

Abstract
A local tissue-specific renin–angiotensin system (local RAS) has been identified in many organs. However, no report has described the role of a local RAS in the hypertrophic differentiation of chondrocytes. To examine the role of a local RAS in the hypertrophic differentiation, we activated angiotensin II type 1 receptor (AT1R) and angiotensin II type 2 receptor (AT2R) separately in the cell line ATDC5, which involves differentiation from mesenchymal stem cells to hypertrophic chondrocytes. Activation of AT1R suppressed and activation of AT2R enhanced the expression of markers of hypertrophic differentiation, including type X collagen, matrix metalloproteinase 13 and runt-related transcription factor 2.
Keywords: Local RAS, ATDC5, Hypertrophic differentiation, Angiotensin II type 1 receptor, Angiotensin II type 2 receptor
Highlights
-
•
Renin–angiotensin system components are expressed during hypertrophic differentiation.
-
•
AT1R is expressed intensely during chondrocyte proliferation.
-
•
AT2R is expressed intensely in the hypertrophic phase.
-
•
The expression of Col.X, MMP13 and Runx2 are repressed by activating AT1R.
-
•
The expression of Col.X, MMP13 and Runx2 are enhanced by activating AT2R.
1. Introduction
Renin was first identified in 1898 by Tigerstedt and Bergman [1], and the renin–angiotensin system (RAS) has been studied extensively since then. Researchers first studied the RAS as a systemic cardiovascular homeostatic system [2]. Thus, inhibitors of the systemic RAS have become important clinical tools in the treatment of renal and cardiovascular diseases such as hypertension, heart failure and diabetic nephropathy [3]. It is now known that an RAS also operates locally. A local tissue-specific RAS (local RAS) has been identified recently in many organs [3]. A local RAS has also been shown to exert a distinct biological action in each organ. For example, in the ovary, components of the local RAS comprise reaction pathways between theca and granulosa cells to inhibit estradiol formation by the theca cells [4]. In the uterus, a local RAS controls uterine blood flow during pregnancy [5]. In the musculoskeletal system, expression of a local RAS has been found in the fracture callus formed in the healing process [6] and in the synovium of individuals with chronic arthritis [7]. Angiotensin II (Ang II) accelerates osteoporosis by activating osteoclasts [8], and treatment with angiotensin-converting enzyme inhibitors is associated with a reduced fracture risk [9]. A local RAS is also expressed in osteoblasts of the fetal rat calvaria [10], suggesting the existence of a local RAS in embryonic osteoblasts. However, it is still unknown whether a local RAS is functional in the hypertrophic differentiation of chondrocytes. Here we investigated the role of a local RAS in hypertrophic differentiation using the ATDC5 cell line. This involves the differentiation of mesenchymal stem cells to form calcified hypertrophic chondrocytes.
2. Materials and methods
2.1. Cell culture
The ATDC5 cell line was obtained from the Riken Cell Bank (Rikagakukenkyusyo, Tsukuba, Japan). Cells were cultured in a 1:1 mixture of Dulbecco's modified Eagle's medium (DMEM) and Ham's F-12 with 5% fetal bovine serum (FBS) and 3 × 10−8 M sodium selenite. The cells were maintained at 37 °C in a 5% CO2 atmosphere and grown as a monolayer. They were first plated at 1.2 × 105 cells per 35 mm well and then allowed to proliferate for about 3 days until they reached confluence. Then, cells were induced to differentiate in a 1:1 mixture of DMEM and Ham's F-12 containing 5% FBS, 10 μg/ml human transferrin, 3 × 10−8 M sodium selenite and 10 μg/ml bovine insulin. The culture medium was replaced every other day. The cells were allocated into 11 groups (groups A–K). Cells allocated to groups A–I were treated with various reagents on Day 14. Cells allocated to group J were treated with various reagents on Day 10. Cells allocated to group K were treated with various reagents on Day 21. Cells assigned to group A were treated with phosphate-buffered saline (PBS) alone as controls. Group B was treated with 0.1 μg/ml Ang II. Group C was treated with 1.0 μg/ml Ang II. Group D was treated with 1.0 μg/ml Ang II and 1.0 μg/ml Olmesartan, an angiotensin II type 1 receptor (AT1R) inhibitor. Group E was treated with 1.0 μg/ml Ang II and 1.0 μg/ml PD123319, an angiotensin II type 2 receptor (AT2R) inhibitor. Group F (control) was cultured without adding any agents. Group G was treated with 0.1 μg/ml Olmesartan; group H was treated with 1.0 μg/ml Olmesartan, and group I was treated with 10 μg/ml Olmesartan. Group J was treated with 1.0 μg/ml Ang II and 1.0 μg/ml PD123319 or with 1.0 μg/ml Ang II and 1.0 μg/ml Olmesartan on Day 10. Group K was treated with 1.0 μg/ml Ang II and 1.0 μg/ml PD123319 or with 1.0 μg/ml Ang II and 1.0 μg/ml Olmesartan on Day 21. Olmesartan was obtained from Daiichi-Sankyo Co., Ltd. (Tokyo, Japan). PD123319 (ditrifluoroacetate) was purchased from Wako Pure Chemical Industries (Osaka, Japan).
