

Abstract
O6-Alkylguanine-DNA alkyltransferase (AGT) is a DNA repair protein which removes alkyl groups from the O-6 position of guanine, thereby providing strong resistance to anticancer agents which alkylate this position. The clinical usefulness of these anticancer agents would be substantially augmented if AGT could be selectively inhibited in tumor tissue, without a corresponding depletion in normal tissue. We report the synthesis of a new AGT inhibitor (5c) which selectively depletes AGT in hypoxic tumor cells.
INTRODUCTION
O6-Alkylguanine-DNA alkyltransferase (AGT, MGMT), a ubiquitous DNA repair protein, effects the stoichiometric removal of alkyl groups from the O-6 position of guanine in DNA.1 Expression of this protein in tumors is the primary mechanism of resistance to guanine O-6 chloroethylating agents such as the N-(2-chloroethyl)-N-nitrosoureas (e.g., BCNU and CCNU) and the sulfonylhydrazine prodrugs synthesized in our laboratory2-5 (e.g., laromustine and KS119), and methylating agents (e.g., DTIC, temozolomide and procarbazine). Inhibition of AGT activity in tumors is currently of great interest to cancer researchers because it can substantially increase the efficacy of antitumor guanine O-6 alkylating agents. The current therapeutic strategy of using an AGT inhibitor such as O6-benzylguanine6 (O6-BG) to sensitize AGT-expressing tumors to the cytotoxic effects of guanine O-6 alkylating agents has one inherent flaw. A global AGT inhibitor not only ablates AGT in tumor tissue where the repair protein is a hindrance to treatment, but it also significantly lowers AGT levels in normal tissues where the protein serves a protective function. For example, although non-toxic doses of O6-BG have been shown in patients to deplete the AGT content of tumors, this action also sensitizes host tissue, necessitating a considerable decrease in the dosage of BCNU, the guanine O-6 alkylating agent used in this trial, because of myelosuppression, leading to an ineffective blood level of BCNU.7,8 Therefore, in order for an AGT inhibitor to have a meaningful impact on cancer therapy involving guanine O-6 alkylating agents it is necessary for it to be delivered selectively or preferentially to the tumor target. This will ensure that the normal tissues are spared without severely compromising therapeutic efficacy.
Repeated studies have demonstrated that oxygen-deficient tumor cells resulting from the inherently abnormal tumor vasculature create an environment conducive to reductive processes. Several anticancer agents, particularly alkylating agents, latentiated as bioreductive prodrugs, have been tested and found to be preferentially toxic to oxygen deficient cells compared to their aerobic counterparts. The substrates examined to date have been predominantly quinones, nitroaromatics and N-oxides (for a review see reference 9). Although azoreduction has been used as a therapeutic strategy in designing prodrugs of nitrogen mustard10,11 and 5-aminosalicylic acid12,13 very little work, if any, has been done on the possible use of hypoxia in solid tumors to activate azo prodrugs for therapeutic purposes. This paper describes the synthesis and evaluation of 6-(benzyloxy)-2-(aryldiazenyl)-9H-purines as prodrugs of O6-BG activated under hypoxic conditions. The 2-amino group in these compounds, which is essential for the AGT inhibitory activity of O6-BG, is latentiated as an azo linkage. Unmasking of the 2-amino group occurs in the hypoxic fraction of a solid tumor via reduction of the azo linkage (Figure 1).14 Once O6-BG is formed it is expected to sensitize not only the hypoxic tumor cells in which it is released, but also the surrounding aerobic tumor cells due to diffusion across membranes.
Figure 1.
