

Abstract
The first regio- and stereocontrolled total synthesis of the bisphenolic, bisquaternary alkaloid (+)-dispegatrine (1) has been accomplished in an overall yield of 8.3% (12 reaction vessels) from 5-methoxy-D-tryptophan ethyl ester (17). A crucial late-stage thallium(III) mediated intermolecular oxidative dehydrodimerization was employed in the formation of the C9-C9′ biaryl axis in 1. The complete stereocontrol observed in this key biaryl coupling step is due to the asymmetric induction by the natural sarpagine configuration of the monomer lochnerine (6) and was confirmed by both the Suzuki and the oxidative dehydrodimerization model studies on the tetrahydro β-carboline (35). The axial chirality of the lochnerine dimer (40) and in turn dispegatrine (1) was established by X-ray crystallography and was determined to be P(S). Additionally, the first total synthesis of the monomeric indole alkaloids (+)-spegatrine (2), (+)-10-methoxyvellosimine (5), (+)-lochnerine (6), lochvinerine (7), (+)-sarpagine (8), and (+)-lochneram (11) were also achieved via the common pentacyclic intermediate 16.
INTRODUCTION
The sarpagine-macroline group is one of the largest groups of structurally related indole alkaloids isolated principally from the Apocynaceae plant family.1 More than 150 members of this group have been isolated to date, many of which exhibit interesting pharmacological activities.1,2 Among them, bisindoles are of special significance because they exhibit more potent bioactivity than their monomeric counterparts.3 However, a vast majority of these alkaloids have been poorly evaluated due to paucity of material available for biological testing. The sarpagine-macroline bases are biogenetically related4 to the clinically important ajmaline alkaloids5 and common to these three classes is the azabicyclo[3.3.1]nonane core, as illustrated in Figure 1.
Figure 1.

SAR of the cycloocta[b]indole framework (I) of the sarpagine-macroline and ajmaline related indole alkaloids active against MptpB.7 EWG: electron withdrawing group.
In continuation of their efforts at the biology-oriented synthesis (BIOS)6 of natural product derived probes, Waldmann et al. targeted the sarpagine-macroline indole alkaloids which led to the cycloocta[b]indole core (I) as a starting point for library design.7 More than a hundred tetracyclic analogues of this core, synthesized by a stereoselective solid-phase synthesis, were investigated for their inhibitory activity in enzymatic assays with various tryrosine phosphatases. The screen yielded an unprecedented class of potent inhibitors of Mycobacterium protein tyrosine phosphatase B (MptpB) with IC50 values in low micromolar range (the most potent MptpB inhibitor of the library has an IC50 value of 4.71±1.14 μM). The MptpB inhibitory activity of I seemed to be greatly influenced by the `S' stereochemistry at C6 and C10 and by the presence of a β-ketoester moiety (see Figure 1). The Nb-benzyl group in I, containing: 1) an oxygen atom either within the aromatic ring system or as a phenol substituent and 2) with m,p-disubstituted electron withdrawing groups were important, to potent MptpB inhibition. MptpA and MptpB secreted by the causative organism of tuberculosis, Mycobacterium tuberculosis, are known to selectively dephosphorylate human host proteins involved in interferon-γ signaling pathways thereby preventing the initiation of host defense mechanisms.8 The tetracyclic core (I) of the sarpagine alkaloids thus may be considered as a promising target for the development of new drug candidates against tuberculosis.
The dried roots and leaves of Rauwolfia verticillata (Lour.) or Lou Fu Mu have been used in Chinese folk medicines for thousands of years as tranquilizers and more recently in the treatment of hypertension and hyperthyroidism.9 Illustrated in Figure 2 is (+)-dispegatrine (1),10 a bisquaternary, bisphenolic sarpagine alkaloid isolated from the water-soluble fraction of the root of R. verticillata (Lour.) Baill var. hainanensis Tsiang along with the monomer (+)-spegatrine (2).11 Bisphenolic, bisquaternary indole alkaloids are very rare.12 Of the 300 or so dimeric indole alkaloids isolated to date, the homodimer 1 and blumeanine13 (not shown) are the only two dimers belonging to this class of alkaloids. Although dispegatrine (1) and its corresponding monomer spegatrine (2) exhibit promising hypotensive activity, the affinities and activities of the dimer 1 on both α1 and α2 adrenergic receptors was about an order of magnitude greater than that of the monomer 2.3e The quaternary alkaloids verticillatine (3)14 and macrospegatrine (4)15 illustrated in Figure 2 are also known to exhibit promising antihypertensive activity.16
Figure 2.

Ring-A oxygenated sarpagine (1–12) indole alkaloids and macroline-sarpagine (4, 13–15) bisindole alkaloids.
A number of biogenetically related ring-A oxygenated monomeric alkaloids such as (+)-10-methoxyvellosimine (5),17 (+)-lochnerine (6),18 (+)-sarpagine (7),19O-acetylsarpagine (8),20 (+)-lochvinerine (9),21 episarpagine (10),20 (+)-lochneram (11)18c,22 and lochnerine Nb-oxide (12)23 have also been reported (Figure 2). Lochnerine (6), which itself has no antitumor activity, when combined with vincristine or daunorubicin (not shown) at subcytotoxic concentrations induced complete inhibition of the vincristine-resistant P388 leukemic cells in vitro.24 Additionally, 6 is known to exhibit promising vasorelaxant actvity.25 Most recently, 6 and 7 were found to be present in some of the new macroline-sarpagine bisindoles (13–15, Figure 2) isolated from Alstonia angustifolia by Kam et al.26 The promising bioactivity of some of the members of these ring-A oxygenated group of indole alkaloids combined with their complex architecture made them very attractive targets.
The total synthesis of dispegatrine (1) and four other monomeric indole alkaloids was recently reported via a general approach for the synthesis of ring-A oxygenated sarpagine alkaloids.27 Such a doubly convergent route could also be employed for the potential synthesis of the complex bisindoles 4, 13–15; the biological activities of which have not yet been fully investigated. In this report, the full details of the development of this general route including the first total synthesis of alkaloids 9 and 11 are reported.
RESULTS AND DISCUSSION
Although dispegatrine (1) was isolated as a single atropodiastereomer and its structure established on the basis of spectroscopic analysis (1H NMR, 13C NMR and mass spectrum comparison to monomer 2),10 the apparent axial chirality at the C9-C9′ biaryl axis in 1 was not determined. The isolation chemists also reported a semisynthesis of 1 in an almost negligible yield of 0.25% (Scheme 1), obtained by an oxidative phenolic dehydrodimerization of 2 in aqueous ammonium acetate with K3Fe(CN)6.10. Since only one atropodiastereomer was reportedly formed, it can be assumed that the complete atropselectivity observed in this coupling is a result of internal asymmetric induction by the natural sarpagine configuration of the monomer 2.
Scheme 1.

Partial Biomimetic Synthesis of (+)-Dispegatrine (1) by Yu et al.10
The retrosynthetic strategy was based on these reports (Scheme 2). A potentially biomimetic intermolecular biaryl coupling could be employed to construct the C9-C9′ bond in 1 via a non-phenolic coupling of 6 or by a phenolic oxidative coupling of 7 or 2. In contrast to the extremely low yield of phenolic coupling of 2,10 a non-phenolic Scholl type oxidative coupling of the methoxy analogue lochnerine (6) could be employed to this end.27 By performing such a late stage biaryl coupling, one could not only avoid the potential formation of atropodiastereomeric intermediates throughout the synthesis, but also take advantage of the existing chiral centers in the sarpagine unit 6 for maximum stereoinduction. The 5-methoxy-D-(+)-tryptophan ethyl ester 17 could be employed as the starting material and the chiral transfer agent for the synthesis of the key pentacyclic framework 16 of the sarpagine alkaloids via the asymmetric Pictet-Spengler,28 which on further functionalization would result in the total synthesis of the target alkaloids.
Scheme 2.

Retrosynthetic Analysis
Enantiospecific Synthesis of the 5-Methoxy-D-(+)-Tryptophan Ethyl Ester (17)
Although a number of synthetic routes to substituted tryptophans exist, principally asymmetric hydrogenation,29 enzymatic synthesis,30 and more recently Negishi coupling;31 attempts to execute these in a practical sense in the laboratory met with only limited success. Eventually, two approaches, the regiospecific bromination32 and the Larock heteroannulation previously employed33 for the synthesis of 5-methoxy-D-tryptophan both utilizing the Schöllkopf chiral auxiliary 18 were reinvestigated.
The palladium-catalyzed heteroannulation of internal alkynes with o-iodoanilines developed by Larock et al.34 has been successfully employed in this research group for the synthesis of optically active 6- and 7-methoxy tryptophans on a multihundred gram scale which in turn have resulted in the total synthesis of a number of alkoxy substituted sarpagine indole alkaloids35 via the asymmetric Pictet-Spengler reaction. Attempts to employ the Larock heteroannulation for the synthesis of 17 under similar conditions, however, resulted in lower yields.32b,33 With the success reported in the synthesis of 4-methoxytryptophan36 via use of the TMS substituted propargylic chiral auxiliary 19 (Scheme 3), it was decided to first test the effect of this alkyne on the regioselectivity of the Larock heteroannulation. If successful, it would provide a shorter and more efficient synthesis of 5-methoxytryptophan 17. Since both regiospecific bromination and Larock heteroannulation employed the Schöllkopf chiral auxiliary, large scale preparation (500 g) of the Schöllkopf chiral auxiliary 18 from L-valine and glycine, based on the earlier procedure,37 was undertaken. The TMS protected chiral auxiliary 19 was then prepared according to the modified procedure by Ma et al.36 in 96–100% de (Scheme 3).
Scheme 3.
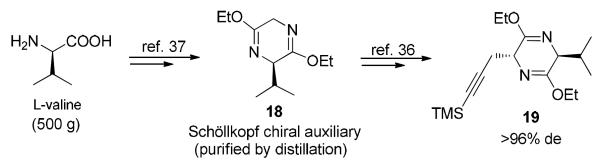
Synthesis of the Propargyl-Substituted Schöllkopf Chiral Auxiliary 19
Larock Heteroannulation
In order to proceed with the Larock heteroannulation, o-iodoaniline 20 was first synthesized based on a published procedure.38 The amino group in p-anisidine was protected with a Boc group followed by a Snieckus ortho lithiation39 with 2.2 equivalents of tert-butyllithium at −78 °C. Initially, deprotonation of the NH moiety took place, after which the next equivalent of t-butyllithium provided the ortho lithiated species stabilized by the Boc group. This process was quenched with diiodoethane to afford the desired Boc protected iodo aniline 20 in 86% yield. With the TMS-substituted alkyne 19 and the iodoaniline 20 in hand, the annulation was carried out according to the recently developed conditions of Ma et al.36 Gratifyingly, in this process the starting aniline 20 was consumed entirely on continued stirring for 36 hours and the Na-H protected indoles 21a and 21b were obtained in a combined yield of 81% (Scheme 4). Hydrolysis of the Schöllkopf chiral auxiliary in 21a (separated by chromatographic separation) with aqueous 2 N HCl in THF, accompanied by concomitant loss of the silyl group provided the optically active 5-methoxy-D-tryptophan ethyl ester (17) in a single step in 86% yield. From a practical point of view, this approach to the synthesis of 17 was disappointing. In spite of replacement of the bulkier TES-substituted alkyne by the TMS alkyne 19, the regioselectivity of this method did not improve. For this reason the earlier regiospecific bromination route32 for the preparation of Na-H-5-methoxy-D-tryptophan ethyl ester (17) was employed. The success of this sequence rested on the ability to scale up the first few steps to multihundred gram levels.