2.2. Quantitative reverse transcription polymerase chain reaction (QRT–PCR)
Total RNA was extracted from ATDC5 cells using TRIzol (Invitrogen, Carlsbad, CA, USA) according to the manufacturer's protocol. Cells to which agents were administered were treated with TRIzol 6 h after the administration. The extracted total RNA was reverse-transcribed using random primers under standard conditions with a high-capacity cDNA reverse transcription kit (Applied Biosystems, Foster City, CA, USA). This first strand cDNA (1:10 dilution) was amplified. QRT–PCR was performed using Perfect real-time SYBR green II (Takara Bio, Inc., Shiga, Japan). PCR amplifications were performed with the Thermal Cycler Dice Real Time PCR System (Takara Bio, Inc.) at 95 °C for 15 s, followed by 40 cycles of 95 °C for 5 s and 60 °C for 30 s. Relative quantification of the gene of interest was normalized to the housekeeping gene for β-actin in the comparative CT method. To quantify the relative expression of each gene, Ct values were normalized against the endogenous reference (ΔCt = Ct target − Ct β-actin) and were compared with a calibrator using the ΔΔCt method (ΔΔCt = ΔCt sample − ΔCt calibrator). We used the average Ct value for untreated ATDC5 cell line as a calibrator. All experiments included negative controls, which consisted of no cDNA for each primer pair. Primer pairs used in this study were as follows: AT1R, forward primer 5′-TGCTCACGTGTCTCAGCATC-3′ and reverse primer 5′-TTTGGCCACCAGCATCGTG-3′; AT2R, forward primer 5′-TAAGCTGATTTATGATAACTGC-3′ and reverse primer 5′-ATATTGAACTGCAGCAACTC-3′; angiotensin-converting enzyme 1 (ACE1), forward primer 5′-AACGAAGCCTACAGACAAGAC-3′ and reverse primer 5′-AGGCATGGAGGTTCAGGTAG-3′; angiotensinogen (ANG), forward primer 5′-TCAGTACAGACAGCACCCTAC-3′ and reverse primer 5′-TGGACTCCAGGCAGCTGAG-3′; type X collagen (Col.X), forward primer 5′- CGCCATAAAGAGTAAAGGGA-3′and reverse primer 5′-ACTTCCATAGCCTGGCTTG-3′; matrix metalloproteinase 13 (MMP13), forward primer 5′-AAGATGTGGAGTGCCTGATG-3′ and reverse primer 5′-TGGGACATATCAGGAGTATAG-3′; runt-related transcription factor 2 (Runx2), forward primer 5′-GTTCAACGATCTGAGATTTGTG-3′ and reverse primer 5′-GGATTTGTGAAGACTGTTATGG-3′; β-actin, forward primer 5′-TTCCAGCCTTCCTTCTTG-3′ and reverse primer 5′-GTCACACTTCATGATGGAATTG-3′.