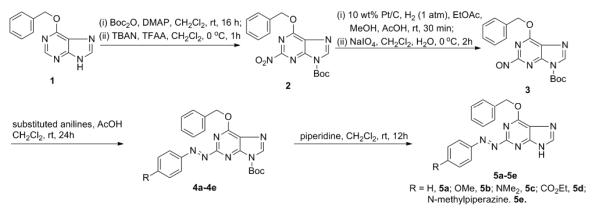
Compounds 5a-e were synthesized as shown. 2-Nitroso-6-benzyloxy-9-Boc-purine (3) was synthesized from 6-benzyloxypurine (1) in 4 steps using published procedures.15,16 Compound 3 was then condensed with the appropriate aniline in dichloromethane in the presence of acetic acid at room temperature to give the corresponding 4a-e.17 Deprotection of the 9-t-Boc group was carried out using an equivalent amount of piperidine in dichloromethane to give the target azo prodrug 5a-e.16
RESULTS AND DISCUSSION
Chemistry
Compounds 5a-e were synthesized as shown in Figure 1. 2-Nitroso-6-benzyloxy-9-Boc-purine (3) was synthesized from 6-benzyloxypurine (1) in 4 steps using published procedures.15,16 Compound 3 was then condensed with the appropriate aniline in dichloromethane in the presence of acetic acid at room temperature to give the corresponding 4a-e.17 Deprotection of the 9-t-BOC group was carried out using an equivalent amount of piperidine in dichloromethane to give the target azo prodrug 5a-e.16
Determination of half-wave reduction potentials
Initially, five compounds were synthesized (5a-e, Fig. 1) and their half-wave reduction potentials determined by differential pulse polarography (Table 1). The E1/2 values versus and Ag/AgCl reference electrode under the tested conditions for compounds 5a-e were −156 mV, −221 mV, −286 mV, −92 mV and −245 mV, respectively. Two candidates (5c and 5e, Figure 1) with the lowest half wave potentials (−286 mV and −245 mV respectively) were chosen for further study. This was done not only to ensure that the candidates were relatively refractory to reductive activation under aerobic conditions, but also to ensure facile activation under hypoxic conditions.
Table 1.
Determination of half-wave reduction potentials.a
| Agent | Concentration | Half-wave reduction potential mV ± SE |
|---|---|---|
| 5d | 10 μM | −92 ± 7 mV |
| 5a | 10 μM | −156 ±1 mV |
| 2-NBP | 50 μM | −213 ± 2 mV |
| 5e | 10 μM | −245 ± 1 mV |
| Nitrofurazone | 50 μM | −279 ± 4 mV |
| 5c | 10 μM | −286 ± 2 mV |
| Mitomycin C | 10 μM | −307 ± 1 mV |
| Misonidazole | 50 μM | −453 ± 3 mV |
| BG-M2 | 50 μM | −475 ± 2 mV |
| Metronidazole | 50 μM | −611 ± 7 mV |
Polarographic half-wave reduction potentials of agents in relation to four well known reference compounds; nitrofurazone a topical bactericide; mitomycin C, a cancer chemotherapeutic agent preferentially targeting hypoxic regions; misonidazole a radiosensitizer, 18F derivatives of which are used for oxygen deficient region imaging; and metronidazole an antibiotic targeting anaerobic bacteria and protozoa. 2-NBP and BG-M2 are prototype O6-BG prodrugs designed to target hypoxic tumors. Half-wave reduction potentials were measured versus an Ag/AgCl reference electrode. All values are the result of at least 3 determinations ± SE
5c sensitizes cancer cells to laromustine
Compounds 5c and 5e were tested in clonogenic assays against DU145 human prostate carcinoma cells, which express relatively high levels of the resistance protein AGT18 (42,000 molecules per cell), to determine their ability to sensitize these cells to laromustine by selective release of O6-BG. Compound 5e, which had a reduction potential slightly higher than 5c, only weakly sensitized DU145 cells to the guanine O-6 alkylator laromustine (Figure 2) under hypoxic conditions and did not sensitize these cells under oxic conditions (Figure 2). 5c was also tested in DU145 cells and demonstrated significant sensitization of these cells to laromustine under conditions of hypoxia with little or no effect seen under oxygenated conditions (Figure 3). No further increase in sensitization was observed at concentrations >10 μM (data not shown) this is likely the result of the limited aqueous solubility of this agent.
Figure 2.
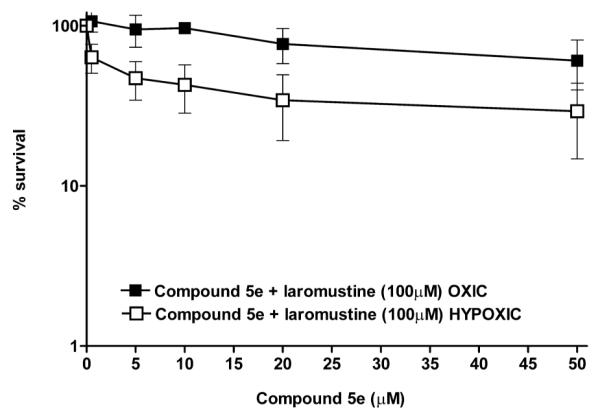
Survival (clonogenic) assays in DU145 human prostate cancer cells pretreated for 4 h with graded concentrations of 5e then dosed with 100 μM laromustine for a total of 24 h. Cells were treated under either oxic (■) or hypoxic (□) conditions before staining and quantification. The horizontal axis indicates the concentration of 5e in μM. The vertical axis indicates the percent survival. All points represent 3 independent determinations ± SEM.