Scheme 4.

Larock Heteroannulation for the Synthesis of 5-Methoxy-D-Tryptophan Ethyl Ester (17)
The commercially available 3-methyl-5-methoxyindole (23)40 was prepared on a 616 g scale via the Japp-Klingmann/Fischer indole protocol developed by Abramovitch and Shapiro41 in two steps from p-anisidine (Scheme 5). Boc protection of the indole Na-H function in 23, followed by allylic bromination32,42 furnished the crude bromide 24 in greater than 90% yield, which was directly alkylated with the anion of the Schöllkopf chiral auxiliary (18) at −78 °C to provide the protected D-tryptophan analogue 25 in 80% yield. Earlier, the Boc group was removed in refluxing xylenes for 7 days;32 however, the much milder TMSOTf/2,6-lutidine system43 was found to be much more effective. Deprotection of the Boc group in 25 was achieved on a 50 g scale at 0 °C in 12 hours to furnish the Schöllkopf analog 26 in 93% yield. Hydrolysis of 26 under aqueous acidic conditions provided the desired optically active Na-H-5-methoxy-D-tryptophan ethyl ester (17) in 85% yield in greater than 98% ee.
Scheme 5.

Improved Regiospecfic Bromination Route for the Synthesis of 5-Methoxy Tryptophan Ethyl Ester (17) on a Large Scale
Synthesis of the 5 Methoxypentacyclic Ketone 16
Analogous to the parent system, the transformation of tryptophan 17 into the desired trans-diester 28b (see Scheme 6) should follow the well-documented trans transfer of chirality in the asymmetric Pictet-Spengler reaction44 in a straightforward fashion; however, this was not the case. The electron rich character of the 5-methoxy indole in 17 facilitated the undesired Pictet-Spengler reaction (with the benzaldehyde imine) as observed for the Na-methyl series;32a moreover, this 5-methoxyindole system 28 was not stable in trifluoroacetic acid (TFA)/CH2Cl2 for extended periods of time. Consequently, modifications were made analogous to the work of Zhao et al.32 to carry out the Pictet-Spengler cyclization and the decarboxylation reaction to achieve optimum yields and very high diastereoselectivity. As illustrated in Scheme 6, tryptophan 17, on treatment with benzaldehyde at room temperature, followed by sodium borohydride reduction (at −10 °C) afforded the corresponding Nb-benzyltryptophan ethyl ester 27 in 93% yield. If the reaction was carried out at room temperature as in the parent system, a 1:1 mixture of 27 and the undesired Pictet-Spengler product 1-phenyl tetrahydro β-carboline (not shown) were observed as reported earlier by Zhao et al.32 However, Pictet-Spengler reaction of tryptophan derivative 27 with the aldehyde, methyl 3-formylpropanoate, in AcOH/CH2Cl2 afforded a mixture of trans- and cis-diesters in nearly quantitative yield. If TFA/CH2Cl2 was employed in this step in place of AcOH/CH2Cl2, decomposition of much of the Nb-benzyl tryptophan 27 was observed. However, on completion of the Pictet-Spengler reaction in acetic acid, one equivalent of TFA was added to epimerize all of the cis isomer into the desired trans-diester 28b in 93% yield. Dieckmann cyclization of 28b was followed by a base mediated hydrolysis/decarboxylation sequence to provide the optically pure tetracyclic ketone 31 (Scheme 6). Based on the work of Wang,45 under normal basic conditions, the δ-lactam 29 was initially formed. Then, in the presence of a large excess of sodium methoxide in refluxing toluene under the same reaction conditions employed for the parent tryptophan system45 for an extended period of time (72 hours), the desired Dieckmann product 30 was obtained in 88% yield (see Scheme 6). The key to success in the Dieckmann cyclization is the controlled stirring rate and temperature for 3 days. A base induced decarboxylation worked smoothly to provide the tetracyclic ketone 31 in 82% yield.
Scheme 6.

Synthesis of the 5-Methoxytetracyclic Ketone 31
Catalytic debenzylation of 31, under acidic conditions furnished the 10-methoxy Nb-H tetracyclic ketone 32 in 92% yield (Scheme 7). Nb-alkylation of the secondary amine 32 with (Z), 1-bromo-2-iodo-2-butene46 under the conditions developed in Milwaukee (dry THF/K2CO3/reflux)32,45 led to the formation of the desired product 33, but the reaction was very sluggish, and led to a formation of a considerable amount of baseline impurities. Since substitution by amines on bromides was clearly an SN2 type process and it is well known that the nucleophilic strength is dependent on the solvent employed in the SN2 reaction, it was decided to increase the nucleophilicity of the amine 32 by employing a more polar aprotic solvent. Dry acetonitrile proved to be the most suitable solvent for this process for the reaction went to completion at room temperature to give 33 in 76% yield. Attempts to increase the yield by adding more bromide were not successful. Initial attempts at executing the key enolate mediated palladium catalyzed cyclization of ketone 33 under the modified conditions47 furnished the desired pentacyclic ketone 16, albeit in lower yields (50%). The lower yields could be due to a combination of the stronger base (t-BuONa) and the electron rich aromatic system in 33, which led to a faster E2 elimination and formation of the unwanted acetylene byproduct (not shown) before the oxidative addition of the vinyl iodide took place. This problem was circumvented by subjecting the vinyl iodide 33 to the much milder conditions [PhOK/Pd(PPh3)4] of Bonjoch et al.48 to furnish the desired pentacyclic ketone 16 in 73% yield.
Scheme 7.

Synthesis of the 5-Methoxypentacyclic Ketone 16
The Regiospecific, Stereospecific Total Synthesis of (+)-Spegatrine (2), (+)-10-Methoxyvellosimine (5), (+)-Lochnerine (6), (+)-Sarpagine (7), (+)-Lochvinerine (11) and (+)-Lochneram (11)
With the key E-ethylidene intermediate 16 in hand conversion into the desired α-aldehyde present in (+)-10-methoxyvellosimine (5) was accomplished by a Wittig-hydrolysis-epimerization sequence (Scheme 8). The one carbon homologation process was achieved by a Wittig reaction, followed by acidic hydrolysis of the corresponding two steroisomeric enol methyl ethers (not shown), to afford the theromodynamically stable α-aldehyde in 5 in 90% yield. The aldehyde function of 5 was then reduced with sodium borohydride to provide (+)-lochnerine (6). As illustrated in Scheme 8, demethylation of (+)-lochnerine (6) was achieved with 5 equivalents of BBr3 in dry CH2Cl2 at −78 °C for 1 hour, after which the solution was allowed to warm to room temperature and stirred for an additional 4 hours. Following work up, (+)-sarpagine (7) was obtained in 80% yield. Subsequent quaternization of the Nb-nitrogen function in 7 with excess methyl iodide provided the Nb-methiodide salt, which on stirring with silver chloride in ethanol49 furnished (+)-spegatrine chloride (2). Lochnerine (6) on Nb-methylation under similar conditions provided (+)-lochneram (11) in 85% yield. The spectral data for the synthetic 2, 5–7 and 11 were in good agreement with that reported for the natural products1a and resulted in the first total synthesis of these alkaloids.
Scheme 8.

Completion of the Total Synthesis of (+)-Spegatrine (2), (+)-10-Methoxyvellosimine (5), (+)-Lochnerine (6), (+)-Sarpagine (7), and (+)-Lochneram (11)
In order to synthesize the β-alcohol at C-16 in lochvinerine (9), the chemoselective hydroboration developed earlier50 was employed. Wittig reaction of the ketone 16 was then carried out with triphenylphosphonium bromide in benzene in the presence of potassium t-butoxide to afford the diene 34 in 90% yield (Scheme 9). Analogous to earlier reports, the 9-BBN reagent was chosen as the hydroborating agent to facilitate attack from the less hindered face of the C16–C17 double bond relative to the C19–C20 site.50 Lochvinerine (9) was obtained as the only detectable diastereomer after the hydroboration-oxidation sequence. The proton NMR spectrum of 9 was in agreement with that of the natural product21 although some difference was observed in chemical shifts of the reported and observed values of protons. This was mainly due to the presence of methanol in the synthetic sample. The synthetic sample retained methanol even after drying the compound under high vacuum for extended periods of time.
Scheme 9.

Completion of the Total Synthesis of (+)-Lochvinerine (9)
Direct/Intermolecular Biaryl Coupling
The biaryl core is an important subunit found in a large number of natural products such as alkaloids, coumarins, flavonoids, lignanes, polyketides tannins and terpenes. In particular, axially chiral biaryls are important as chiral ligands in asymmetric synthesis. Because of their interesting biological activity and complex architecture, natural and unnatural biaryls are considered as attractive synthetic targets.51 For this reason considerable efforts have been made in recent decades for the efficient asymmetric synthesis of biaryls both in the intramolecular and the intermolecular mode and have been the subject of many reviews.51,52 Particularly noteworthy is the review by Bringmann et al.51a which focusses on enantioselective methods employed for the total synthesis of complex biaryl natural products.
Within the area of intermolecular biaryl synthesis, the two most common approaches employed are: (1) oxidative coupling52c and (2) reductive coupling such as Ullmann,52,54 Suzuki,52,55 Stille52,56 or C- H activation52,57 etc. Oxidative phenolic coupling is a powerful and economical method for the synthesis of biaryl compounds and is especially suited to natural product synthesis since many of the biosynthetic routes to biaryls involve such a coupling.58 Although considerable progress has been made in carrying out the intramolecular oxidative couplings, in the absence of activating groups, the position of the newly formed C-C bond is determined by the electronic and steric preferences of the substrate and can lead to regioisomeric reaction products in complex substrates especially in the intermolecular mode.52c,53c The coupling mechanism is believed to involve either one two-electron or two one-electron oxidations to form an aryl–aryl coupled dimer through the ortho and/or para positions.52c,59 In the case of phenols bearing no ortho- and para-substituents, the ortho-ortho, ortho-para and para-para coupling reactions occur, giving rise to often unseparable mixtures. In some cases competing and/or subsequent oxidation to form quinones occurs, either from the coupled product or from the original substrate. Thus it is not always predictable which of the possible products will be formed predominantly, consequently, thorough optimization of the oxidizing agent/reaction conditions is required for successful transformations.
