

1. DISEASE CHARACTERISTICS
1.1 Name of the disease (synonyms)
Hereditary diffuse gastric cancer (HDGC); Familial diffuse gastric cancer (FDGC); FDGC and cleft lip with or without cleft palate; and E-cadherin associated hereditary gastric cancer (GC).
1.2 OMIM# of the disease
OMIM#137215
1.3 Name of the analysed genes or DNA/chromosome segments:
Gene: CDH1; cadherin 1, and type 1 E-cadherin (epithelial)
Cytogenetic location: 16q22.1
Genomic coordinates (GRCh37): 16:68 771 194–68 869 443 (from NCBI)
1.4 OMIM# of the gene(s)
OMIM*192090
1.5 Mutational spectrum
Mutation-detection rate: approximately 45% of all families fulfilling the strict HDGC.1
De novo events: these are not precisely known because patients and parents have not yet been systematically tested. CDH1 germline mutations occurred in 7.2% of apparently sporadic early-onset GC patients invariably with diffuse or mixed histology of GC. From these, proven CDH1-mutation pathogenicity has been assigned only to 2.3% of the cases that were recurrently diagnosed before 35 years old.2
Genomic rearrangements: Deletions of one or multiple CDH1 exons together or not with UTR sequences (5′- and 3′-untranslated regions) occur in 3.8% of all tested HDGC families.1
Nearly 100 different CDH1 germline alterations have been described in more than 120 families. These alterations are distributed throughout the coding, splice-site sequences of the gene, as well as throughout all protein functional domains.
About 15% of all alterations described have recurrently appeared in one-third of all HDGC families, suggesting derivation of common ancestors and/or mutation hotspots. So far, 12 families have been proven to share a common ancestor (carrying four mutations and one large deletion).1, 3
Small frameshift insertions and deletions, which occur in ∼30% of all families described so far, are the most frequent mutation type found in CDH1-associated families, followed by splice-site mutations that occur in ∼25%. Nonsense mutations occur in ∼20% and missense mutations occur in another 20% of the families. Large deletions account for ∼4% of all families, whereas in-frame deletions and germline-promoter methylation are very rare events, accounting for 1% of CDH1-associated families.
In terms of the predicted impact of all these alterations on the protein structure and function, 80% of the families encompass alterations that potentially result in protein, due to introduction of premature stop codons, truncation or even complete lack of expression, due to germline CDH1-promoter hypermethylation, as well as complete or partial deletion of the promoter region of the gene together or not with exon 1 and 21. The remaining 20% of the alterations are not expected to lead to protein loss of expression as their impact is expected to change or remove a single amino acid, in the case of missense mutations or in-frame deletions, respectively. For these mutations, functional studies are essential to assess their pathogenicity.4
1.6 Analytical methods
Stepwise analyses:
Clinical selection: CDH1-mutation analysis should be considered in a family with at least two GC cases, one confirmed diffuse GC (DGC) aged <50, or; three confirmed DGC cases in first- or second-degree relatives independent of age, or; personal or family history of DGC and lobular breast cancer (LBC), one diagnosed before the age of 50, or; or an isolated patient with DGC aged <40. A careful clinical examination, documented histopathology, and selection of an affected individual in the family, as the proband for genetic screening, are mandatory features for performing cost-effective and trustworthy mutation analysis and genetic counseling.5
Germline-mutation analysis:
Sequencing of all coding regions of exons and intron–exon boundaries for point and small indel mutation detection.
In some centers, pre-screening of the gene by DHPLC and SSCP (genomic level).
Screening for exon deletions and duplications by multiplex ligation-dependent probe amplification (MLPA).
In case of detection of an unclassified missense variant, assessment of its pathogenicity by:
Sequencing of at least 50 (100 chromosomes) cancer-free and bona-fide controls.
In silico assessment of putative effect on splicing, the protein product, and its function.
Functional analysis by a reference centre (IPATIMUP, Porto Portugal), to test in vitro the impact of the variant in cell-invasion, cell-cell adhesion, and sub-cellular localization.