Figure 3.
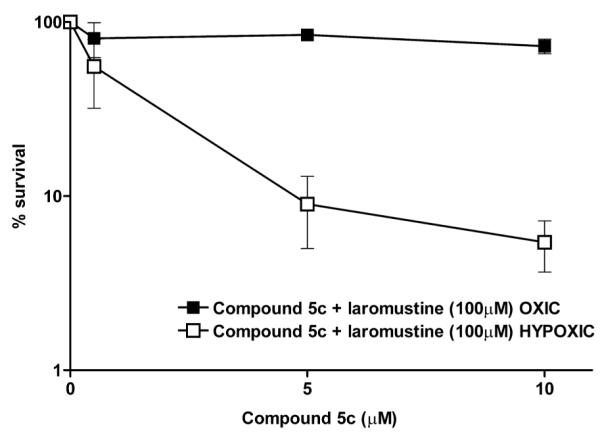
Survival (clonogenic) assays using DU145 cells pretreated for 4 h with graded concentrations of 5c then exposed to 100 μM laromustine for a total of 24 h. Cells were treated under either oxic (■) or hypoxic (□) conditions before staining and quantification. The X axis indicates the concentration of 5c in μM. The Y axis indicates the percent survival. All points represent 3 independent determinations ± SEM.
5c selectively produces O6-benzylguanine under hypoxic conditions
Compound 5c, which sensitizes DU145 cells to laromustine was further studied to determine the levels of O6-BG released under oxic and hypoxic conditions. 5c was incubated under aerobic conditions and under hypoxic conditions in the presence of EMT6 mouse mammary tumor cells (1 × 107/ml) at 37°C and samples were taken at hourly intervals and analyzed by HPLC as described in Figure 4. Over the course of 3 h approximately 50% of the initial starting amount of 50 μM of the prodrug 5c was converted to O6-BG under hypoxic conditions with essentially no production of O6-BG detected under oxic conditions. Under hypoxic conditions in EMT6 cells, the loss of prodrug exceeded the production of O6-BG, likely due to loss from non-reductive metabolism or precipitation. Studies of other O6-BG prodrugs designed by this laboratory have yielded similar incomplete conversions.19 5c was also tested for the ability to generate when incubated with DU145 cells. As can be seen in Figure 5, DU145 cells converted over 84% of the initial input of 5c to O6-BG under hypoxic conditions with only minimal production of O6-BG under oxic conditions. These studies using high cell densities indicate that significant quantities of the AGT inhibitor O6-BG are released selectively under hypoxic conditions by the novel azo prodrug 5c. The yields of O6-BG obtained under hypoxic conditions following reductive fragmentation of 5c are significantly higher than those obtained from 2-(4-nitrophenyl)propan-2-yl (6-(benzyloxy)-9H-purin-2-yl)carbamate (BG-M2) under similar conditions.19 A significant decrease in the concentration of 5c was seen with both cell lines under aerobic conditions, but this was not associated with the production of O6-BG and was likely the result of parental material lost to precipitation or non-reductive routes of metabolism.
Figure 4.
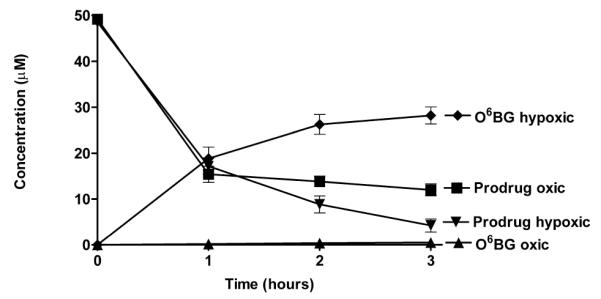
The generation of O6-BG, and parental prodrug loss (5c) over a 3 h time interval. Prodrug was incubated with 107/mL of EMT6 mouse mammary tumor cells and samples were analyzed at 1 h intervals by HPLC. The X axis indicates time in h. The Y axis indicates the concentration in μM. All points are the results of at least 3 determinations ± SEM.
Figure 5.
