

Abstract
Osteopetrosis, a rare congenital genetic disease characterized by increased bone density due to impaired bone resorption by osteoclasts. It is classified into three forms: Infantile malignant autosomal recessive (AR) osteopetrosis, intermediate (AR) osteopetrosis and autosomal dominant (AD) osteopetrosis. Incidence of infantile malignant AR is 1/2,00,000 and if untreated has a fatal outcome. The condition is commonly diagnosed in infancy with symptoms of significant hematologic abnormalities with bone marrow failure, hepatosplenomegaly, macrocephaly with frontal bossing and bone fractures. Because of rarity of this type of malignant infantile form of osteopretrosis, we like to report this case of malignant infantile osteopetrosis who presented with bronchopneumonia, anemia with melaena at 2 months 15 days of age.
Keywords: Fatal, genetic, hepatosplenomegaly, infancy, malignant infantile osteopetrosis
Introduction
Osteopetrosis is a rare congenital genetic disorder characterized by increased bone density due to impaired bone resorption by osteoclasts.[1] The autosomal dominant (AD) form of osteopetrosis is usually asymptomatic. It is diagnosed incidentally or may exhibit mild symptoms in late childhood or adult life, but is compatible with long term survival.[2] Autosomal recessive (AR) malignant infantile osteopetrosis (MIOP), Online Mendelian Inheritance in men (OMIM)(259700), is uncommon however, it is a severe fatal disorder with an average incidence of 1:200,000-1:300,000.[1] This disease is caused by defector mutation in gene and 14 out of 20 MIOP patients had gene mutations.[1] This condition is most commonly diagnosed soon after birth or within the1st year of life with severe symptoms of abnormal bone remodeling, including significant hematologic abnormalities with bone marrow failure and extramedullary hematopoiesis, resulting in hepatosplenomegaly, a characteristic macrocephaly with frontal bossing, exophthalmus, bonefractures, and failure to thrive.[1,3] We present a case of MIOP who presented with bronchopneumonia, anemia with melaena at 2 months 15 days of age.
Case Report
A 2 months 15 days old hindu, female baby born to non-consanguineous couple presented with fever, cough, hurried respiration and melaena since 4 days. There was prominent frontal bossing, depressed nasal bridge and wide opened anterior fontenellae 4 cm × 5 cm and posterior 0.5 cm × 0.5 cm. She was febrile (100°F), RR 60/min, PR 120/min and blood pressure of 90/64mm of Hg. She was pale, otherwise skin and mucous membrane were normal and her head circumference 38 cm (>10th centile), weight 4 kg (<5th centile), and length 48 cm (<5th centile). Abdominal examination revealed firm hepatomegaly and 7 cm spleenomegaly. Respiratory system revealed crepitations and ronchi. Her detailed ophthalmic examination including fundus was normal. Other systems examination was unremarkable. Investigations revealed: Haemoglobin 9 g/dl, TC 39, 400/mm3 with differential count of N-32%, L-42%, M-0%, E-05%, B-0%, Promyelocyte 06%, Myelocyte 7%, Metamyelocyte 05%, ESR 25 mm in 1st hr and platelet count 66,000/mm3. Peripheral Smear revealed erythrocytosis showing moderate degree of anisopoikilocytosis, majority of Red Blood Cells are normocytic and normochromic. Nucleated RBC's seen (12/100WBC's), along with good number of polychromoatophilic RBC's. Tear drop cells also seen. Leucocytes are increased with shift to left, platelets are reduced. Her RBC's indices are: MCH 24.6 pg, MCV-78.2, MCHC-31.5%, RDW-19.4 and reticulocyte count-6.5%. Impression: Leukoerythroblastic blood film. Her bloodsugar, renal functions and LFT's were normal. Her serum calcium 8.0 mg/dl, phosphorus 2.9 mg/dl, Alkalinephosphatase (AP) 3077 U/L, and Lactate dehydrogenase (LDH) 1840U/L. Quantitative Buffy Coat smear (QBC) for malarial parasite and HIV status were negative. Her cranial and abdominal ultrasound was normal. X-ray showed increased density in all the bones (bone in bone appearance and) [Figures 1 and 2]. She was diagnosed as a case of MIOP with anemia and bronchopneumonia. She was treated with antibiotics, blood transfusion, calcium, and vitamin D.