2.3. Western blot analysis
ATDC5 cells were homogenized in sodium dodecyl sulfate (SDS) buffer (4% SDS, 125 mM tris–glycine, 10% 2-mercaptoethanol, 2% bromophenol blue in 30% glycerol) and subjected to polyacrylamide gel electrophoresis in the presence of SDS followed by electrotransfer onto polyvinylidene difluoride membranes (Hybond-P; Amersham Pharmacia Biotech, Buckinghamshire, UK). Cells to which agents were administered were homogenized in SDS buffer 36 h after the administration. The membranes were blocked overnight with Block Ace (Dainippon Sumitomo Pharma, Osaka, Japan) and treated with a primary antibody overnight at 4 °C. Primary antibodies were diluted to 1:10,000 with PBS. The membranes were then treated with horseradish peroxidase-conjugated secondary antibodies for 1 h while being shaken gently at room temperature. Secondary antibodies were diluted 1:50,000 with PBS. Detection was realized by enhanced chemiluminescence (ECL) with an ECL Plus western blotting detection system (Amersham Pharmacia Biotech, Buckinghamshire, UK) and a charge-coupled-device-based chemiluminescent analyzer, LAS 4000 (GE Healthcare UK Ltd., Buckinghamshire, UK). Relative expression levels were quantified by normalizing western blot signals to the housekeeping gene for actin. Primary antibodies were as follows: anti-actin goat polyclonal antibody (Santa Cruz Biotechnology, Dallas, TX, USA, sc-1616); anti-ANG rabbit monoclonal antibody (Abcam, Cambridge, UK, ab108334); anti-ACE1 goat polyclonal antibody (Santa Cruz Biotechnology, sc-12187); anti-AT1R goat polyclonal antibody (Santa Cruz Biotechnology, sc-31181); anti-AT2R rabbit polyclonal antibody (Santa Cruz Biotechnology, sc-9040); and anti-Col.X rabbit polyclonal antibody (EMD Millipore, Billerica, MA, USA, #234196-500UL). Secondary antibodies were as follows: anti-goat IgG horseradish peroxidase-conjugated donkey antibody (Santa Cruz Biotechnology, sc-2020) for actin, ACE1 and AT1R detection; and anti-rabbit-IgG horseradish peroxidase-conjugated goat antibody (Santa Cruz Biotechnology, sc-2004) for ANG, AT2R and Col.X detection.
2.4. Statistical analysis
All experiments were performed eight times. Results are presented as the mean ± SD and were processed using Stat View 5.0 statistical software. Differences between results were evaluated using Student's t test or Dunnett's test, and P < 0.05 was considered statistically significant.
3. Results
We first examined the chronological mRNA expression levels of ANG, ACE1, AT1R and AT2R in ATDC5 cells without any agents (group F) using QRT–PCR analysis. The mRNA expression of ANG began to increase in the proliferating phase and maintained expression during the hypertrophic phase with a peak on Day 14 (Fig. 1A). The mRNA expression of AT1R increased intensely in the proliferating phase with a peak on Day 10 (Fig. 1B). Thus, AT1R was still expressed in the early period of hypertrophy. The mRNA expression of ACE1 began to increase on Day 10 and kept increasing (Fig. 1C). The mRNA expression of AT2R began to increase on Day 14 and increased abruptly on Day 21 (Fig. 1D). Thus, AT2R was expressed predominantly in the hypertrophic phase. On Day 14, when both AT1R and AT2R were expressed, the relative mRNA expression of AT1R was about 25 times that of AT2R (Fig. 1E). We also examined the chronological protein synthesis of local RAS components without any agents using western blot analysis (group F). ANG, AT1R and ACE1 were expressed as synthesized proteins in both the proliferating and hypertrophic phases. However, AT2R was not produced during proliferation but was produced intensely in the hypertrophic phase as synthesized protein (Fig. 1F). Thus, we confirmed that ANG, ACE1, AT1R and AT2R were expressed in the early period of the hypertrophic phase. Then, to examine the function of the local RAS in hypertrophic differentiation, we administered specific AT1R and AT2R inhibitors to ATDC5 cells as described above. Because the results of QRT–PCR analysis for the local RAS components suggested that both AT1R and AT2R were expressed on Day 14, we chose this as the day for administration. The mRNA expression of Col.X was downregulated in a concentration-dependent manner with a significant difference from the control cells (group A) when adding Ang II on Day 14 (groups B and C; Fig. 2A). Ang II treatment upregulated the mRNA expression of Col.X with a significant difference from the control cells (group A) treated with Olmesartan (group D; Fig. 2B). On the other hand, Ang II downregulated the expression of Col.X with a significant difference from the control cells (group A) treated with PD123319 (group E; Fig. 2C). We also examined the protein synthesis of Col.X with various agents using western blot analysis on Day 14. Ang II upregulated the expression of Col.X with a significant difference from the control cells (group A) treated with Olmesartan (group D; Fig. 2D and E). On the other hand, Ang II treatment downregulated the expression of Col.X with a significant difference from the control cells (group A) treated with PD123319 (group E; Fig. 2D and E). We also examined the side effects of adding Olmesartan. We administered 0.1, 1.0 and 10 μg/ml Olmesartan on Day 14. Adding the two lower doses did not interfere with the expression of Col.X (groups G and H; Fig. 3). However, treatment with 10 μg/ml Olmesartan without adding AngII upregulated the expression of Col.X with a significant difference from the control cells (group I; Fig. 3). We also administered agents on Days 10 and 21. On Day 10, Ang II treatment downregulated the expression of Col.X with a significant difference from the control cells treated with PD123319 (group J; Fig. 4A). However, Ang II treatment made no significant change to cells treated with Olmesartan on Day 10 (group J; Fig. 4A). On Day 21, Ang II treatment made no significant change to cells treated with PD123319 (group K; Fig. 4B). However, Ang II treatment upregulated the expression of Col.X with a significant difference from the control cells treated with Olmesartan (group K; Fig. 4B). Additionally, we examined the mRNA expression levels of MMP13 and Runx2 using QRT–PCR analysis. Ang II upregulated the mRNA expressions of MMP13 and Runx2 with a significant difference from the control cells (group A) treated with Olmesartan (group D; Fig. 5A and C). On the other hand, Ang II downregulated the expressions of MMP13 and Runx2 with significant differences from the control cells (group A) treated with PD123319 (group E; Fig. 5B and D).