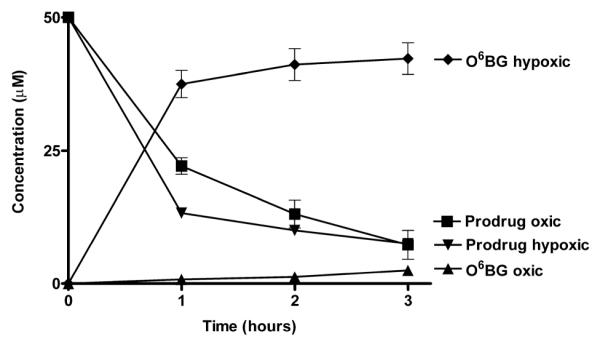
The generation of O6-BG and parental prodrug (5c) loss over a 3 h time interval. Prodrug was incubated with 107/mL of DU145 human prostate carcinoma cells and samples were analyzed at 1 h intervals by HPLC. The X axis indicates time in h. The Y axis indicates the concentration in μM. All points are the results of at least 3 determinations ± SEM.
Conclusion
Compound 5c, conceived as a hypoxia-activated prodrug of O6-BG, has the following desirable properties: (a) Compound 5c has little or no activity under normoxic conditions in tissue culture as measured by its ability to sensitize AGT expressing DU145 cells to the cytotoxic effects of laromustine, a DNA guanine O-6 alkylating agent. However, significant sensitization of DU145 cells to laromustine occurs under hypoxic conditions. (b) Reduction of 5c under hypoxic conditions by DU145 and EMT6 cells results in excellent yields of O6-BG. In fact, the yields of O6-BG obtained were significantly higher, under similar conditions, than that obtained with our previously designed O6-BG prodrugs in many cases. The yields of O6-BG from compound 5c using EMT6 cells were approximately 10-fold greater than from 2-(4-nitrophenyl)propan-2-yl (6-(benzyloxy)-9H-purin-2-yl)carbamate (BG-M2) and comparable to those from, the more readily reduced, 2-nitro-6-benzyloxypurine (2-NBP). In the case of DU145 cells the yields of O6-BG from 5c exceeded those from either BG-M2 or 2-NBP by > 40-fold. The high hypoxic azo-reduction dependent activation of 5c by DU145 cells is particularly noteworthy as this cell line tends to feebly activate nitro-reduction dependent prodrugs. The poor water-solubility of 5c is a potential drawback and we are in the process of synthesizing analogs of this agent which are considerably more water-soluble, yet retain the desirable characteristics of 5c, with the intent of conducting in vivo studies on suitable compounds.
Experimental Section
Chemical Syntheses
Melting points were determined on a Thomas-Hoover Unimelt melting point apparatus and are uncorrected. 1H NMR spectra were recorded on a Bruker Avance DPX-400 spectrometer (400 MHz) with tetramethylsilane as an internal standard. High resolution mass spectra were recorded on a Bruker Daltonics 9.4 T APEXQe FT-ICR & Ultimate mass Spectrometer. Column chromatography was conducted with Merck silica gel 60 (230-400 mesh). Thin layer chromatography was performed on EM pre-coated silica gel sheets containing a fluorescent indicator. All the reported test compounds possess a purity of at least 95% as determined by HPLC.
tert-Butyl-6-(benzyloxy)-2-(phenyldiazenyl)-9H-purine-9-carboxylate (4a). To a solution of aniline (0.28 g, 3 mmol) in CH2Cl2 (10 mL) containing acetic acid (0.5 mL), was added 2-nitroso-6-benzyloxy-9-Boc-purine (3) (0.71 g, 2 mmol) at room temperature. The reaction mixture was stirred for 24 h. The solvent was removed by vacuum, and the residue was purified by flash chromatography using CH2Cl2:EtOAc (20:1) as eluent, which gave 4a as a yellow solid (0.43g, 61%); m.p. 230-232 °C; 1H NMR (400 MHz, CDCl3) δ 8.45 (s, 1H), 8.20-8.01 (m, 2H), 7.68-7.50 (m, 5H), 7.45-7.30 (m, 3H), 5.80 (s, 2H), 1.71 (s, 9H).