The Lewis- acid catalyzed direct oxidative non-phenolic coupling of two unfunctionalized arenes or the Scholl reaction60 is one of the oldest C-C bond forming reactions and has been extensively used for the intramolecular oxidative dehydrodimerization of branched phenylene precursors to the corresponding polycyclic aromatic hydrocarbons (PAHs) such as triphenylenes, hexa-peri-hexabenzocoronenes (HBCs) etc.61 and for the synthesis of hexaalkoxytriphenylenes.62 Various strong acids (Brønsted acids/Lewis acids) and metal salts are generally used in combination with an oxidant (O2, KMnO4, RNO2).60 Variants of the reaction include oxidants such as: FeCl3,63 CuCl2, or Cu(OTf)2 with AlCl3,64 Tl(O2CCF3)3/BF3·OEt2,65 Pb(OAc)4/BF3·OEt2,66 MoCl5,67 SbCl5,68 and (CF3CO2)2IIIIC6H5 [phenyliodine(III) bis(trifluoroacetate): PIFA]/BF3·OEt2 in CH2Cl2.69 Electrochemical oxidations of electron-rich aryl compounds have been reported, but are not commonly used.70 Based on the observations of King et al.71 the outcome of the Scholl reaction of substituted substrates follows the directing group effects observed in electrophilic aromatic substitution. Alkoxy and alkyl groups are effective o,p- directors, whereas deactivating m-directors (e.g. NO2) suppress the reaction rate. A variety of electron-rich substrates such as alkyl/alkoxy substituted aryl compounds have been successfully dimerized using the above mentioned combinations. While a majority of these C-C bond forming reactions are reported in the intramolecular mode, very few cases are reported in the intermolecular mode, albeit in substrates with simpler systems, or in substrates with substitution patterns such as to prevent side reactions.
Among the reductive coupling processes, the Suzuki reaction is the most widely used method often affording good to high yields of the coupled products with predictable regioselectivity.55 However, the advantages of employing pre-activated aryl components can be offset by the requirement for their independent preparation in cases where multiple synthetic steps are required, especially on a large scale. Also since no standard procedures are available, each new catalyst and substrate requires time consuming optimization of the particular reaction conditions.
Reductive Coupling-Model Reaction
As per the retrosynthetic plan, both oxidative (phenolic and Scholl-type) or reductive cross coupling could be employed to form the C9-C9′ bond in 1. Although biomimetic oxidative phenolic coupling still stands as a practical method for the synthesis of many biaryls, the negligible yield obtained during the oxidative phenolic coupling of 2 by Lin et al.10 made it the weakest method of choice. In order to avoid the potential problems associated with phenolic oxidative couplings it was thus decided to employ a non-phenolic coupling reaction for the construction of the biaryl axis in 1. A non-phenolic oxidative coupling of the sarpagine alkaloid (+)-lochnerine (6), which contains all the chiral information required, could be attempted to this end. The quaternary alkaloid lochneram (11) could also serve the same purpose but could affect the isolation/purification process due to it polar nature. The non-phenolic coupling could either be a reductive cross-coupling or a Scholl type direct oxidative coupling.
Taking into consideration the pros and cons of both types of coupling reactions, it was decided to first study the biaryl coupling on a more robust model indole substrate. By doing so one would be able to compare the results of both reactions on the same substrate and then select the best method for coupling the sarpagine alkaloid 6. It would also give one a chance to study the effect of oxidative dimerization conditions on the more sensitive 5-methoxy sarpagine substrate, especially due to lack of literature precedent for such transformations. Additionally, the supposition that the natural sarpagine framework was indispensable for complete asymmetric induction in the formation of the biaryl axis could also be assessed. Tetrahydro β-carboline 35, the Na-methyl analogue of 28b, was chosen as the model substrate for this purpose. In order to perform any type of reductive coupling reaction (such as the Ullmann, Suzuki, Stille etc.) on 35, it was necessary to first synthesize the corresponding aryl halide coupling partner.
Synthesis of the Aryl Halide Coupling Partners 36a/37a
Due to the moderate reactivity of iodine, electrophilic iodination of aromatic compounds requires the use of an appropriate oxidant for efficient transformation. Numerous methods employing iodonium donating agents have been developed over the years; many of which employ harsh reaction conditions and longer reaction times.72 Initially, a milder method for iodination with N-iodosuccinimide (NIS) and catalytic TFA, reported by Colobert et al.73 was employed for iodination of the model substrate 35. Although the reaction proceeded at room temperature in acetonitirile, longer reaction times (16 hours) were required for complete conversion of 35 and the regiosiosmers 36a and 36b were obtained in a combined yield of 80% (Scheme 10). For synthesis of the corresponding aryl bromide, an NBS/TFA system provided the optimum yield of the desired product 37a. Recently Moorthy et al.74 reported an expedient protocol for iodination of a variety of electron rich and electron-poor aromatic compounds using an IBX-I2 redox couple. The high yields and very short reaction times were attributed to the rapid generation of 4 equivalents of iodonium ions per equivalent of IBX, thereby increasing the rate of the reaction even for electron poor substrates. As illustrated in Scheme 10, iodination of the model substrate 35 under these conditions resulted in a much faster (8 hours) and a cleaner reaction, with the desired product 36a formed in 82% yield. The regiochemistry of iodination in 36a was also confirmed by X-ray analysis (See SI).
Scheme 10.

Electrophilic Halogenation of 35
Attempted Ullman Coupling Reaction
With the appropriate aromatic halides 36a and 37a in hand, the Ullmann reductive coupling54 to form the C7-C7′ bond was first investigated. Similar to other reductive coupling processes, various protocols of the Ullmann-type coupling procedures have been developed; however, very few are suitable for atropselective synthesis of biaryls mainly due to the harsh conditions (multi-hour reactions at temperatures >100 °C) required to obtain high yields of the desired biaryl. Such harsher conditions could lead to racemization of the biaryls and are thus not useful.55e Illustrated in Table 1 are the various attempts to perform the Cu-mediated Ullmann coupling reaction of 36a and 37a. In spite of using up to 8 equivalents of activated Cu-powder at very high temperatures, either no conversion or complete decomposition of the starting materials 36a and 37a was observed (Table 1, entries 3–5). A copper(I)-thiophene-2-carboxylate (CuTC) protocol developed by Liebskind et al.75 was also tested on both the aryl iodide 36a and bromide 37a (Table 1, entries 2 and 6) but failed to provide the desired product. After the failure of the Ullmann reaction to dimerize the model substrate 36a/37a, the Suzuki reductive coupling process55 was next attempted. To accomplish this, the aryl boronate ester coupling partner from the corresponding aryl halide had to be prepared.
Table 1.
Attempted Copper-Mediated Ullmann Coupling Reaction of 36a and 37a
| Entry | Reaction Conditions | Results |
|---|---|---|
| 1. | Cu powder (2.5 equiv),53b DMF, 100 °C, 12 h | SM only |
| 2. | CuTC (3.0 equiv), N-methyl 2-pyrrolidine, 70 °C, 72 h | SM only |
| 3. | Activated Cu powder (8 equiv), DMF, reflux, 72 h | SM only |
| 4. | Activated Cu powder (8 equiv), N-methyl 2-pyrrolidine, sealed tube, sand bath, 140 °C, 26 h | SM only |
| 5. | Activated Cu powder (8 equiv), N-methyl 2-pyrrolidine, sealed tube, sand bath, 140 °C, 72 h | decomposition |
| 6. | CuTC (8 equiv), N-methyl 2-pyrrolidine, sealed tube, sand bath, 140 °C, 26–72 h | decomposition |
CuTC: copper(I)-thiophene-2-carboxylate; SM: Starting materials 36a or 37a.
Synthesis of the Aryl Boronic Ester Coupling Partner
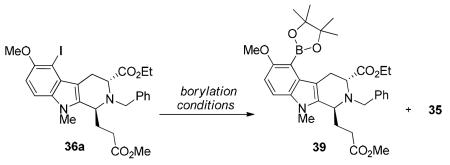
The palladium catalyzed cross coupling reaction of (Bpin)2 with haloarenes developed by Miyaura et al.55d,e provided a direct procedure for the synthesis of aryl boronic esters from aryl halides. Application of these borylation conditions in the system under study here furnished the desired aryl boronate 39 in 55% yield, accompanied by a large amount of the hydrodehaolgenation species 35 (Table 2, entry1). Performing the reaction at a lower temperature with Pd(PPh3)4 as the catalyst, did decrease the amount of the byproduct 35 but also left some starting material (36a) unreacted (as observed on TLC). Based on the initial findings of Masuda et al.76 and reports by Buchwald et al.77 and Baudoin et al.,78 it was found that a palladium-catalyzed coupling of pinacolborane with aryl halides in presence of a tertiary amine as a base, especially Et3N, prevented the pinacolborane from acting as a hydride donor and thus avoided formation of the undesirable Ar-H byproduct (35 in this case). As illustrated in Table 2 (entries 3–8), a careful optimization of the reaction by varying the palladium source and the ligand, finally led to a combination of Pd(OAc)2 with the more electron-rich and bulkier ligand 2-(dicyclohexylphosphanyl)biphenyl (DCPB) to provide 93% yield of the arylboronate 39 (Table 2, entry 8).
Table 2.