In case of detection of a putative splice-site mutation (missense and intronic):
RNA analysis to test inactivation of a splice site or activation of cryptic splicing leading to skipping of fragments or complete exons, or retention of intronic portions in the mRNA, frequently generate premature termination of the protein.
Future perspective: sequencing of the coding regions or the whole-gene locus by next-generation sequencing technologies. Use of reliable in vitro tools to test the effect of splice-site mutations, when RNA from CDH1 mutation carriers is not available.
1.7 Analytical validation
Databases such as HGMD (http://www.hgmd.org/); LOVD (http://www.lovd.nl); and ENSEMBL genome browser (http://www.ensembl.org/) should be used to collect information on CDH1 mutations previously identified, their relationship with a phenotype of HDGC or early-onset diffuse GC, and importantly to assess whether a novel unclassified variant has been detected. In the latter case, only those with a proven pathogenic role should be acknowledged.
Diagnostic testing must be carried out within a certified and accredited laboratory, prepared to decide on the eligibility of the sample to be screened, to evaluate the results obtained, and to write a report with relevant and comprehensive information.
1.8 Estimated frequency of the disease (Incidence at birth (‘birth prevalence') or population prevalence)
Incidence at birth: nearly 0%.
Population prevalence: <0.1 per 100 0006; GLOBOCAN at http://globocan.iarc.fr/).
Prevalence among GC patients: Less than 1%. From all GCs that present a family history, 1–3% will be related to E-cadherin susceptibility, depending on the population analysed (high or low GC incidence geographical areas).
1.9 If applicable, prevalence in the ethnic group of investigated person
Not applicable.
1.10 Diagnostic setting

Comment: A prenatal diagnosis is very rarely requested and, given that the penetrance of the disease in HDGC families carrying deleterious CDH1 mutations is incomplete, and thus about 20% of all mutation carriers may never develop clinical disease, this subject should be a theme of extensive discussion during genetic counselling.
2. TEST CHARACTERISTICS
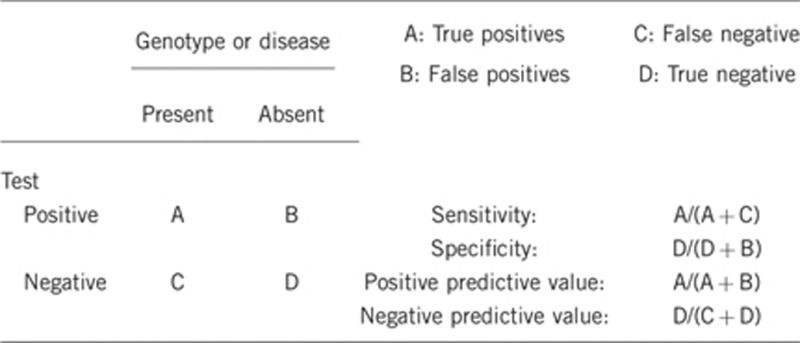
2.1 Analytical sensitivity
(proportion of positive tests if the genotype is present)
It has not been determined, nevertheless, we estimate that it will be high (50–75%), with a combination of direct sequencing and MLPA analyses of the coding regions. Nevertheless, it has been published that >50% of CDH1-alteration negative HDGC probands displayed germline CDH1 allele-specific expression imbalance re-enforcing the existence of unreported defects at the CDH1 locus.7
2.2 Analytical specificity
(proportion of negative tests if the genotype is not present)
It has not been determined, nevertheless, we estimate that it will be >99%, with a combination of direct sequencing and MLPA analyses of the coding regions.
2.3 Clinical sensitivity
(proportion of positive tests if the disease is present)
The clinical sensitivity can be dependent on variable factors, such as age or family history. In such cases, a general statement should be given, even if a quantification can only be made case by case.
If the currently accepted clinical criteria are used for patients/families selection, which considers mainly early-onset of diffuse GC and/or family history of this disease, one can expect that the clinical sensitivity will be lower than the mutation-detection rate among HDGC families, which is <45% of all HDGC families when families from different geographic backgrounds are combined. Something that should be pointed out is the fact that if considering a geographical area with low incidence of GC, the mutation-detection rate (clinical sensitivity) will be higher than when considering a geographical area with high or moderate incidence of GC.1, 5
2.4 Clinical specificity
(proportion of negative tests if the disease is not present)
The clinical specificity can be dependent on variable factors, such as age or family history. In such cases, a general statement should be given, even if a quantification can only be made case by case.