Fig. 1.
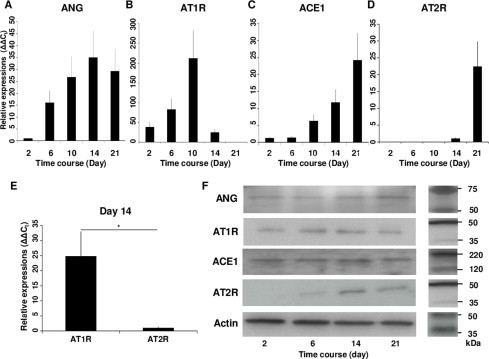
Expression of local RAS components during the hypertrophic differentiation of ATDC5 cell line. (A) ANG began to be expressed in the proliferating phase and maintained expression during the hypertrophic phase. (B) AT1R was expressed intensely during proliferation. (C and D) ACE1 (C) and AT2R (D) were expressed intensely in the hypertrophic phase. (E) On Day 14 when both AT1R and AT2R were expressed, the relative expression of AT1R was about 25 times that of AT2R. (F) Western blot analysis showed that ANG, AT1R and ACE1 were expressed in both the proliferating and hypertrophic phases and that AT2R was expressed intensely in the hypertrophic phase. *P < 0.05 between treatments. Abbreviations: ANG, angiotensinogen; AT1R, angiotensin II type 1 receptor; ACE1, angiotensin-converting enzyme 1; AT2R, angiotensin II type 2 receptor.
Fig. 2.
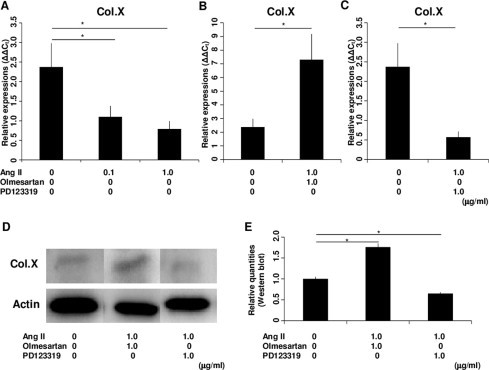
Expression of Col.X in the ATDC5 cell line treated with various agents on Day 14. (A) Ang II downregulated the mRNA expression of Col.X in a concentration-dependent manner. (B) When cells were treated with Olmesartan, Ang II upregulated the mRNA expression of Col.X. (C) When cells were treated with PD123319, Ang II downregulated the mRNA expression of Col.X. (D) Western blot analysis showed that Ang II upregulated the expression of Col.X when cells were treated with Olmesartan and that Ang II downregulated the expression of Col.X when cells were treated with PD123319. (E) Western blotting detection of Col.X showed significant differences between treatments. The molar concentration ratios of antagonists to agonist were 2.32 (1.0 μg/ml Olmesartan/1.0 μg/ml AngII) and 1.77 (1.0 μg/ml PD123319/1.0 μg/ml AngII). *P < 0.05 between treatments. Abbreviations: Col.X, type X collagen; Ang II, angiotensin II.
Fig. 3.
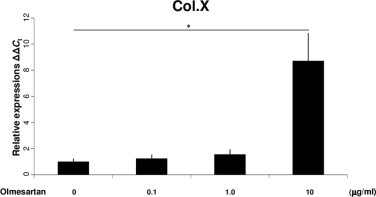
Expression of Col.X in the ATDC5 cell line treated with Olmesartan on Day 14. Adding 0.1 and 1.0 μg/ml Olmesartan made no significant changes to the mRNA expression of Col.X. Adding 10 μg/ml Olmesartan upregulated the mRNA expression of Col.X. *P < 0.05 between treatments. Abbreviation: Col.X, type X collagen.
