artery occlusion in the rat. J Neurol Sci. 1994;125:3–10. doi: 10.1016/0022-510x(94)90234-8. [DOI] [PubMed] [Google Scholar]