Synthesis of the Aryl Boronic Ester Coupling Partner 39
| Boron Sourcea | Pd Source | Ligandb | Basec | Solvent | Temp. (Time) | Resultsd | |
|---|---|---|---|---|---|---|---|
| 1 | (Bpin)2 (4 equiv) | Pd(dppf)Cl2 (10 mol %) | - | KOAc | DMSO | 100 °C (10 h) | 39(55%) + 35 (30%) |
| 2 | (Bpin)2 (4 equiv) | Pd(PPh3)4 (10 mol %) | - | KOAc | DMSO | 80 °C (15 h) | 36a + 39 + 35 e |
| HBpin (3 equiv) | Pd(MeCN)2Cl2 (5 mol %) | DCPB (20 mol %) | Et3N | dioxane | 110 °C (3 h) | 36a + 39 e | |
| 4 | HBpin (3 equiv) | Pd(MeCN)2Cl2 (10 mol %) | DCPB (40 mol %) | Et3N | dioxane | 110 °C (3 h) | 36a +39 e |
| 5 | HBpin (3 equiv) | Pd(MeCN)2Cl2 (5 mol %) | XPhos (20 mol %) | Et3N | dioxane | 100 °C (3 h) | 36a + 39 + 35 e |
| 6 | HBpin (3 equiv) | Pd(OAc)2 (5 mol %) | DPEPhos (10 mol %) | Et3N | dioxane | 100 °C (3 h) | 39 (78%) + 35 (15%) |
| 7 | HBpin (3 equiv) | Pd(OAc)2 (5 mol %) | DCPB (20 mol %) | Et3N | dioxane | 110 °C (3 h) | 39 (80%) + 35 (10%) |
| 8 | HBpin (3 equiv) | Pd(OAc)2 (5 mol %) | DCPB (20 mol %) | Et3N | dioxane | 100 °C (3 h) | 39 (93%) + 35 (3%) |
(Bpin)2: Bis(pinacolato) diboron; HBpin: Pinacol borane;
DCPB: 2-(Dicyclohexylphosphanyl)biphenyl; DPEPhos: Bis(2-phenylphosphinophenyl)ether; XPhos: 2-Dicyclohexylphosphino-2′,4′,6′-triisopropylbiphenyl;
3 equiv of base was employed;
isolated yields;
not separated.
Suzuki -Miyaura Coupling (SMC)
With the appropriate aromatic halides (36a and 37a) and aryl boronate ester 39 in hand, the Suzuki coupling reaction of these di-ortho substituted coupling partners was next attempted. The synthesis of hindered biaryls via the Suzuki reaction, especially of substrates containing large ortho substituents, and/or ortho, ortho' substituents has been shown to be a challenging task.73,77 The difficulty in such transformations can be increased in cases where the substrate has electron-donating groups thereby slowing down the oxidative-addition process. More recently the bulky and electron-rich, monodentate dialkylbiarylphospine ligands, synthesized by Buchwald et al.77 were shown to improve the efficiency of such couplings. The superior activity of the catalysts derived from the biarylphosphine ligands was attributed to a combination of electronic and steric properties that enhanced the rates of oxidative addition, transmetalation, and reductive elimination steps in the catalytic cycle.
Outlined in Table 3 are the different catalyst systems that were employed to carry out the Suzuki-coupling reactions of the aryl halides 36a and 37a with the boronate ester 39. Based on the various synthetic applications79 a general reaction system of Pd(OAc)2/dialkyl-biarylphospine ligand in a 1:4 ratio was employed with different solvents and bases (Table 3). Initial couplings with 20 mol % of the DCPB, XPhos and DavePhos ligands, in presence of a milder base (K3PO4), resulted in mixtures of hydrodehaolgenation byproduct 35 and unreacted starting material 36a/37a (Table 3, entries 1–5). Employment of the more efficient, electron-rich and bulkier SPhos79 ligand also yielded similar result (Table 3, entry 6). It was possible that a combination of basic reaction conditions at higher temperature was responsible for the extensive hydrodehaolgenation. In order to circumvent this it was decided to employ THF as the co-solvent. The low boiling point and higher dielectric constant of THF provided a more homogenous reaction medium at a lower temperature. This change reduced the excessive hydrodehaolgenation taking place and a combined yield of 30% was obtained in favor of the M(R)-atropdiastereomer (Table 3, entry 7). Stirring the reaction mixture for a longer time led to complete conversion of the starting material 36a, and a 54% combined yield of the atropodiastereomers was observed (Table 3, entry 8). Use of Pd2(dba)3 also provided similar results (Table 3, entry 9). At this point no further optimization was performed. Subsequently, the more direct Scholl-type oxidative dehydrodimerization of the model substrate 35 was attempted.
Table 3.
Suzuki-Miyaura Coupling (SMC) Reaction
![]()
| ArX | Pd Sourcea,b | Ligandc,d | Basee,f | Solventg | Temp. (°C)/Time (h) | Results | |
|---|---|---|---|---|---|---|---|
| 1 | 36a | Pd(OAc)2 | DCPB | K3PO4 | dioxane:H2O | 100/24 | 35 + 36a |
| 2 | 37a | Pd(OAc)2 | DCPB | K3PO4 | dioxane:H2O | 100/24 | 35 + 37a |
| 3 | 36a | Pd(OAc)2 | DCPB | CsF | dioxane | 100/24 | 35 + 36a |
| 4 | 36a | Pd(OAc)2 | XPhos | K3PO4 | Tol:H2O | 100/24 | 35 + 36a |
| 5 | 36a | Pd(OAc)2 | DavePhos | K3PO4 | Tol:H2O | 100/24 | 35 + 36a |
| 6 | 36a | Pd(OAc)2 | SPhos | K3PO4 | Tol:H2O | 100/24 | 35 + 36a |
| 7 | 36a | Pd(OAc)2 | SPhos | K3PO4 | THF:H2O | 50/24 | 38a (20%)+38b (10%) + 35 (15%) + 36a (13%) |
| 8 | 36a | Pd(OAc)2 | SPhos | K3PO4 | THF:H2O | 50/48 | 38a (37%) + 38b (18%) + 35 (20%) |
| 9 | 36a | Pd2(dba)3 | SPhos | K3PO4 | THF:H2O | 50/48 | 38a (38%) + 38b (19%) + 35 (20%) |
5 mo % of Pd(OAc)2;
2.5 mol % of Pd2(dba)3;
Ligands: DCPB: 2-(Dicyclohexylphosphanyl)biphenyl; XPhos: 2-Dicyclohexylphosphino-2′,4′,6′-triisopropylbiphenyl; DavePhos: 2-Dicyclohexylphosphino-2′-(N,N-dimethylamino)biphenyl; SPhos: 2-Dicyclohexylphosphino-2′6′-dimethoxybiphenyl;
20 mol % of ligand;
2 equiv. of K3PO4;
8 equiv. of CsF;
solvent:H2O/10:1.
Scholl Type Oxidative Dehdyrodimerization- Model Reaction
With samples of the atropdiastereomeric biaryls (38a and 38b) obtained from the Suzuki-Miyaura coupling on the model substrate 35 in hand, the desired products could now be identified from a potential mixture of regioisomers which could arise during the oxidative coupling of the model substrate. Since one was examining the effect of existing chiral centers on the atropselectivity, it was decided to use achiral catalysts for this process. Of the various protocols of the Scholl type oxidation discussed earlier, it was decided to carry out a hypervalent iodine(III) mediated protocol developed by Kita et al.69,80 and a thallium(III) mediated oxidative dehydrodimerization approach developed by Taylor et al.65,81 Both methods employ a combination of a two electron oxidant and a Lewis acid (BF3·Et2O) in polar aprotic solvents such as CCl4, CH2Cl2 or MeCN, at temperatures ranging from −78 °C to room temperature. Although hypervalent iodine(III) has been extensively used for intramolecular coupling reactions,69,82 there has been little progress made in the intermolecular mode.78a,83 Thallium(III) has also been successfully employed intramolecularly for the synthesis of isoquinoline alkaloids,84 lignans85 and colchinol derivatives86 as well as aporphine alkaloids.87 Effective oxidative dimerizations of 2-substituted indoles,88 and indolocarbazoles89 by thallium(III) trifluoroacetate have also been reported. The intermolecular oxidative dimerization of a polysubstituted indole core by Keller et al.90 provided a suitable precedent for which to effect the oxidative dimerization in the system under study here.
Illustrated in Table 4 are the results of the intermolecular oxidative dehydrodimerization reaction on the model tetrahydro β-carboline 35. Addition of PIFA (0.65 to 1.02 equiv) at 0 °C provided very little formation of the desired dimers 38a and 38b (Table 4, entries 1 & 2). The same trend was observed for the thallium trifluoroacetate mediated oxidation (Table 4, entry 7), although all the PIFA-oxidations seemed to produce a lot of baseline and colored impurities. A lower reaction temperature of −40 °C (both the reagents and the substrate added at the same temperature) seemed to have an immediate effect on the reaction and higher yields were obtained in both PIFA (Table 4, entry 4) and the thallium mediated oxidations (Table 4, entry 8), more so in the latter case. A decrease in the reaction temperature to −78 °C (Table 4, entry 5) or replacing PIFA with phenyliodine(III) diacetate [PIDA (−40 °C)] resulted in considerable amount of starting material 35 (Table 4, entry 6) remaining unreacted. A similar effect was observed upon the use of the milder Tl(III) acetate as the oxidant (Table 4, entry 10). Although some starting material 35 (11%) remained unreacted, a combined yield of 67% was obtained in favor of 38b, the axial chirality of which was determined to be P(S) by X-ray analysis.27 Importantly, the model non-phenolic oxidative coupling reaction was completely regioselective (without any preactivation) and the combination of the milder Tl(III) acetate with BF3·Et2O, which provided the optimum yield (Table 4, entry 10), could now be attempted to construct the desired C9-C9' bond in 1. It was also clear that an oxidative biaryl coupling at a later stage in the synthesis, especially on the natural sarpagine framework, was essential for ensuring complete atropselectivity in the process. This was a key finding from the lack of stereospecificity in the model work.
Table 4.
Optimization of the Non-Phenolic Oxidative Coupling on the Model Substrate 35
| Oxidant (equiv) | Lewis Acid (equiv) | Temp (Time) | Solvent | Results | ||
|---|---|---|---|---|---|---|
| % Yield | M:P | |||||
| 1 | PIFA (0.6) | BF3·Et2O (2.5 equiv) | 0 °C - rt (2 h) | DCM | nd | nd |
| 2 | PIFA (1.02) | BF3·Et2O (4 equiv) | 0 °C - rt (8 h) | DCM | 11 | 3 : 2 |
| 3 | PIFA (1.02) | BF3·Et2O (4 equiv) | −0 °C - rt (1.5 h) | DCM | 20 | 3 : 2 |
| 4 | PIFA (0.8) | BF3·Et2O (3 equiv) | −40 °C (0.5 h) | DCM | 30 | 4 : 1 |
| 5 | PIFA (0.8) | BF3·Et2O (3 equiv) | −78 °C (0.5 h) | DCM | 25a | 4 : 1 |
| 6 | PIDA (0.8) | BF3·Et2O (3 equiv) | −40 °C (2.5 h) | DCM | nd | nd |
| 7 | Tl(OCOCF3)3 (0.8) | BF3·Et2O (3 equiv) | rt (15 min) | MeCN | nd | nd |
| 8 | Tl(OCOCF3)3 (0.8) | BF3·Et2O (3 equiv) | −40 °C (20 min) | MeCN | 38 | 2 : 3 |
| 9 | Tl(OCOCF3)3 (0.5) | BF3·Et2O (2.5 equiv) | −78 °C (40 min) | MeCN | 30 | 3 : 7 |
| 10 | Tl(OCOCH3)3 (0.7) | BF3·Et2O (3 equiv) | −40 °C (1.25 h) | MeCN | 67a | 3 : 7 |
PIFA: phenyliodine(III) bis(trifluoroacetate); PIDA: Phenyliodine(III) diacetate.