Fig. 4.
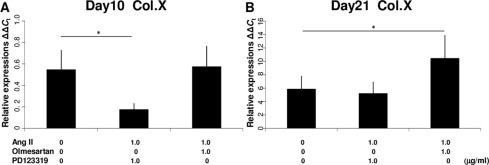
Expression of Col.X in the ATDC5 cell line treated with various agents on Days 10 and 21. (A) When cells were treated with PD123319, Ang II downregulated the mRNA expression of Col.X on Day 10. When cells were treated with Olmesartan, adding Ang II made no significant changes in the mRNA expression of Col.X on Day 10. (B) When cells were treated with PD123319, adding Ang II made no significant changes to the mRNA expression of Col.X on Day 21. When cells were treated with Olmesartan, Ang II upregulated the mRNA expression of Col.X on Day 21. The molar concentration ratios of antagonists to agonist were 2.32 (1.0 μg/ml Olmesartan/1.0 μg/ml AngII) and 1.77 (1.0 μg/ml PD123319/1.0 μg/ml AngII). *P < 0.05 between treatments. Abbreviations: Col.X, type X collagen; Ang II, angiotensin II.
Fig. 5.
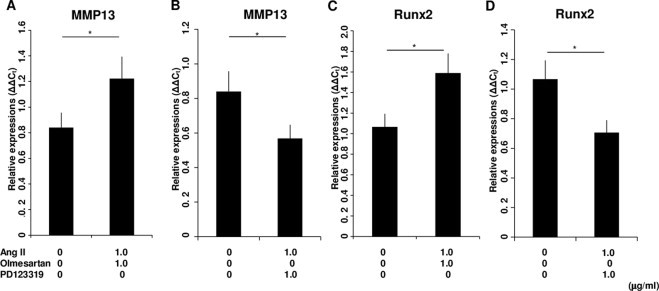
Expression of MMP13 and Runx2 in ATDC5 cells treated with various agents on Day 14. (A) When cells were treated with Olmesartan, Ang II upregulated the mRNA expression of MMP13. (B) When cells were treated with PD123319, Ang II downregulated the mRNA expression of MMP13. (C) When cells were treated with Olmesartan, Ang II upregulated the mRNA expression of Runx2. (D) When cells were treated with PD123319, Ang II downregulated the mRNA expression of Runx2. The molar concentration ratios of antagonists to agonist were 2.32 (1.0 μg/ml Olmesartan/1.0 μg/ml AngII) and 1.77 (1.0 μg/ml PD123319/1.0 μg/ml AngII). *P < 0.05 between treatments. Abbreviations: MMP13, matrix metalloproteinase 13; Runx 2, runt-related transcription factor 2; Ang II, angiotensin II.
4. Discussion
The existence of a specific local RAS has been reported in many tissues [3]. However, no report has described the role of a local RAS in the hypertrophic differentiation of chondrocytes. In a previous study, it was confirmed that AT1R is expressed in cultured osteoblasts [11]. Activating AT1R inhibited differentiation and bone formation in osteoblasts of the rat calvaria [10]. Unlike AT1R, no significant function was found for AT2R in such target cells using a specific blocker [10]. However, AT2R has a reciprocal function to the function of AT1R in many other local and systemic RAS pathways [12]. For example, AT2R receptor exerts an antiproliferative effect in vascular smooth muscle, counteracting the growth action of AT1R [13]. It was also reported that AT2R can bind directly to AT1R and thereby antagonizes its function [14]. Therefore, we tested the hypothesis that AT2R could have a function opposite to that of AT1R in the hypertrophic differentiation of chondrocytes.