Compounds 4b-4e were synthesized using procedures analogous to the one described above
tert-Butyl-6-(benzyloxy)-2-((4-methoxyphenyl)diazenyl)-9H-purine-9-carboxylate (4b). Compound 4b was obtained in a 60% yield; m.p. 143-144 °C; 1H NMR (400 MHz, CDCl3) δ 8.42 (s, 1H), 8.20-8.09 (m, 2H), 7.67-7.56 (m, 2H), 7.45-7.32 (m, 3H), 7.11-7.02 (m, 2H), 5.80 (s, 2H), 3.93 (s, 3H), 1.71 (s, 9H).
tert-Butyl-6-(benzyloxy)-2-((4 (dimethylamino)phenyl)diazenyl)-9H-purine-9-carboxylate (4c). Compound 4c was obtained in a 55% yield. m.p. 226-227 °C; 1H NMR (400 MHz, CDCl3) δ 8.38 (s, 1H), 8.10 (t, J = 9.2 Hz, 2H), 7.60 (d, J = 6.9 Hz, 2H), 7.44-7.30 (m, 3H), 6.77 (d, J = 9.2 Hz, 2H), 5.81 (s, 2H), 3.15 (s, 6H), 1.71 (s, 9H).
tert-Butyl-6-(benzyloxy)-2-((4 (ethoxycarbonyl)phenyl)diazenyl)-9H-purine-9-carboxylate (4d). Compound 4d was obtained in a 52% yield; m.p. 138-140 °C; 1H NMR (400 MHz, CDCl3) δ 8.46 (s, 1H), 8.28-8.09 (m, 3H), 7.59 (d, J = 7.7 Hz, 2H), 7.43-7.29 (m, 3H), 5.79 (s, 2H), 4.43 (q, J = 7.1 Hz, 2H), 1.71 (s, 9H), 1.43 (t, J = 7.1 Hz, 3H).
tert-Butyl-6-(benzyloxy)-2-((4-(4-methylpiperazin-1-yl)phenyl)diazenyl)-9H-purine-9-carboxylate (4e). Compound 4e was obtained in a 50% yield; m.p. 192-194 °C; 1H NMR (400 MHz, CDCl3) δ 8.40 (s, 1H), 8.09 (d, J = 9.2 Hz, 2H), 7.67-7.54 (m, 2H), 7.43-7.29 (m, 3H), 6.98 (d, J = 9.2 Hz, 2H), 5.80 (s, 2H), 3.53-3.43 (m, 4H), 2.63-2.53 (m, 4H), 2.37 (s, 3H), 1.71 (s, 9H).
6-(Benzyloxy)-2-(phenyldiazenyl)-9H-purine (5a). To a solution of tert-butyl 6-(benzyloxy)-2-(phenyldiazenyl)-9H-purine-9-carboxylate (4a) (0.43 g, 1 mmol) in CH2Cl2 (5 mL) was added dropwise piperidine (0.08 g, 1 mmol) at room temperature. The mixture was stirred overnight. The precipitate was collected, washed with CH2Cl2 and ether to give the target molecule as a yellow solid (0.30 g, 90%); m.p. 242-243 °C; 1H NMR (400 MHz, DMSO-d6) δ13.74 (s, 1H), 8.55 (s, 1H), 8.05-7.98 (m, 2H), 7.74-7.56 (m, 5H), 7.41 (dt, J = 21.6, 7.1 Hz, 3H), 5.71 (s, 2H). HRMS, calculated for C18H14N6O, m/z: 331.1302 [(M+H)+], found, 331.1303. HPLC: tr = 35.26 min (97.1% ).
Compounds 5b-5e were synthesized using procedures analogous to the one described above
6-(Benzyloxy)-2-((4-methoxyphenyl)diazenyl)-9H-purine (5b). Compound 5b was obtained in an 88% yield. m.p. 223-224 °C; 1H NMR (400 MHz, DMSO-d6) δ 13.61 (s, 1H), 8.51 (s, 1H), 8.07-7.93 (m, 2H), 7.69-7.55 (m, 2H), 7.48-7.32 (m, 3H), 7.26-7.14 (m, 2H), 5.70 (s, 2H), 3.91 (s, 3H). HRMS, calculated for C19 H16N6O2, m/z: 361.1408 [(M+H)+], found, 361.1407.
4-((6-(Benzyloxy)-9H-purin-2-yl)diazenyl)-N,N dimethylaniline (5c). Compound 5c was obtained in an 80% yield; m.p. 245-246 °C; 1H NMR (400 MHz, DMSO-d6) δ 13.52 (s, 1H), 8.44 (s, 1H), 7.88 (d, J = 9.1 Hz, 2H), 7.61 (d, J = 7.0 Hz, 2H), 7.40 (dt, J = 21.5, 7.1 Hz, 3H), 6.88 (d, J = 9.2 Hz, 2H), 5.68 (s, 2H), 3.11 (s, 6H). HRMS, calculated for C20H19 N7O, m/z: 374.1724 [(M+H)+], found, 374.1716.