Figure 1.
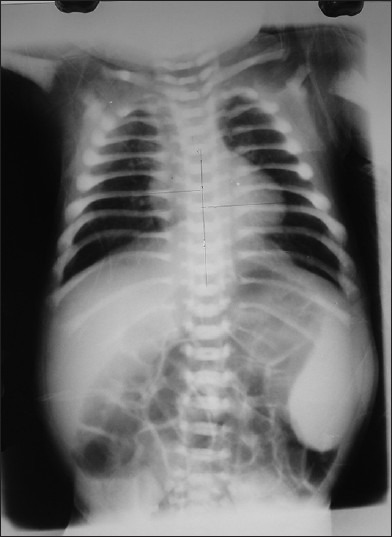
X-ray showing increased density in all the bones (bone in bone appearance)
Figure 2.
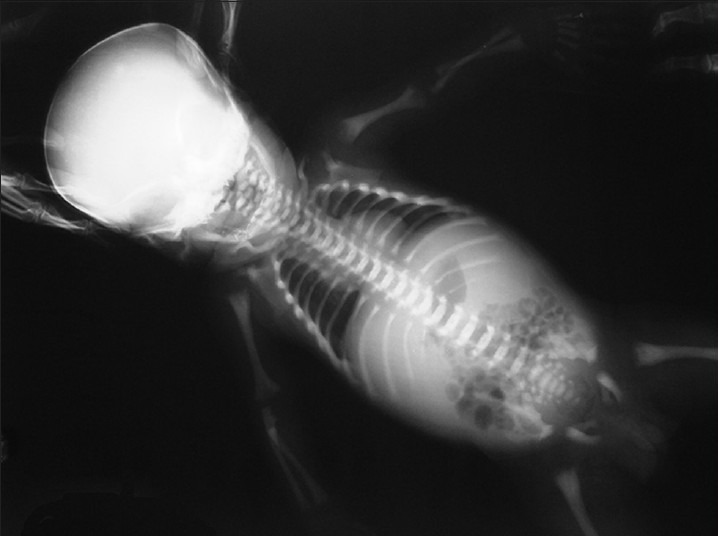
X-ray showing increased density in all the bones (bone in bone appearance)
Discussion
MIOP is a AR form which if untreated has a fatal outcome.[3] The diagnosis of MIOP is based on typical clinical and hematological parameters and characteristic radiological changes of increased bone density. The characteristic radiological feature of osteopetrosis is generalized sclerosis of bone.[2] Our child had classical characteristics of MIOP and increased bone density was found in all the bones. Most of the cases present within 1st year of life.[1,3] The age at the time of diagnosis was between 3 months and 18 months in a study by Phadke et al.[2] The mean age at the time of diagnosis was 3.9 months (range 15 days to 9.5 months) in a study by Mazzolari et al.[1] Out of 8 children 7 were diagnosed after 6 months of age in a study from India.[3] Usually, MIOP presents with anemia, thrombocytopenia, hepatosplenomegaly, visual impairment due to optic atrophy, and deafness within a year of life.[1,2,4] The major clinical features derived from bony overgrowth of the marrow space and compression of optic and auditory nerve.[1] The commonest clinical presentation was due to optic nerve compression in various studies.[1,4] Even Phadke et al. Observed optic atrophy as the common finding in their cases.[3] However, the most common presenting symptoms were pallor and listlessness in anotherstudy[2] However, our child presented with anemia and gross hepatosplenomegaly at 2 months 15 days but without vision defects. The risk of developing a visual defect in the 1st year of life is about 75% and surgical decompression of optic nerve may restore vision.[2] The hematological manifestations in MIOP are due to obliteration of marrow cavity leading to leukoerythroblastic bonemarrow.[2] Cells of promyeloid series may be found due to ineffective marrow function, which was found in our case. Hepatosplenomegaly develops because of extramedullary hematopoiesis. Hypersplenism may lead to thrombocytopenia, who may present with bleeding similar to our case. Anemia and thrombocytopenia is a constant feature in most of the studies.[2,3] The risk of developing hematological impairment in the 1st year of life is about 75% and its onset within 3 months of life is indicative of a poor outcome.[4] MIOP should be considered in the differential diagnosis of anemia with hepatosplenomegaly in an infant otherwise he/she may be wrongly diagnosed as a leukemia or other hemolytic anemia.[2] Increased risk of infections because of unrecognized immunologic abnormalities in patients with MIOP was observed.