Ang II acts via AT1R and AT2R [12]. These receptors are members of the 7-transmembrane-spanning G protein-coupled receptor superfamily (GPCRs) [15]. To activate these receptors separately, we administered Ang II and Olmesartan or Ang II and PD123319 to the ATDC5 cell line on Day 14. Olmesartan is a well-known strong AT1R blocker and also has an inverse agonist activity for AT1R [16,17]. To determine the concentrations of Olmesartan needed, we examined the separate influence of adding Olmesartan. Adding 10 μg/ml Olmesartan upregulated the expression of Col.X without the addition of AngII. We thought that this interference might arise from Olmesartan's inverse agonist activity for AT1R. Inverse agonist activity is defined as the ability to block the agonist-independent weak signal transduction of GRCPs [18]. AT1R is one of the GPCRs [16]. AT1R also induces agonist-independent continuous GRCP signal transduction [19]. Namely, the ATDC5 cell line might be affected by weak internal signals from AT1R, which suppress hypertrophic changes. By blocking these continuous suppressive signals of AT1R for hypertrophy by inverse agonist activity, treatment with 10 μg/ml Olmesartan might induce hypertrophic changes. However this treatment might fully block the AT1R, we did not choose it. We thought that the use of 10 μg/ml Olmesartan might complicate the experimental situation because of its own inverse agonist activity. On the other hand, we could not find any separate influence when adding 0.1 and 1.0 μg/ml Olmesartan. Needing to handle ATDC5 cells without interference from inverse agonist activity, we chose the dose of 1.0 μg/ml Olmesartan. We aimed to make the concentration of PD123319 the same as Olmesartan, because we wanted to equalize the concentrations of these two blockers. Indeed, the molar concentration ratios of agonists to antagonists were 2.32 (1.0 μg/ml Olmesartan/1.0 μg/ml AngII) and 1.77 (1.0 μg/ml PD123319/1.0 μg/ml AngII), respectively. Under these concentration ratios, the receptors might be only partially blocked. On this point, we thought that complete blocking was not essential. In previous studies, it was shown that AT1R and AT2R dominated each other [12,14]; thus, we thought that the relationship of AT1R to AT2R resembled a seesaw. We considered that even partial blocking and using agonists could change the ‘balance’ of the seesaw. Needing to obtain pure stimulation with AT1R or AT2R, we considered using agonists with strong selectivity for one receptor, such as Compound 21 [20]. However, we did not use this approach. Eventually, we decided that using AngII with blockers was more physiologically actual than using strong selective artificial agonists. Under the strong blockade of AT1R by Olmesartan, Ang II might mainly activate AT2R. On the other hand, PD123319 is a well-known AT2R blocker. Under the PD123319's strong blockade of AT2R, Ang II might primarily activate AT1R. When AT1R was activated, the mRNA expression levels of Col.X, MMP13 and Runx2 were downregulated. On the other hand, when AT2R was activated, these expression levels were enhanced. Thus, AT1R acted to suppress hypertrophic chondrocyte differentiation, whereas AT2R activated it. In other words, we confirmed that AT1R and AT2R showed reciprocal functions in hypertrophic differentiation. The results with administering agents on Day 10 indicated that AT1R can suppress Col.X expression without the dominance of AT2R, because this was not expressed on Day 10. Likewise, the results with administering agents on Day 21 indicated that AT2R can upregulate Col.X expression without requiring the dominance of AT1R.
Furthermore, the mRNA expression level of Col.X was downregulated by adding only Ang II on Day 14. Ang II activated both AT1R and AT2R. The relative expression of AT1R was much stronger than that of AT2R at the mRNA level. However, we could not determine whether AT1R was more strongly expressed than AT2R at the protein level. There must also be differences in the efficiency of translation of mRNA between AT1R and AT2R. Even in such a situation, the 25-fold differences at mRNA level might not been reversed during translation. We also must consider the receptor's affinities to Ang II. In this study, we did not measure the affinity of AngII for AT1R and AT2R in the ATDC5 cell line. However, it has been reported that AngII's affinity for AT1R and AT2R is almost equal in other cells [21]. On the other hand, there is no report that the AT1R and AT2R expressed on ATDC5 cells are different from other cells in terms of ligand affinity. Therefore, we assumed that the affinities for AT1R and AT2R in ATDC5 cells might be similar. In this way, the signals arising by activating AT1R might have priority over the signals produced by the activation of AT2R. However, this is still only an hypothesis. In this study, our primary focus was on receptor functions. Considering the environment in which chondrocytes live in vivo, it is very uncertain as to whether Ang II is present locally. Chondrocytes usually exist in nonvascular areas. In these areas, Ang II, which is usually supplied by blood, might be at a low level. Therefore, Ang II might be supplied by an autocrine system. Because we confirmed that the substrate (ANG) and the enzyme (ACE1) were expressed, autocrine-produced Ang II might exist in the chondrocyte environment. However, we consider that the mainly functional ligand of AT receptors on chondrocytes must not be autocrine-derived Ang II. We suspect that articular motion is the main cause. Thus, mechanical stress can activate AT1R without the involvement of Ang II [22]. Moreover, shear stress can also activate AT1R [23]. Considering the environment that surrounds chondrocytes, we think that mechanical stress is the most reasonable cause. Therefore, we do not think that the presence or absence of Ang II was important in our study. In any event, we needed to generate isocratic receptor activation to examine it quantitatively. Therefore, we used Ang II in our study instead of mechanical stress; however, we should consider using in vivo mechanical stimulation in future studies.