Ethyl4-((6-(benzyloxy)-9H-purin-2-yl)diazenyl)benzoate (5d). Compound 5d was obtained in a 90% yield. m.p. 228-230°C 1H NMR (400 MHz, DMSO-d6) δ 13.80 (s, 1H), 8.58 (s, 1H), 8.28-8.18 (m, 2H), 8.15–8.06 (m, 2H), 7.66-7.56 (m, 2H), 7.50-7.35 (m, 3H), 5.71 (s, 2H), 4.38 (q, J = 7.1 Hz, 2H), 1.37 (t, J = 7.1 Hz, 3H). HRMS, calculated for C21H18N6O3, m/z: 403.1513 [(M+H)+], found, 403.1513.
6-(Benzyloxy)-2-((4-(4-methylpiperazin-1-yl)phenyl)diazenyl)-9H-purine (5e). Compound 5e was obtained in a 74% yield. m.p. 219-221°C; 1H NMR (400 MHz, DMSO-d6) δ 13.57 (s, 1H), 8.46 (s, 1H), 7.88 (d, J = 9.2 Hz, 2H), 7.61 (d, J = 6.9 Hz, 2H), 7.47-7.33 (m, 3H), 7.13 (d, J = 9.2 Hz, 2H), 5.68 (s, 2H), 3.47-3.39 (m, 4H), 2.49-2.41 (m, 4H), 2.24 (s, 3H). HRMS, calculated for C23H24N8O, m/z:, 429.2146 [(M+H)+], found, 429.2133. HPLC: tr = 17.55 min (99.9%).
Cell Culture
DU145 human prostate carcinoma cells were cultured in alpha-MEM medium supplemented with 10% fetal bovine serum. EMT6 mouse mammary carcinoma cells were cultured in DMEM supplemented with 10% fetal bovine serum. Both cell lines were maintained at 37°C in a 5% CO2 atmosphere.
Cytotoxicity assays
Cells survival (clonogenic) assays were performed as previously described.5,20,21 DU145 cells were plated in 25 cm2 plastic flasks at a density of 2 × 105 cells and used when near confluent.5 Cells were pretreated for 4 h with graded concentrations of 5c or 5e under oxic or hypoxic conditions prior to the addition of 100 μM of laromustine for a total incubation time of 24 h at 37°C. Hypoxia was generated by the direct depletion of oxygen in sealed flasks using the glucose oxidase (2 units/ mL, Sigma G6641) and catalase (120 units/mL, Sigma C1345) dual enzyme system as previously described.5,20,21
Determination of half-wave reduction potentials (E1/2)
The E1/2 values were determined by differential pulse polarography (DPP). The supporting electrolyte was 80% by volume 100 mM potassium chloride and 50 mM potassium phosphate (pH 7.0) and 20% by volume of CH3CN in all cases. Agents were added as 1% by volume solutions in DMSO. The E1/2 values of six reference compounds were also measured. Dissolved oxygen was removed by purging with nitrogen. DPP voltammograms were generated using a Princeton Applied Research electrochemical trace analyzer model 394, with a model 303A static mercury drop electrode (Princeton Applied Research, Oak Ridge, TN, U.S.) utilizing a platinum counter electrode and an Ag/AgCl reference electrode. Voltammograms were taken from 0 to −900 mV at a scan rate of 2 mV/s using a pulse amplitude of 50 mV. The E1/2 value was determined from the peak current potential (EP) using the following equation: E1/2 = EP-pulse amplitude/2.22
Cell dependent O6-BG generation
Cell suspensions (107 cells/mL) were treated with 5c (50 μM) under oxic or hypoxic conditions in DMEM (EMT6), or alpha-MEM (DU145) media containing 10% FBS. Plastic flasks (25 cm2) with shallow 4-mL layers were employed for oxic studies and were shaken to maintain aeration. The glucose oxidase/catalase/glucose system was used to generate hypoxic conditions before the addition of 5c; the mixtures were stirred gently in sealed tubes. Using this system, oxygen is depleted in ~3 min and H2O2 is removed rapidly by a large excess of catalase.5,20,21 It is expected that this low transient exposure to H2O2 will have no significant effect on the reduction of 5c during these 1 h incubations. At various time intervals, samples were withdrawn and the cellular and medium components were precipitated by mixing with an equal volume of acetonitrile for 20 min at room temperature followed by centrifugation at 10000 × g for 15 min. The supernatant was then analyzed by HPLC for 5c and O6-BG.