It has not been determined, nevertheless, we estimate that it will be >99%.
2.5 Positive clinical predictive value
(life-time risk to develop the disease if the test is positive)
Penetrance in proven mutation carriers is incomplete, and the estimated cumulative risk of diffuse GC and LBC has been calculated. When considering a broad group of CDH1-mutation carriers, the estimated lifetime risks of diffuse GC was >80% in both men and women by age 80 and of LBC was 60% in women by age 805. The combined risk of GC and breast cancer in women has been calculated to be 90% by age 80.8
2.6 Negative clinical predictive value
(probability not to develop the disease if the test is negative)
Assume an increased risk based on family history for a nonaffected person. Allelic and locus heterogeneity may need to be considered.
Index case in that family had been tested:
If a pathogenic germline mutation has been found in the index case, and a first- or second-degree relative does not carry this mutation, the negative clinical predictive value is >99%. A very rare scenario would be a phenocopy.
Index case in that family had not been tested:
It is important to highlight that a negative result in a healthy individual from a family with history of HDGC, where a causative mutation has not been identified, does not have any meaning for relatives. Therefore, this approach can best be avoided.
3. CLINICAL UTILITY
3.1 (Differential) diagnostics: The tested person is clinically affected
(To be answered if in 1.10 ‘A' was marked)
3.1.1 Can a diagnosis be made other than through a genetic test?

3.1.2 Describe the burden of alternative diagnostic methods to the patient
The diagnosis of HDGC can only be made by genetic testing. However, the diagnosis of diffuse GC in a clinically affected person can be established by endoscopic biopsy sampling of the gastric mucosa and subsequent histological scrutiny, which is a burdensome examination. On the other hand, the diagnosis of diffuse GC in a clinically unaffected person can be established by total prophylactic gastrectomy and subsequent histological examination, which is also a burdensome procedure. Nevertheless, almost 100% of these prophylactic gastrectomies performed in mutation carriers revealed the presence of microscopic cancer foci, and have proven to be curative in asymptomatic CDH1 germline-mutation carriers.5
Alternative burdenless diagnostic methods are not available for a clinically affected person.
3.1.3 How is the cost effectiveness of alternative diagnostic methods to be judged?
Not applicable.
3.1.4 Will disease management be influenced by the result of a genetic test?

3.2 Predictive Setting: The tested person is clinically unaffected but carries an increased risk based on family history
(To be answered if in 1.10 ‘B' was marked)
3.2.1 Will the result of a genetic test influence lifestyle and prevention?
If the test result is positive (please describe):
CDH1-mutation-positive patients with normal gastric biopsies should be advised to undergo gastrectomy once the genetic testing results are known and once individuals are older than 20 years. The timing of this operation may also vary according to the preferences, as well as the physical and psychological fitness of the individual. In patients going forward for gastrectomy, a baseline endoscopy should be performed before surgery to look for macroscopic tumour and in order to inform the data on endoscopic detection of microscopic lesions. For individuals in whom gastrectomy is not currently being pursued (for example, through patient choice), annual gastroscopy with multiple blind biopsies should be offered in order to ensure that there is no evidence of clinically significant lesions and for research purposes.5
For females, annual breast cancer surveillance is recommended starting at age 35 to detect breast cancer at an early stage. Some women might choose for prophylactic mastectomy.5, 11
In some cases, a positive result may influence family planning, namely considering offspring.
If the test result is negative (please describe)
In a family with a detected CDH1 mutation, discharge from intensive screening program that will contribute to psychological relief.
3.2.2 Which options in view of lifestyle and prevention does a person at risk have if no genetic test has been done (please describe)?