The yield is based on recovered starting material 36.
%Yield is based on isolation of both the diasteromers.
nd: not determined.
Thallium(III) Mediated Oxidative Coupling of (+)-Lochnerine (6)
As illustrated in Scheme 11, the monomeric sarpagine alkaloid lochnerine (6) was subjected to the modified conditions of the Tl(III) mediated oxidative dimerization. A combination of Tl(O2CCH3)3 (0.65 equiv) and BF3·Et2O (3.0 equiv) in acetonitrile at −40 °C afforded the key C9-C9' biaryl 40 as the sole atropdiastereomer in 60% yield (based on 12% recovered starting material) with complete regioselectivity. The free indole Na-H, highly basic Nb-nitrogen and the primary hydroxyl function at C-17 remained unaffected. The formation of the biaryl 40, accompanied by the competing electrophilic aromatic thallation byproduct 41 at C-9 is in complete agreement with the detailed mechanistic studies by Kochi et al.91 Increasing the equivalents of Tl(O2CCH3)3 led to increased conversion of indole 6 to the organothallium byproduct 41 and baseline impurities. X-ray crystallographic analysis of 40 established the axial chirality at the C9-C9' bond as P(S).27 The natural sarpagine configuration in indole 6 imparted complete stereocontrol in the key biaryl coupling step thereby forming a single atropdiastereomer 40. This would be in agreement with a potential biomimetic coupling in the plant since none of the other atropodiastereomer was reported during isolation and semisynthesis by Yu et al.10
Scheme 11.

Thallium(III) Mediated Oxidative Coupling of (+)-Lochnerine (6)
b.r.s.m: based on recovered starting material
The Regiospecific, Stereospecific Total Synthesis of (+)-Dispegtarine (1)
Completion of the total synthesis of (+)-dispegatrine (1) was then achieved in two more steps. As illustrated in Scheme 12, the C10-C10' methoxy groups in 40 were demethylated with 9 equivalents of BBr3/CH2Cl2 at −78 °C to furnish the sarpagine dimer 42 in 80% yield. The highly zwitterionic nature of 42 was evident from the 1H NMR of the compound which showed a large downfield shift of the C-3 and C-21 protons as is generally observed in Nb-quaternary sarpagine compounds. Due to this reason, the Nb-methylation of the highly polar dimer 42 was sluggish and very little formation of 1 was observed at room temperature. Eventually, heating the reaction mixture in a sealed tube at 40 °C led to complete conversion of the starting material 42 into the Nb-methyl salt of dispegatrine (1). A LRMS (FAB) of the sample at this stage was taken to confirm the formation of the desired product 1. Further treatment with AgCl/MeOH at room temperature completed the total synthesis of P-(+)-dispegatrine (1). The synthetic material 1 exhibited NMR spectra (1H) that compared favorably with reported values.10 The coupling constants and splitting pattern in the 1H NMR spectrum were in excellent agreement with the literature values,10 except for the proton chemical shifts for carbon atoms at C-3,3' and 5,5' which were observed with natural material with D2O.27 To obtain better spectroscopic data, it was decided to synthesize the bismethyl ether of 1, by subjecting the dimer 40 to Nb-quaternization first. Complete conversion to the bisquaternary salt 43 was achieved with a large excess of MeI/MeOH, both at room temperature or 40 °C. Analogous to blumeanine (isolated as its diacetate),13 chromatographic purification and isolation of this bisquaternary salt 43 was much easier in comparison to (+)-dispegatrine (1). The 2D NMR correlation experiments were then carried out on the dimer 43 to establish the position of the H-3,3' and H-5,5' protons.27 These NMR experiments were in complete agreement with the assigned positions of H-3,3' and H-5,5'.27 In the absence of an authentic sample92 [for circular dichroism (CD) analysis or thin layer chromatography (TLC) comparison], it is impossible to unequivocally report that synthetic 1 is identical to the natural product even though the 1H NMR is in good agreement.93 However, the fact that the biomimetic coupling by Yu et al.10 gave only the natural isomer and our oxidative coupling gave the P-atropodiastereomer from similar scaffolds strongly suggests that they are the same.
Scheme 12.

Completion of the Total Synthesis of (+)-Dispegatrine (1)
CONCLUSION
In conclusion, the regio- and diastereospecific doubly convergent first total synthesis of the P-atropodiastereomer of the dimeric indole alkaloid (+)-dispegatrine (1) has been accomplished from 5-methoxy-D-tryptophan methyl ester (17). p-Anisidine was converted into 17 on a large scale by employing the modified regiospecific bromination procedure. The stereospecific conversion of 17 into the optically active sarpagine framework 16 was achieved by the asymmetric Pictet-Spengler reaction and it was then employed to complete the first total synthesis of the monomeric 10-oxysubstituted alkaloids (+)-spegatrine (2), (+)-10-methoxyvellosimine (5), (+)-lochnerine (6), (+)-sarpagine (7), lochvinerine (9) and (+)-lochneram (11). Intermolecular non-phenolic oxidative dimerizations of highly functionalized substrates are very rare and the work described in this paper provides an efficient method to carry out such couplings, thereby providing an alternative to the phenolic oxidative couplings which often times produces complex mixtures with sensitive substrates. Both reductive cross coupling (Suzuki) and direct oxidative coupling were first studied on the electron-rich indole model substrate 35 and provided excellent insight into the regioselectivity and atropselectivity of the process. Based on the results of the model study, advantage was taken of the natural sarpagine configuration in lochnerine (6), a thallium(III)acetate mediated oxidative dimerization of which provided the atropodiastereomer P-40 exclusively. Completion of the total synthesis of 1 was then achieved in two more steps from 40. The total synthesis of the bisquaternary alkaloid 1 is thus notable for its brevity, principally because it employed lochnerine (6) as the substrate for the key biaryl coupling, inspite of the presence of the free indole Na-H, free hydroxyl group at C-17 and a highly basic Nb-nitrogen function. In addition, the use of thallium(III) acetate as the oxidant for direct oxidative dehydrodimerization of electron-rich substrates such as 6 and 35 has expanded the scope of this oxidant in intermolecular hetero-biaryl synthesis.
EXPERIMENTAL SECTION
The experimental details for the synthesis of alkaloids 1, 2, 5–7 and compounds 16, 38a, 38b and 40–43 are contained in the SI of reference 27b. General procedures for borylation (entries 1–7, Table 2) and SMC (entries 1–8, Table 3) are analogous to the preparation for 39 and 38a,b respectively. For general experimental considerations see SI.
3-(((2R,5S)-3,6-Diethoxy-5-isopropyl-2,5-dihydropyrazin-2-yl)methyl)-5-methoxy-2-(trimethylsilyl)-1H-indole (21a) and the regioisomer 2-(((2R,5S)-3,6-diethoxy-5-isopropyl-2,5-dihydropyrazin-2-yl)methyl)-5-methoxy-1H-indole (21b)
In a round-bottom flask (2 L) equipped with a magnetic stir were added tert-butyl (2-iodo-4-methoxyphenyl)carbamate 20 (1 g, 2.86 mmol), the internal alkyne 19 (1.016 g, 3.15 mmol), palladium(II) acetate (38.5 mg, 0.171 mmol), potassium carbonate (989 mg, 7.16 mmol), lithium chloride (133 mg, 3.150 mmol) and DMF (20 mL). The reaction mixture was degassed under vacuum (argon) and then heated at 100 °C under a slow stream of argon for 36 h. The mixture was cooled to rt, and then EtOAc (400 mL) was added to the solution, after which it was then filtered through celite to remove the Pd black and inorganic salts. The solution which resulted was diluted with additional EtOAc (20 mL), and it was then washed with water (5 × 15 mL), brine (15 mL), and dried (Na2SO4). The solvent was removed under reduced pressure, and the residue was purified (short flash column) to give the 5-methoxy indole 21a, accompanied by the byproduct 21b. Column chromatography of the crude mixture in EtOAc/hexanes provided the desired indole 21a (873 mg, 68%) and the regioisomer 21b (140 mg, 13%).
21a: 1H NMR (300 MHz, CDCl3) δ 7.85 (s, 1H), 7.24 (d, 1H, J = 8.8 Hz), 7.17 (d, 1H, J = 2.3 Hz), 6.85 (dd, 1H, J = 8.8, 2.4 Hz), 4.31 – 3.97 (m, 5H), 3.91 (t, 1H, J = 3.4 Hz), 3.86 (s, 3H), 3.52 (dd, 1H, J = 14.2, 3.7 Hz), 2.87 (dd, 1H, J = 14.2, 9.4 Hz), 2.35 – 2.25 (m, 1H), 1.31 (t, 3H, J = 7.1 Hz), 1.19 (t, 3H, J = 7.1 Hz), 1.06 (d, 3H, J = 6.8 Hz), 0.70 (d, 3H, J = 6.8 Hz), 0.42 (s, 9H); 13C NMR (75 MHz, CDCl3) δ 163.7 (C), 162.8 (C), 153.5 (C), 134.8 (C), 133.5 (C), 129.9 (C), 122.5 (C), 112.5 (CH), 111.1 (CH), 102.0 (CH), 60.6 (CH2), 60.5 (CH2), 60.4 (CH), 58.5 (CH), 55.9 (CH3), 31.9 (CH2), 31.5 (CH), 19.1 (CH3), 16.5 (CH3), 14.2 (2 × CH3), −0.6 (3 × CH3); EIMS (m/e, relative intensity) 443 (M+•, 26), 232 (100), 212 (39), 190 (13), 169 (34), 73 (20); HRMS (EI-trisector) m/z: Calcd for C24H37N3O3Si 443.2604, Found 443.2590.
21b: 1H NMR (300 MHz, CDCl3) δ 9.08 (s, 1H), 7.18 (d, 1H, J = 8.7 Hz), 7.03 (d, 1H, J = 2.4), 6.79 (dd, 1H, J = 8.7, 2.4 Hz), 6.22 (br,s, 1H), 4.30 – 4.14 (m, 5H), 3.89 (t, 1H, J = 5.6 Hz), 3.86 (s, 3H), 3.42 (dd, 1H, J = 14.7, 3.2 Hz), 3.01 (dd, 1H, J = 14.7, 8.7 Hz), 2.30 – 2.19 (m, 1H), 1.41 – 1.34 (m, 6H), 1.02 (d, 3H, J = 6.9 Hz), 0.75 (d, 3H, J = 6.8 Hz); 13C NMR (75 MHz, CDCl3) δ 197.8 (C), 161.9 (C), 153.8 (C), 137.9 (C), 131.0 (C), 128.7 (C), 111.0 (CH), 110.8 (CH), 101.9 (CH), 100.6 (CH), 61.0 (CH), 60.9 (CH2), 60.8 (CH2), 55.8 (CH), 55.7 (CH3), 32.7 (CH2), 32.2 (CH), 18.9 (CH3), 16.9 (CH3), 14.3 (CH3), 14.2 (CH3); EIMS (m/e, relative intensity) 371 (M+•, 32), 342 (7), 211 (36), 169 (38), 160 (100), 145 (13), 117 (26); HRMS (EI-trisector) m/z: Calcd for C21H29N3O3 371.2209, Found 371.2200.