In conclusion, activating AT1R suppresses and activating AT2R enhances the hypertrophic differentiation of chondrocytes. Namely, a local RAS can serve to modulate the hypertrophic differentiation of chondrocytes through activating AT1R or AT2R.
Contributions
All authors contributed equally to the work.
Conflict of interests
All authors declare no conflict of interests.
Acknowledgements
We gratefully acknowledge Katsumi Okumoto MT, Life Science Research Institute, Kinki University, for technical advice. We also thank Eri Matsuki, Naoko Ohoshi, Department of Orthopaedic Surgery, Faculty of Medicine, Kinki University, and Kanae Shigi, Division of Cell Biology for Regenerative Medicine, Institute of Advanced Clinical Medicine, Kinki University, for technical assistance in the experiments. This work was supported in part by the Grant-in-Aid from the Ministry of Education, Culture, Sports, Science, and Technology of Japan (JSPS KAKENHI Grant Number 25462389).
Footnotes
This is an open-access article distributed under the terms of the Creative Commons Attribution-NonCommercial-No Derivative Works License, which permits non-commercial use, distribution, and reproduction in any medium, provided the original author and source are credited.
References
- 1.Tigerstedt R., Bergman P.G. Niere und Kreislauf. Skand. Arch. Physiol. 1898;8:223–271. [in German] [Google Scholar]
- 2.Marks L.S., Maxwell M.H. Tigerstedt and the discovery of renin. An historical note. Hypertension. 1979;1:384–388. doi: 10.1161/01.hyp.1.4.384. [DOI] [PubMed] [Google Scholar]
- 3.Paul M., Poyan Mehr A., Kreutz R. Physiology of local renin–angiotensin systems. Physiol. Rev. 2006;86:747–803. doi: 10.1152/physrev.00036.2005. [DOI] [PubMed] [Google Scholar]
- 4.Morris R.S., Francis M.M., Do Y.S., Hsueh W.A., Lobo R.A., Paulson R.J. Angiotensin II (AII) modulation of steroidogenesis by luteinized granulosa cells in vitro. J. Assist. Reprod. Genet. 1994;11:117–122. doi: 10.1007/BF02332088. [DOI] [PubMed] [Google Scholar]
- 5.Hagemann A., Nielsen A.H., Poulsen K. The uteroplacental renin–angiotensin system: a review. Exp. Clin. Endocrinol. 1994;102:252–261. doi: 10.1055/s-0029-1211289. [DOI] [PubMed] [Google Scholar]
- 6.Garcia P., Schwenzer S., Slotta J.E., Scheuer C., Tami A.E., Holstein J.H. Inhibition of angiotensin-converting enzyme stimulates fracture healing and periosteal callus formation – role of a local renin–angiotensin system. Br. J. Pharmacol. 2010;159:1672–1680. doi: 10.1111/j.1476-5381.2010.00651.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Price A., Lockhart J.C., Ferrell W.R., Gsell W., McLean S., Sturrock R.D. Angiotensin II type 1 receptor as a novel therapeutic target in rheumatoid arthritis: in vivo analyses in rodent models of arthritis and ex vivo analyses in human inflammatory synovitis. Arthritis Rheum. 2007;56:441–447. doi: 10.1002/art.22335. [DOI] [PubMed] [Google Scholar]
- 8.Shimizu H., Nakagami H., Osako M.K., Hanayama R., Kunugiza Y., Kizawa T. Angiotensin II accelerates osteoporosis by activating osteoclasts. FASEB J. 2008;22:2465–2475. doi: 10.1096/fj.07-098954. [DOI] [PubMed] [Google Scholar]
- 9.Rejnmark L., Vestergaard P., Mosekilde L. Treatment with beta-blockers, ACE inhibitors, and calcium-channel blockers is associated with a reduced fracture risk: a nationwide case–control study. J. Hypertens. 2006;24:581–589. doi: 10.1097/01.hjh.0000203845.26690.cb. [DOI] [PubMed] [Google Scholar]