ACKNOWLEDGMENT
This work was supported in part by U.S. Public Health Service Grants CA090671, CA122112 and CA129186 from the National Cancer Institute and a grant from the National Foundation for Cancer Research.
REFERENCES
- (1).Pegg AE. Mammalian O6-alkylguanine-DNA alkyltransferase: regulation and importance in response to alkylating carcinogenic and therapeutic agents. Cancer Res. 1990;50:6119–6129. [PubMed] [Google Scholar]
- (2).Penketh PG, Shyam K, Baumann RP, Remack JS, Brent TP, Sartorelli AC. 1,2-Bis(methylsulfonyl)-1-(2-chloroethyl)-2-[(methylamino)carbonyl]hydrazine (VNP40101M). I. Direct inhibition of O6-alkylguanine-DNA alkyltransferase (AGT) by electrophilic species generated by decomposition. Cancer Chemother. Pharmacol. 2004;53:279–287. doi: 10.1007/s00280-003-0740-7. [DOI] [PubMed] [Google Scholar]
- (3).Baumann RP, Shyam K, Penketh PG, Remack JS, Brent TP, Sartorelli AC. 1,2-Bis(methylsulfonyl)-1-(2-chloroethyl)-2-[(methylamino)carbonyl]hydrazine (VNP40101M). II. Role of O6-alkylguanine-DNA alkyltransferase in cytotoxicity. Cancer Chemother. Pharmacol. 2004;53:288–295. doi: 10.1007/s00280-003-0739-0. [DOI] [PubMed] [Google Scholar]
- (4).Ishiguro K, Shyam K, Penketh PG, Sartorelli AC. Role of O6-alkylguanine-DNA alkyltransferase in the cytotoxic activity of cloretazine. Mol. Cancer Ther. 2005;4:1755–1763. doi: 10.1158/1535-7163.MCT-05-0169. [DOI] [PubMed] [Google Scholar]
- (5).Baumann RP, Penketh PG, Ishiguro K, Shyam K, Zhu YL, Sartorelli AC. Reductive activation of the prodrug 1,2-bis(methylsulfonyl)-1-(2-chloroethyl)-2-[[1-(4-nitrophenyl)ethoxy]carbonyl]hydrazine (KS119) selectively occurs in oxygen-deficient cells and overcomes O6-alkylguanine-DNA alkyltransferase mediated KS119 tumor cell resistance. Biochem. Pharmacol. 2010;79:1553–1561. doi: 10.1016/j.bcp.2009.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Dolan ME, Pegg AE. O6-Benzylguanine and its role in chemotherapy. Clin. Cancer Res. 1997;3:837–847. [PubMed] [Google Scholar]
- (7).Quinn JA, Pluda J, Dolan ME, Delaney S, Kaplan R, Rich JN, Friedman AH, Reardon DA, Sampson JH, Colvin OM, Haglund MM, Pegg AE, Moschel RC, McClendon RE, Provenzale JM, Guruangan S, Tourt-Uhlig S, Herndon JE, II, Bigner DD, Freidman HS. Phase II trial of carmustine plus O6-benzylguanine for patients with nitrosourea-resistant recurrent or progressive malignant glioma. J. Clin. Oncol. 2002;20:2277–2283. doi: 10.1200/JCO.2002.09.084. [DOI] [PubMed] [Google Scholar]
- (8).Schilsky RL, Dolan ME, Bertucci D, Ewesuedo RB, Vogelzang NJ, Mani S, Wilson LR, Ratain MJ. Phase I clinical and pharmacological study of O6-benzylguanine followed by carmustine in patients with advanced cancer. Clin. Cancer Res. 2000;6:3025–3031. [PubMed] [Google Scholar]
- (9).de Groot FMH, Damen DWP, Scheeren HW. Anti-cancer prodrugs for application in monotherapy: targeting hypoxia, tumor-associated enzymes and receptors. Curr. Med. Chem. 2001;8:1093–1122. doi: 10.2174/0929867013372634. [DOI] [PubMed] [Google Scholar]
- (10).Ross WCJ, Warwick GP. Reduction of cytotoxic azo compounds by hydrazine and by the xanthine oxidase-xanthine system. Nature. 1955;176:298–299. doi: 10.1038/176298a0. (1955) [DOI] [PubMed] [Google Scholar]
- (11).Ross WCJ, Warwick GP. Aryl-2-halogenoalkylamines. Part XVI. The preparation of 4-[di-(2-chloroalkyl)amino]azobenzenes. J. Chem. Soc. 1956:1364–1369. [Google Scholar]