3-(((2R,5S)-3,6-Diethoxy-5-isopropyl-2,5-dihydropyrazin-2-yl)methyl)-5-methoxy-1H-indole (26)
To a solution of 25 (55 g, 0.116 mol) in dry CH2Cl2 (536 mL) under nitrogen was added 2,6-lutidine (38 g, 0.353 mol) and TMSOTf (31.4 g, 0.141 mol) dropwise at 0 °C. The solution which resulted was stirred at 0 °C for 30 min and then allowed to warm to rt (12 h). The reaction mixture was then poured into a cold saturated aq solution of NaHCO3 (300 mL). The organic layer was separated and the combined organic layers were washed with brine (240 mL) and dried (Na2SO4). After removal of solvent under reduced pressure, the residue was purified by chromatography (hexanes/ethyl acetate, 6/1) to afford 26 as a colorless oil (38 g, 93%). The 1H NMR spectra was in excellent agreement with the literature values.32
(R)-Ethyl-2-(benzylamino)-3-(5 methoxy-1H-indol-3-yl)propanoate (27)
To a solution of tryptophan ethyl ester 17 (29 g, 110.6 mmol) in dry ethanol (500 mL) at 0 °C under nitrogen was added benzaldehyde (24.07 g, 226.89 mmol) and anhydrous Na2SO4 (78.6 g, 553.4 mmol). The solution was stirred at 0 °C for 5 h, cooled to −10 °C, and treated portionwise with NaBH4 (4.44 g, 117.2 mmol) over a period of 3 h keeping the temperature below −5 °C to prevent epimerization at C-3 and the formation of unwanted tetrahydro β-carboline. After the mixture was allowed to stir for an additional 1 h, ice water (15 mL) was added and the mixture was allowed to warm to room temperature. The methanol was removed under reduced pressure and the aq residue was extracted with EtOAc (3 × 360 mL). The combined organic layers were washed with brine and dried (Na2SO4). After removal of the solvent under reduced pressure the residue was purified by flash chromatography (EtOAc : hexanes, 3 : 1) to afford the Na-H, Nb-benzyl-7-methoxy-D-tryptophan ethyl ester 27 as an oil in 90% (35 g) yield. FTIR (CHCl3) 3405, 2930, 1725 cm−1; 1H NMR (300 MHz, CDCl3) δ 7.98 (br, 1H), 7.32 – 7.22 (m, 6H), 7.04 (dd, 2H, J = 15.1, 2.2 Hz), 6.86 (dd, 1H, J = 8.8, 2.3 Hz), 4.12 (q, 2H, J = 7.1 Hz), 3.87 (d, 1H, J = 14.6 Hz), 3.85 (s, 3H), 3.69 (dd, 2H, J = 13.4, 7.2 Hz), 3.16 (dd, 1H, J = 13.5, 6.0 Hz), 3.09 (dd, 1H, J = 13.5, 6.0 Hz), 1.95 (br, 1H), 1.18 (t, 3H, J = 7.2 Hz); 13C NMR (75.5 MHz, CDCl3) δ 174.9, 154.0, 139.8, 131.4, 128.3, 128.2, 128.0, 127.0, 123.6, 112.4, 111.8, 111.2, 100.7, 61.3, 60.6, 55.9, 52.2, 29.5, 14.2; EIMS (m/e, relative intensity) 352 (M+•, 100), 279 (50), 236 (11); HRMS (ESI-TOF) m/z: (M + H)+ Calcd for C21H25N2O3 353.1865, Found 353.1856.
(1S,3R)-Ethyl-2-benzyl-6-methoxy-1-(3-methoxy-3-oxopropyl)-2,3,4,9-tetrahydro-1H-pyrido[3,4-b]indole-3-carboxylate (28b)
To a round,bottom flask (500 mL) that contained a solution of optically active Na-H, Nb-benzyl-D-tryptophan ethyl ester 27 (21 g, 59.6 mmol) in dry CH2Cl2 (300 mL) was added the aldehyde, 4-oxobutyric acid methyl ester (17.23 g, 148.4 mmol), and AcOH (3.6 g, 59.9 mmol) at 0 °C. The reaction mixture which resulted was stirred at rt overnight. TFA (6.8 g, 59.6 mmol) in dry CH2Cl2 (75 mL) was then added at 0 °C. The reaction mixture which resulted was stirred at rt for 4 days and then cooled in an ice bath and brought to pH 8 with an aq solution of NH4OH (14%). The aq layer was separated and extracted with CH2Cl2 (3 × 200 mL). The combined organic layers were washed with brine, dried (K2CO3) and the solvent was removed under reduced pressure. The residue which resulted was purified by flash chromatography (silica gel, EtOAc : hexanes, 1 : 4) to provide 28b (25 g, 93%) as a white crystalline solid. [α]D20 −41.08 (c 2.6, MeOH); 1H NMR (600 MHz, CDCl3) δ 7.92 (s, 1H), 7.38 (d, 2H, J = 7.2 Hz), 7.33 (t, 2H, J = 7.2 Hz), 7.28 (d, 1H, J = 7.8 Hz), 7.24 (d, 1H, J = 8.4 Hz), 7.02 (d, 1H, J = 2.4 Hz), 6.86 (dd, 1H, J = 9.0, 2.4 Hz), 4.33 – 4.27 (m, 1H), 4.26 – 4.20 (m, 1H), 4.01 (dd, 1H, J = 9.0, 4.8 Hz), 3.90 (s, 3H), 3.88 (d, 2H, J = 13.8 Hz), 3.59 (d, 1H, J = 13.8 Hz), 3.53 (s, 3H), 3.13 (dd, 1H, J = 15.6, 9.6 Hz), 3.02 (dd, 1H, J = 15.6, 4.8 Hz), 2.45 (dt, 1H, J = 16.8, 7.2 Hz), 2.34 (dt, 1H, J = 16.8, 6.6 Hz), 2.11 – 2.06 (m, 1H), 2.01 – 1.95 (m, 1H), 1.34 (t, 3H, J = 7.2 Hz); 13C NMR (150 MHz, CDCl3) δ 174.3 (C), 173.0 (C), 154.1 (C), 139.4 (C), 135.1 (C), 131.3 (C), 129.2 (2 × CH), 128.2 (2 × CH), 127.4 (C), 127.1 (CH), 111.6 (2 × CH), 107.3 (C), 100.4 (CH), 60.8 (CH2), 56.7 (CH), 56.0 (CH3), 54.6 (CH), 53.3 (CH2), 51.5 (CH3), 29.6 (CH2), 29.0 (CH2), 21.1 (CH2), 14.4 (CH3); EIMS (m/e, relative intensity) 450 (M+•, 59), 418 (26), 377 (51), 363 (100), 327 (50), 285 (22), 227 (13); HRMS (ESI-TOF) m/z: (M + H)+ Calcd for C26H31N2O5 451.2233, Found 451.2222; Anal. Calcd for C26H30N2O5: C, 69.31; H, 6.71; N, 6.22. Found: C, 69.87; H, 6.74; N, 6.98.
(6S,10S)-Methyl-12-benzyl-9-hydroxy-2-methoxy-6,7,10,11-tetrahydro-5H-6,10-epiminocycloocta[b]indole-8-carboxylate (30)
To a solution of the trans diester 28b (10 g, 22.1 mmol) in dry toluene (400 mL), which had been predried by azeotropic removal of H2O by a Dean Stark Trap (refluxed 6 h) under argon, was added sodium hydride (8.8 g of 60% NaH dispersion in mineral oil, 221.9 mmol) at 0 °C. Dry methanol (18.0 mL, 443.9 mmol) was added carefully to the above mixture dropwise at 0 °C (a large amount of H2 was evolved at this point). The mixture which resulted was allowed to warm to rt for 0.5 h, and then heated to reflux for an additional 72 h (Note: The top of the flask was covered with aluminum foil to prevent carbonization of the intermediate lactam 29). The reaction mixture was then allowed to cool to rt and quenched with ice cold H2O (200 mL). The organic layer was separated, and the aq layer was then extracted with CH2Cl2 (3 × 400 mL). The organic layers were combined, washed with brine, and dried (Na2SO4). The solvent was removed under reduced pressure and the mineral oil was separated by decantation. The residue which resulted was purified by flash chromatography (silica gel, EtOAc/hexanes, 1 : 4) to provide the Na-H, β-ketoester 30 (7.9 g, 88%) as a yellow colored solid. FTIR (CHCl3) 3398, 2922, 1659, 1621, 1441 cm−1; 1H NMR (600 MHz, CDCl3) δ 12.03 (s, 1H), 7.58 (s, 1H), 7.40 (d, 2H, J = 7.2 Hz), 7.36 (t, 2H, J = 7.8 Hz), 7.32 (d, 1H, J = 7.2 Hz), 7.22 (d, 1H, J = 8.4 Hz) 6.99 (d, 1H, J = 2.4 Hz), 6.85 (dd, 1H, J = 9.0, 2.4 Hz), 4.02 (d, 1H, J = 5.4 Hz), 3.89 (s, 3H), 3.86 (d, 1H, J = 13.2 Hz), 3.81 (d, 1H, J = 6.0 Hz), 3.76 (d, 1H, J = 13.8 Hz), 3.70 (s, 3H), 3.19 (dd, 1H, J = 15.6, 6.0 Hz), 2.91 (d, 1H, J = 15.6 Hz), 2.85 (dd, 1H, J = 15.6, 5.4 Hz), 2.35 (d, 1H, J = 15 Hz); 13C NMR (150 MHz, CDCl3) δ 172.6 (C), 171.7 (C). 154.2 (C), 138.3 (C), 134.3 (C), 130.7 (C), 128.8 (2 × CH), 128.5 (2 × CH), 127.5 (C), 127.3 (CH), 111.7 (CH), 111.6 (CH), 106.3 (C), 100.4 (CH), 94.4 (C), 56.0 (CH3), 56.0 (CH2), 55.2 (CH), 51.5 (CH3), 49.8 (CH), 28.9 (CH2), 22.1 (CH2); EIMS (m/e, relative intensity) 404 (M+•, 62), 372 (28), 289 (100), 199 (66), 156 (45); HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C24H25N2O4 405.1814, Found 405.1788.