[1] Infact our baby presented to us with bronchopneumonia. In a study by Fattore et al. Bone AP was low in AD variety of osteopetrosis whereas it was high in AR variety.[5] Our child also had A P values of 3500U/L and elevation of LDH. However, other studies did not show much elevation of AP.[2,3] AP (mean: 921 U/L; range: 156-2583 U/L) were markedly increased and they were within the normal range only in four subjects in a study by Mazzolari et al.[1] In a study by Srinivasan et al. Symptomatic hypocalcemia was observed in the 1st month of life in 8 infants with MIOP[6] but our child had normal calcium levels. In a study by Phadke et al. None of them had hypocalcemia and only one had alkaline phosphatase levels >1500 U/L.[2] Clinical outcome with paratharmone, calcitrol, interferon gamma and prednisone therapy has been found to be inconsistent and variable.[1,2] An accurate diagnosis is essential in view of availability of curative treatment (Bon emarrow transplant) and for genetic counseling as the risk of recurrence in siblings is 25%.[1,2] Most of the children die during infancy or early child hood without curative treatment by bonemarrow transplantation.[3] To conclude MIO Prema insun recognized as a cause of anemia with hepatospleenomegaly in less than 6 months of life and often results in diagnostic delay. Early diagnosis is important in the context of bonemarrow transplantation.
Footnotes
Source of Support: Nil
Conflict of Interest: None declared.
References
- 1.Mazzolari E, Forino C, Razza A, Porta F, Villa A, Notarangelo LD. Asingle-center experience in 20 patients with infantile malignant osteopetrosis. Am J Hematol. 2009;84:473–9. doi: 10.1002/ajh.21447. [DOI] [PubMed] [Google Scholar]
- 2.Phadke SR, Gupta A, Pahi J, Pandey A, Gautam P, Agarwal SS. Malignant recessive osteopetrosis. Indian Pediatr. 1999;36:69–74. [PubMed] [Google Scholar]
- 3.Phadke SR, Fischer B, Gupta N, Ranganath P, Kabra M, Kornak U. Novel mutations in Indian patients with autosomal recessive infantile malignant osteopetrosis. Indian J Med Res. 2010;131:508–14. [PubMed] [Google Scholar]
- 4.Gerritsen EJ, Vossen JM, van LooI H, Hermans J, Helfrich MH, Griscelli C, et al. Autosomal recessive osteopetrosis: Variability offinding sat diagnosis and during the natural course. Pediatrics. 1994;93:247–53. [PubMed] [Google Scholar]
- 5.Del Fattore A, Peruzzi B, Rucci N, Recchia I, Cappariello A, Longo M, et al. Clinical, genetic, and cellular analysis of 49 osteopetrotic patients: Implications for diagnosis and treatment. J Med Genet. 2006;43:315–25. doi: 10.1136/jmg.2005.036673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Srinivasan M, Abinun M, Cant AJ, Tan K, Oakhill A, Steward CG. Malignant infantile osteopetrosis presenting with neonatal hypocalcaemia. Arch Dis Child Fetal Neonatal Ed. 2000;83:F21–3. doi: 10.1136/fn.83.1.F21. [DOI] [PMC free article] [PubMed] [Google Scholar]