- (12).Carceller E, Salas J, Merlos M, Giral M, Ferrando R, Escamilla I, Ramis J, Garcia-Rafanell J, Forn J. Novel azo derivatives as prodrugs of 5-aminosalicylic acid and amino derivatives with potent platelet activating factor antagonist activity. J. Med. Chem. 2001;44:3001–3013. doi: 10.1021/jm010852p. [DOI] [PubMed] [Google Scholar]
- (13).Chourasia MK, Jain SK. Pharmaceutical approaches to colon targeted drug delivery systems. J. Pharm. Pharmaceu. Sci. 2003;6:33–66. [PubMed] [Google Scholar]
- (14).Goeptar AR, Scheerens H, Vermeulen NPE. Oxygen and xenobiotic reductase activities of cytochrome P450. Crit. Rev. Toxicol. 1995;25:25–65. doi: 10.3109/10408449509089886. [DOI] [PubMed] [Google Scholar]
- (15).Chae M-Y, McDougall MG, Dolan ME, Swenn K, Pegg AE, Moschel RC. Substituted O6-benzylguanine derivatives and their inactivation of human O6-alkylguanine-DNA alkyltransferase. J. Med. Chem. 1994;37:342–347. doi: 10.1021/jm00029a005. [DOI] [PubMed] [Google Scholar]
- (16).Wanner MJ, Koch M, Koomen G-J. Synthesis and antitumor activity of methyltriazene prodrugs simultaneously releasing DNA-methylating agents and the antiresistance drug O6-benzylguanine. J. Med. Chem. 2004;47:6875–6883. doi: 10.1021/jm049556d. [DOI] [PubMed] [Google Scholar]
- (17).Davey MH, Lee VY, Miller RD, Marks TJ. Synthesis of aryl nitroso derivatives by tert-butyl hypochlorite oxidation in homogeneous media. Intermediates for the preparation of high-hyperpolarizability chromophore skeletons. J. Org. Chem. 1999;64:4976–4979. doi: 10.1021/jo990235x. [DOI] [PubMed] [Google Scholar]
- (18).Ishiguro K, Shyam K, Penketh PG, Sartorelli A. Development of an O6-alkylguanine DNA alkyltransferase assay based on covalent transfer of the benzyl moiety from [benzene-(3)H] O6-benzylguanine to the protein. Anal. Biochem. 2008;383:44–51. doi: 10.1016/j.ab.2008.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Zhu R, Liu MC, Luo MZ, Penketh PG, Baumann RP, Shyam K, Sartorelli AC. 4-nitrobenzyloxycarbonyl derivatives of O(6)-benzylguanine as hypoxia-activated prodrug inhibitors of O(6)-alkylguanine-DNA alkyltransferase (AGT), which produces resistance to agents targeting the O-6 position of DNA guanine. J. Med. Chem. 2011;54:7720–7728. doi: 10.1021/jm201115f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Baumann RP, Ishiguro K, Penketh PG, Shyam K, Zhu R, Sartorelli AC. KS900: A hypoxia-directed, reductively activated methylating antitumor prodrug that selectively ablates O(6)-alkylguanine-DNA alkyltransferase in neoplastic cells. Biochem. Pharmacol. 2011;81:1201–1210. doi: 10.1016/j.bcp.2011.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Baumann RP, Penketh PG, Seow HA, Shyam K, Sartorelli AC. Generation of oxygen deficiency in cell culture using a two-enzyme system to evaluate agents targeting hypoxic tumor cells. Radiat. Res. 2008;170:651–660. doi: 10.1667/RR1431.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Zhu R, Seow HA, Baumann RP, Ishiguro K, Penketh PG, Shyam K, Sartorelli AC. Design of a hypoxia-activated prodrug inhibitor of O(6)-alkylguanine DNA alkyltransferase. Bioorg Med. Chem. Lett. 2012;22:6242–6247. doi: 10.1016/j.bmcl.2012.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]