In case of 50% risk to carry a CDH1 mutation that runs in the family, the same options as those presented for CDH1-mutation carriers that chose not to have a prophylactic gastrectomy. No data are available on the outcome of prophylactic gastrectomies done in asymptomatic people at risk not screened for CDH1 mutations. It is nevertheless expected that given the current mutation-detection rate for HDGC patients, as well as the autosomal pattern of inheritance of the disease (50% chance of not inheriting the mutant allele), such options would be ineffectual in about half of these persons at risk.
People at risk that chose not to be tested for CDH1 mutations, but have a family history of HDGC, may discuss with clinicians and genetic counsellors the implementation of lifestyle and preventive measures described above for CDH1-mutation carriers. The cost/benefit for such patients should be strongly debated, mainly in what concerns invasive procedures, such as prophylactic surgery.
3.3 Genetic risk assessment in family members of a diseased person
(To be answered if in 1.10 ‘C' was marked)
3.3.1 Does the result of a genetic test resolve the genetic situation in that family?
Yes, it will define an autosomal-dominant inheritance if the mutation is known in the family.
3.3.2 Can a genetic test in the index patient save genetic or other tests in family members?
Yes, if the result is negative or uncertain, testing of family members should not be recommended. Recommendation for screening applies only to mutation carriers and persons at risk.
3.3.3 Does a positive genetic test result in the index patient enable a predictive test in a family member?
Yes.
3.4 Prenatal diagnosis
(To be answered if in 1.10 ‘D' was marked)
3.4.1 Does a positive genetic test result in the index patient enable a prenatal diagnosis?
Technically feasible, generally either not recommended or not well accepted by parents, because of an incomplete penetrance and adult onset of the disease. This may vary among different countries, namely specific laws and ethical values.
4. IF APPLICABLE, FURTHER CONSEQUENCES OF TESTING
Please assume that the result of a genetic test has no immediate medical consequences. Is there any evidence that a genetic test is nevertheless useful for the patient or his/her relatives? (Please describe)
Support for family life organisation.
Efficiency of subsequent clinical management.
For many patients, prove of diagnosis is a value itself – irrespective of a medical benefit – because the disease and its cause can clearly be identified.
Although there is no cure for HDGC syndrome, the diagnosis helps to guide appropriate medical management. For asymptomatic carriers, this information is particularly important because it will implicate guidance through options of prophylactic gastrectomy or frequent endoscopy surveillance. Treatment for patients with clinical presentation of the disease is similar to that of sporadic GC patients.
An affected person/asymptomatic mutation carrier can learn that his or her children may develop the disease, but also that they have 50% probability of being noncarriers.
Acknowledgments
This work was supported by EuroGentest2 (Unit 2: ‘Genetic testing as part of health care'), a Coordination Action under FP7 (Grant Agreement Number 261469) and the European Society of Human Genetics. This study was also supported by the Portuguese Foundation for Science and Technology (FCT) (Projects: PTDC/SAU-GMG/110785/2009 and PTDC/SAU-ONC/110294/2009); and by Calouste Gulbenkian Foundation.
The authors declare no conflict of interest.