(6S,10S)-2-Methoxy-7,8,10,11-tetrahydro-5H-6,10-epiminocycloocta[b]indol-9(6H)-one (32)
To a solution of the β-ketoester 30 (18 g, 0.045 mol) in 1,4-dioxane (350 mL) was added 33% aq KOH (350 mL). The reaction mixture which resulted was heated to reflux for 48 h. The solution was allowed to cool to rt and the 1,4-dioxane was removed under reduced pressure. The mixture that remained was extracted with CH2Cl2 (3 × 20 mL). The organic layer was separated, washed with brine, and dried (Na2SO4). The solvent was removed under reduced pressure and the residue was purified by flash chromatography (hexanes : EtOAc, 3 : 1) to afford the Nb-benzyl tetracyclic ketone 31 (12.6 g, 82%) which was taken forward to the next step without subjecting it to any analytical characterization.
To a solution of the tetracyclic ketone 31 (8.94 g, 25.8 mmol) in dry EtOH (50 mL) was added a saturated solution of EtOH/HCl(g) dropwise until the solid completely dissolved. The solvent was removed under reduced pressure to furnish an HCl salt of the Nb-benzyl tetracyclic ketone 31. Then EtOH was added to the salt and removed under reduced pressure. This process was repeated 3 times to remove excess hydrogen chloride. The HCl salt of 31 was degassed under reduced pressure at rt and back filled with argon (2 times). Dry Pd/C (10% by wt, 1.99 g, 1.54 mmol) was added to the above HCl salt followed by slow addition of dry ethanol (100 mL). The mixture was degassed under reduced pressure at rt and back filled with H2 (3 times). The mixture which resulted was allowed to stir at rt under an atmosphere of hydrogen (1 atm) for 12 h. After analysis by TLC (silica gel plate was exposed to NH3 vapors) indicated the absence of starting material 31, the catalyst was removed by filtration through celite and the solid washed with EtOH (3 × 15 mL). The organic layers were combined and the solvent was removed under reduced pressure to give a yellow residue, which was dissolved in a mixture of CHCl3 (200 mL) and ice water, after which the solution was brought to pH 8 by addition of a solution of aq NH4OH (14%). The aq layer was extracted with CHCl3 (3 × 100 mL). The combined organic layers were washed with brine (200 mL) and dried (K2CO3). The solvent was removed under reduced pressure to afford the crude product, which was purified by chromatography on silica gel (EtOAc : hexanes, 5 : 1) to provide the Nb-H tetracyclic ketone 32 (6.1 g, 92% yield) as a yellow colored oil. 1H NMR (600 MHz, CDCl3) δ 7.87 (br, 1H), 7.29 (s, 1H), 7.23 (d, 1H, J = 9.0 Hz), 6.93 (d, 1H, J = 2.4 Hz), 6.86 (dd, 1H, J = 9.0, 2.4 Hz), 4.33 (m, 1H), 3.98 (d, 1H, J = 6.6 Hz), 3.88 (s, 3H), 3.12 (dd, 1H, J = 16.2, 6.6 Hz), 2.82 (d, 1H, J = 16.8 Hz), 2.54 – 2.44 (m, 2H), 2.21 – 2.14 (m, 2H); 13C NMR (150 MHz, CDCl3) δ 211.0 (C), 154.3 (C), 134.9 (C), 130.7 (C), 127.4 (C), 112.0 (CH), 111.6 (CH), 107.5 (C), 100.3 (CH), 59.9 (CH), 55.9 (CH3), 46.3 (CH), 35.2 (CH2), 32.2 (CH2), 26.0 (CH2); HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C15H17N2O2 257.1290, Found 257.1302.
(6S,10S)-12-((Z)-2-iodobut-2-en-1-yl)-2-methoxy-7,8,10,11-tetrahydro-5H-6,10-epimino cycloocta[b]indol-9(6H)-one (33)
To a solution of the Na-H, Nb-H tetracyclic ketone 32 (6.0 g, 23.4 mmol) and molecular sieves (5.0 g) in anhydrous acetonitrile (250 mL) under an inert atmosphere was added K2CO3 (12.9 g, 93.7 mmol) and Z-1-bromo-2-iodo-2-butene46 (7.9 g, 30.4 mmol) and the mixture which resulted was stirred at rt for 8 h. Analysis by TLC (silica gel, CHCl3 : EtOH, 4 : 1) indicated the absence of tetracyclic ketone 32. The solids were removed by filtration and washed with EtOAc (3 × 100 mL). The combined organic layers were concentrated under reduced pressure to provide a light yellow residue. Purification of the crude product by flash chromatography (silica gel, EtOAc/hexanes, 1 : 9) provided the Nb-Z-2′-iodo-2′-butenyl, tetracyclic ketone 33 (7.76 g, 76%) as a yellow colored solid. FTIR (CHCl3) 2929, 1707 cm−1; 1H NMR (300 MHz, CDCl3) δ 7.89 (s, 1H), 7.24 (d, 1H, J = 8.7 Hz), 6.94 (d, 1H, J = 2.3 Hz), 6.86 (dd, 1H, J = 8.7, 2.4 Hz), 5.85 (q, 1H, J = 6.3 Hz), 4.03 (d, 1H, J = 2.6 Hz), 3.88 (s, 3H), 3.72 (d, 1H, J = 6.5 Hz), 3.39 (dd, 2H, J = 17.5, 13.9 Hz), 3.11 (dd, 1H, J = 16.7, 6.6 Hz), 2.69 (d, 1H, J = 16.7 Hz), 2.56 – 2.46 (m, 2H), 2.16 – 1.99 (m, 2H), 1.82 (d, 3H, J = 6.4 Hz); 13C NMR (75 MHz, CDCl3) δ 210.2 (C), 154.2 (C), 133.0 (C), 132.7 (CH), 130.8 (C), 127.2 (C), 111.9 (CH), 111.6 (CH), 108.5 (C), 106.7 (C), 100.3 (CH), 64.1 (CH), 63.4 (CH2), 55.9 (CH3), 49.9 (CH), 34.5 (CH2), 30.4 (CH2), 21.7 (CH3), 20.6 (CH2); EIMS (m/e, relative intensity) 436 (M+•, 52), 408 (6), 379 (100), 309 (10), 281 (23); HRMS (EI-trisector) m/z: Calcd for C19H21IN2O2 436.0648, Found 436.0663.
(+)-Lochneram (11)
To a stirred solution of 6 (100 mg, 0.308 mmol) in freshly distilled MeOH (2 mL) at 0 °C was added MeI (2 mL) and the reaction was allowed to warm to rt in the dark (24 h) until disappearance of the starting material 6 (TLC, silica gel). The solvent and excess MeI was removed under reduced pressure to provide the crude Nb-methiodide salt. The solvent was removed under reduced pressure and the residue passed through a short column of activated neutral alumina using CHCl3/MeOH (16:1) as eluant to provide (+)-lochneram (11, 122 mg) in 85% yield as a clear oil. 1H NMR (600 MHz, CD3OD) δ 7.31 (d, 1H, J = 9.0 Hz), 7.04 (br, s, 1H), 6.87 (d, 1H, J = 8.4 Hz), 5.68 (q, 1H, J = 6.6 Hz), 4.91 (1H, peak is embedded in CD3OD peak), 4.46 (d, 1H, J = 15.6 Hz), 4.25 (d, 1H, J = 15.6 Hz), 3.85 (s, 3H), 3.58 (d, 3H, J = 7.8 Hz), 3.31 (d, 1H, J = 4.8 Hz, part of the peak is embedded in CD3OD peak), 3.15 – 3.10 (m, 5H), 2.55 (t, 1H, J = 12.0 Hz), 2.21 – 2.17 (m, 2H), 1.74 (d, 3H, J = 6.6 Hz); 13C NMR (150 MHz) δ 154.5 (C), 132.4 (C), 131.7 (C), 127.7 (C), 126.5 (C), 120.7 (CH), 112.6 (CH), 112.0 (CH), 100.3 (C), 99.8 (CH), 65.4 (CH), 64.4 (CH2), 62.4 (CH), 61.0 (CH2), 54.8 (CH3), 46.7 (CH3), 43.6 (CH), 32.0 (CH2), 26.0 (CH), 23.9 (CH2), 11.6 (CH3); HRMS (ESI-TOF) m/z: Calcd for C21H27N2O2 (M)+ 339.2073; Found 339.2057. The spectral data for 11 were identical to those reported in the literature.1a,22
(+)-Lochvinerine (9)
A mixture of anhydrous potassium tert butoxide (313 mg, 0.279 mmol) and methyl-triphenylphosphonium bromide (911 mg, 0.25 mmol) in dry benzene (14 mL) was allowed to stir at rt for 1 h. The pentacyclic ketone 16 (124 mg, 0.40 mmol) in THF (5 mL) was then added into the above orange-colored solution dropwise at rt. The mixture which resulted was stirred at rt for 4 h. The mixture was diluted with EtOAc (50 mL), washed with H2O (3 × 10 mL) as well as brine (25 mL), and dried (K2CO3). The solvent was removed under reduced pressure and the oil that resulted was chromatographed (silica gel, CHCl3/MeOH; 15:1) to provide the olefin 34 (111 mg, 92%). 1H NMR (300 MHz, CDCl3) δ 7.81 (s, 1H), 7.18 (d, 1H, J = 8.7 Hz), 6.95 (d, 1H, J = 2.4 Hz), 6.79 (dd, 1H, J = 8.7, 2.5 Hz), 5.27 (q, 1H, J = 6.7 Hz), 4.86 – 4.84 (m, 2H), 4.15 (dd, 1H, J = 9.8, 2.2 Hz), 3.87 – 3.85 (s, 4H), 3.70 (br, s, 2H), 3.31 (d, 1H, J = 2.5 Hz), 3.13 (dd, 1H, J = 15.3, 5.4 Hz), 2.96 (dd, 1H, J = 15.3, 1.5 Hz), 2.14 (ddd, 1H, J = 12.0, 10.1, 1.7 Hz), 1.93 – 1.85 (m, 1H), 1.66 (d, 3H, J = 6.8 Hz); 13C NMR (75 MHz, CDCl3) δ 153.9 (C), 152.8 (C), 138.6 (C), 137.6 (C), 131.3 (C), 127.9 (C), 114.6 (CH), 111.4 (CH), 110.9 (CH), 105.1 (CH2), 104.7 (C), 100.5 (CH), 56.6 (CH), 55.9 (CH2), 55.9 (CH3), 50.5 (CH), 36.7 (CH), 36.3 (CH2), 26.3 (CH2), 12.3 (CH3); EIMS (m/e, relative intensity) 306 (M+•, 100), 291 (16), 265 (12), 251 (10), 198 (40), 183 (16), 156 (10), 77 (10). This material was employed directly in the next step without any further characterization.