- 10.Hagiwara H., Hiruma Y., Inoue A., Yamaguchi A., Hirose S. Deceleration by angiotensin II of the differentiation and bone formation of rat calvarial osteoblastic cells. J. Endocrinol. 1998;156:543–550. doi: 10.1677/joe.0.1560543. [DOI] [PubMed] [Google Scholar]
- 11.Bandow K., Nishikawa Y., Ohnishi T., Kakimoto K., Soejima K., Iwabuchi S. Low-intensity pulsed ultrasound (LIPUS) induces RANKL, MCP-1, and MIP-1beta expression in osteoblasts through the angiotensin II type 1 receptor. J. Cell. Physiol. 2007;211:392–398. doi: 10.1002/jcp.20944. [DOI] [PubMed] [Google Scholar]
- 12.Steckelings U.M., Kaschina E., Unger T. The AT2 receptor – a matter of love and hate. Peptides. 2005;26:1401–1409. doi: 10.1016/j.peptides.2005.03.010. [DOI] [PubMed] [Google Scholar]
- 13.Nakajima M., Hutchinson H.G., Fujinaga M., Hayashida W., Morishita R., Zhang L. The angiotensin II type 2 (AT2) receptor antagonizes the growth effects of the AT1 receptor: gain-of-function study using gene transfer. Proc. Natl. Acad. Sci. U.S.A. 1995;92:10663–10667. doi: 10.1073/pnas.92.23.10663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.AbdAlla, S., Lother, H., Abdel-tawab, A.M., Quitterer, U. (2001) The angiotensin II AT2 receptor is an AT1 receptor antagonist. J. Biol. Chem. 276, 39721–39726. [DOI] [PubMed]
- 15.Alexander S.P.H., Mathie A., Peters J.A. Guide to Receptors and Channels (GRAC), 4th edition. Br. J. Pharmacol. 2009;158:S1–S254. doi: 10.1111/j.1476-5381.2009.00499.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Miura S., Fujino M., Hanzawa H., Kiya Y., Imaizumi S., Matsuo Y. Molecular mechanism underlying inverse agonist of angiotensin II type 1 receptor. J. Biol. Chem. 2006;281:19288–19295. doi: 10.1074/jbc.M602144200. [DOI] [PubMed] [Google Scholar]
- 17.Miura S., Fujimoto M., Saku K. Angiotensin II receptor blocker as an inverse agonist. Curr. Hypertens. Rev. 2005;1:115–121. [Google Scholar]
- 18.Seifert R., Wenzel-Seifert K. Constitutive activity of G-protein-coupled receptors: cause of disease and common property of wild-type receptors. Naunyn Schmiedebergs Arch. Pharmacol. 2002;366:381–416. doi: 10.1007/s00210-002-0588-0. [DOI] [PubMed] [Google Scholar]
- 19.Feng Y.H., Miura S., Husain A., Karnik S.S. Mechanism of constitutive activation of the AT1 receptor: influence of the size of the agonist switch binding residue Asn(111) Biochemistry. 1998;37:15791–15798. doi: 10.1021/bi980863t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Steckelings U.M., Larhed M., Hallberg A., Widdop R.E., Jones E.S., Wallinder C. Non-peptide AT2-receptor agonists. Curr. Opin. Pharmacol. 2011;11:187–192. doi: 10.1016/j.coph.2010.11.002. [DOI] [PubMed] [Google Scholar]
- 21.Mizuno M., Sada T., Ikeda M., Fukuda N., Miyamoto M., Yanagisawa H. Pharmacology of CS-866, a novel nonpeptide angiotensin II receptor antagonist. Eur. J. Pharmacol. 1995;285:181–188. doi: 10.1016/0014-2999(95)00401-6. [DOI] [PubMed] [Google Scholar]
- 22.Zou Y., Akazawa H., Qin Y., Sano M., Takano H., Minamino T. Mechanical stress activates angiotensin II type 1 receptor without the involvement of angiotensin II. Nat. Cell. Biol. 2004;6:499–506. doi: 10.1038/ncb1137. [DOI] [PubMed] [Google Scholar]
- 23.Barauna V.G., Magalhaes F.C., Campos L.C., Reis R.I., Kunapuli S.P., Costa-Neto C.M. Shear stress-induced Ang II AT1 receptor activation: G-protein dependent and independent mechanisms. Biochem. Biophys. Res. Commun. 2013;434:647–652. doi: 10.1016/j.bbrc.2013.04.005. [DOI] [PubMed] [Google Scholar]