References
- Oliveira C, Senz J, Kaurah P, et al. Germline CDH1 deletions in hereditary diffuse gastric cancer families. Hum Mol Genet. 2009;18:1545–1555. doi: 10.1093/hmg/ddp046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corso G, Pedrazzani C, Pinheiro H, et al. E-cadherin genetic screening and clinico-pathologic characteristics of early onset gastric cancer. Eur J Cancer. 2011;47:631–639. doi: 10.1016/j.ejca.2010.10.011. [DOI] [PubMed] [Google Scholar]
- Kaurah P, MacMillan A, Boyd N, et al. Founder and recurrent CDH1 mutations in families with hereditary diffuse gastric cancer. JAMA. 2007;297:2360–2372. doi: 10.1001/jama.297.21.2360. [DOI] [PubMed] [Google Scholar]
- Suriano G, Oliveira C, Ferreira P, et al. Identification of CDH1 germline missense mutations associated with functional inactivation of the E-cadherin protein in young gastric cancer probands. Hum Mol Genet. 2003;12:575–582. doi: 10.1093/hmg/ddg048. [DOI] [PubMed] [Google Scholar]
- Fitzgerald RC, Hardwick R, Huntsman D, et al. International Gastric Cancer Linkage Consortium: hereditary diffuse gastric cancer: updated consensus guidelines for clinical management and directions for future research. J Med Genet. 2010;47:436–444. doi: 10.1136/jmg.2009.074237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliveira C, Seruca R, Carneiro F. Hereditary gastric cancer. Best Pract Res Clin Gastroenterol. 2009;23:147–157. doi: 10.1016/j.bpg.2009.02.003. [DOI] [PubMed] [Google Scholar]
- Pinheiro H, Bordeira-Carriço R, Seixas S, et al. Allele-specific CDH1 downregulation and hereditary diffuse gastric cancer. Hum Mol Genet. 2010;19:943–952. doi: 10.1093/hmg/ddp537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pharoah PD, Guilford P, Caldas C. International Gastric Cancer Linkage Consortium: incidence of gastric cancer and breast cancer in CDH1 (E-cadherin) mutation carriers from hereditary diffuse gastric cancer families. Gastroenterology. 2001;121:1348–1353. doi: 10.1053/gast.2001.29611. [DOI] [PubMed] [Google Scholar]
- Oliveira C, Moreira H, Seruca R, de Oliveira MC, Carneiro F. Role of pathology in the identification of hereditary diffuse gastric cancer: report of a Portuguese family. Virchows Arch. 2005;446:181–184. doi: 10.1007/s00428-004-1156-4. [DOI] [PubMed] [Google Scholar]
- Barber ME, Save V, Carneiro F, et al. Histopathological and molecular analysis of gastrectomy specimens from hereditary diffuse gastric cancer patients has implications for endoscopic surveillance of individuals at risk. J Pathol. 2008;216:286–294. doi: 10.1002/path.2415. [DOI] [PubMed] [Google Scholar]
- Kluijt I, Sijmons RH, Hoogerbrugge N, et al. Familial gastric cancer: guidelines for diagnosis, treatment and periodic surveillance. Fam Cancer. 2012;11:363–369. doi: 10.1007/s10689-012-9521-y. [DOI] [PubMed] [Google Scholar]
- Kurian AW, Sigal BM, Plevritis SK. Survival analysis of cancer risk reduction strategies for BRCA1/2 mutation carriers. J Clin Oncol. 2010;28:222–231. doi: 10.1200/JCO.2009.22.7991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoogerbrugge N, Kamm YJ, Bult P, et al. The impact of a false-positive MRI on the choice for mastectomy in BRCA mutation carriers is limited. Ann Oncol. 2008;19:655–659. doi: 10.1093/annonc/mdm537. [DOI] [PubMed] [Google Scholar]
- Kaurah P, Fitzgerald R, Dwerryhouse S, Huntsman DG. Pregnancy after prophylactic total gastrectomy. Fam Cancer. 2010;9:331–334. doi: 10.1007/s10689-009-9316-y. [DOI] [PubMed] [Google Scholar]
- Fitzgerald RC, Caldas C. Clinical implications of E-cadherin associated hereditary diffuse gastric cancer. Gut. 2004;53:775–778. doi: 10.1136/gut.2003.022061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huntsman DG, Carneiro F, Lewis FR, et al. Early gastric cancer in young, asymptomatic carriers of germ-line E-cadherin mutations. N Engl J Med. 2001;344:1904–1909. doi: 10.1056/NEJM200106213442504. [DOI] [PubMed] [Google Scholar]
- Carneiro F, Huntsman DG, Smyrk TC, et al. Model of the early development of diffuse gastric cancer in E-cadherin mutation carriers and its implications for patient screening. J Pathol. 2004;203:681–687. doi: 10.1002/path.1564. [DOI] [PubMed] [Google Scholar]
- Kluijt I, Siemerink EJ, Ausems MG, on behalf of the Dutch Working Group on Hereditary Gastric Cancer et al. CDH1-related hereditary diffuse gastric cancer syndrome: Clinical variations and implications for counseling. Int J Cancer. 2011;131:367–376. doi: 10.1002/ijc.26398. [DOI] [PubMed] [Google Scholar]