To a solution of olefin 34 (124 mg, 0.40 mmol) in THF (12 mL) was added 9-BBN (0.5 M in THF, 5 mL, 2.43 mmol) dropwise at 0 °C. The solution was allowed to warm to rt and stirred for 1.5 h. The reaction mixture was then cooled to 0 °C and NaBO3·4H2O (1.1g, 7.28 mmol) was added and the reaction temperature allowed to warm to rt. The mixture that resulted was stirred for 2 h at rt, diluted with CH2Cl2 (200 mL), washed with H2O (3 × 50 mL) as well as brine (100 mL) and dried (K2CO3). The solvent was removed under reduced pressure and the residue was chromatographed (silica gel, CHCl3/MeOH; 9:1) to provide lochvinerine 9 (98 mg, 75%) as a clear oil. 1H NMR (300 MHz, CDCl3) 10.2 (br, s, 1H), 7.30 (d, 1H, part of the peak is embedded in CDCl3 peak), 6.87 (d, 1H, J = 2.2 Hz), 6.80 (dd, 1H, J = 8.8, 2.3 Hz), 5.16 (q, 1H, J = 6.8 Hz), 4.43 (d, 1H, J = 5.8 Hz), 3.88 – 3.84 (m, 4H), 3.71 (d, 1H, J = 17.6 Hz), 3.52 (dd, 1H, J2 = 7.2 Hz, part of the peak is embedded in MeOH peak), 3.50 (1H is embedded in MeOH peak), 3.11 (d, 1H, J = 16.6 Hz), 3.36 – 3.24 (m, 2H), 2.93 (br, s, 1H), 2.35 – 2.33 (m, 1H), 2.19 (s, 1H), 2.03 – 1.95 (m, 2H), 1.63 (d, 3H, J = 6.5 Hz); 13C NMR (75 MHz, CDCl3) δ 154.0 (C), 140 – 132 (2 quaternary carbons not observed), 131.7 (C), 125.7 (C), 117.3 (CH), 112.5 (CH), 111.9 (CH), 104.2 (C), 100.2 (CH), 60.3 (CH2), 55.7 (CH3), 54.9 (CH2), 53.9 (CH), 50.6 (CH), 40.9 (CH), 26.5 (CH2), 25.7 (CH), 21.4 (CH2), 12.7 (CH3); HRMS (ESI-TOF) m/z: (M + H)+ Calcd for C20H25N2O2 325.1916; Found 325.1920. The spectral data for 9 were identical to those reported in the literature.21
(1S,3R)-Ethyl-2-benzyl-5-iodo-6-methoxy-1-(3-methoxy-3-oxopropyl)-9-methyl-2,3,4,9-tetrahydro-1H-pyrido[3,4-b]indole-3-carboxylate (36a)
To a solution of 35 (750 mg, 1.61 mol) in acetonitrile (20.7 mL) cooled to 0 °C, TFA (2.3 mL) was added dropwise, followed by IBX (226 mg, 0.807 mol) and I2 (497 mg, 1.77 mol). The reaction mixture was stirred at 0 °C for 30 min and then allowed to warm to rt (8 h). The reaction mixture was diluted with ethyl acetate (30 mL), cooled in an ice bath and adjusted to pH 8 with a solution of cold aq NH4OH (10%), followed by treatment with a saturated aq solution of Na2S2O3 (35 mL) to remove excess iodine. The organic layer was separated, washed with brine (3 × 10 mL), dried (Na2SO4) and concentrated under reduced pressure to give a reddish brown oil. Flash column chromatography (silica gel, ethyl acetate/hexanes; 2 : 8) provided 36a (780 mg, 82%) as a white crystalline solid and the regioisomer 36b (76 mg, 8%) as a light yellow colored oil.
36a: 1H NMR (300 MHz, CDCl3) δ 7.38 – 7.34 (m, 3H), 7.32 (br,s, 1H), 7.30 – 7.28 (m, 1H), 7.22 (d, 1H, J = 8.7 Hz), 6.90 (d, 1H, J = 8.7 Hz), 4.40 – 4.23 (m, 2H), 4.02 (dd, 1H, J = 11.2, 4.8 Hz), 3.94 (s, 3H), 3.89 (d, 1H, J = 13.4 Hz), 3.80 (dd, 1H, J = 10.6, 3.1 Hz), 3.74 – 3.63 (m, 4H), 3.49 – 3.44 (m, 4H), 3.38 (d, 1H, J = 13.2 Hz), 2.65 (ddd, 1H, J = 17.5, 9.5, 5.4 Hz), 2.40 (dt, 1H, J = 17.5, 5.3 Hz), 2.06 – 1.95 (m, 1H), 1.93 – 1.81 (m, 1H), 1.39 (t, 3H, J = 7.1 Hz); 13C NMR (75 MHz, CDCl3) δ 173.8 (C), 172.7 (C), 152.3 (C), 139.2 (C), 137.8 (C), 133.9 (C), 129.2 (2 × CH), 128.9 (C), 128.1 (2 × CH), 126.9 (CH), 109.2 (CH), 107.9 (CH), 107.7 (C), 60.8 (CH2), 58.3 (CH3), 55.8 (CH), 53.3 (CH), 52.6 (CH2), 51.2 (CH3), 29.8 (CH3), 29.6 (CH2), 29.3 (CH2), 22.2 (CH2), 14.3 (CH3) (One of the quaternary carbon atoms is embedded in the above carbons); EIMS (m/e, relative intensity) 590 (M+•, 14), 517 (55), 503 (100), 377 (20), 339 (22), 303 (14); HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C27H32IN2O5 591.1356, Found 591.1332. The structure was confirmed by X-ray analysis (see SI).
36b: EIMS (m/e, relative intensity) 590 (M+•, 14), 517 (55), 503 (100), 377 (20), 339 (22), 303 (14); HRMS (ESI-TOF) m/z: (M + H)+ Calcd for C27H32IN2O5 591.1356, Found 591.1254. The identity of the regioisomer 36b was confirmed by comparison of the crude 1H NMR with 36a. HRMS was also performed on the same sample. No further characterization was carried out.
(1S,3R)-Ethyl-2-benzyl-5-bromo-6-methoxy-1-(3-methoxy-3-oxopropyl)-9-methyl-2,3,4,9-tetrahydro-1H-pyrido[3,4-b]indole-3-carboxylate (37a)
To a solution of indole 35 (500 mg, 1.07 mol) in acetonitrile (40 mL), cooled to 0 °C, TFA (0.12 mL, 1.61 mol) was added dropwise and this was followed by NBS (229 mg, 1.29 mol). The reaction mixture was stirred at 0 °C for 30 min and then allowed to warm to rt (18 h). The reaction mixture was diluted with ethyl acetate (30 mL), cooled in an ice bath and adjusted to pH 8 with cold aq NH4OH solution (10%). The organic layer was separated, washed with brine (3 × 10 mL), dried (Na2SO4) and concentrated under reduced pressure to give a reddish brown oil. Column chromatography (silica gel, ethyl acetate/hexanes; 1 : 4) provided the 9-bromoindole 37a (380 mg, 65%) and the regioisomer 37b (68 mg, 13%).
37a: 1H NMR (300 MHz, CDCl3) δ 7.40 – 7.24 (m, 5H), 7.19 (d, 1H, J = 8.7 Hz), 6.94 (d, 1H, J = 8.8), 4.41 – 4.22 (m, 2H), 4.02 (dd, 1H, J = 11.2, 4.9 Hz), 3.95 (s, 3H), 3.89 (d, 1H, J = 13.2 Hz), 3.79 (dd, 1H, J = 10.8, 3.1 Hz), 3.62 (s, 3H), 3.57 (d, 1H, J = 4.9 Hz), 3.48 – 3.36 (m, 5H), 2.64 (ddd, 1H, J = 17.4, 9.5, 5.3 Hz), 2.40 (dt, 1H, J = 17.5, 5.3 Hz), 2.06 – 1.95 (m, 1H), 1.93 – 1.80 (m, 1H), 1.39 (t, 3H, J = 7.1 Hz); 13C NMR (75 MHz, CDCl3) δ 173.8 (C), 172.7 (C), 150.0 (C), 139.2 (C), 137.7 (C), 134.0 (C), 129.2 (2 × CH), 128.1 (2 × CH), 126.9 (CH), 126.3 (C), 108.9 (CH), 108.1 (CH), 107.0 (C), 103.0 (C), 60.8 (CH2), 58.2 (CH3), 56.0 (CH), 53.2 (CH), 52.6 (CH2), 51.2 (CH3), 29.8 (CH3), 29.3 (CH2), 27.7 (CH2), 22.1 (CH2), 14.3 (CH3); EIMS (m/e, relative intensity) 544 (M+•, 5), 542 (M+•, 5), 471 (14), 469 (17), 457 (99), 455 (100), 377 (36); HRMS (ESI-TOF) m/z: (M + H)+ Calcd for C27H32BrN2O5 543.1488, Found 543.1503.
37b: The identity of the regioisomer 37b was confirmed by comparison of the crude 1H NMR to that of 36b. No further characterization was carried out.
(1S,3R)-Ethyl-2-benzyl-6-methoxy-1-(3-methoxy-3-oxopropyl)-9-methyl-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-2,3,4,9-tetrahydro-1H-pyrido[3,4-b]indole-3-carboxylate (39) [Entry 8, Table 2]
General procedure for entries 1 – 7 is the same as described below.
To a resealable Schlenk tube possessing a Teflon screw valve were added 36a (118 mg, 0.2 mmol), Pd(OAc)2 (2.2 mg, 0.01 mmol) and 2-(dicyclohexylphosphanyl)biphenyl (DCPB, 14 mg, 0.04 mmol). The Schlenk tube was capped with a rubber septum and then evacuated and backfilled with argon (this sequence was carried out a total of three times). Freshly degassed 1,4-dioxane (2.5 mL) was added via a syringe through the septum, followed by the addition of dry Et3N (0.11 mL, 80 mg, 0.80 mmol) and pinacol borane (0.09 mL, 77 mg, 0.6 mmol). The septum was then replaced with a Teflon screw valve under a positive argon pressure and the Schlenk tube was sealed. The reaction mixture was heated to 100 °C and stirred at that temperature for 3 h. At the end of this period the reaction mixture was cooled to rt, diluted with EtOAc (10 mL) and was passed through a short pad of celite. The celite pad was further washed with EtOAc (20 mL) and the combined filtrates were washed with water (20 mL) and brine (20 mL). The organic extracts were dried (Na2SO4) and concentrated under reduced pressure. The crude residue thus obtained was purified by flash chromatography on a silica gel column, eluted with 4: 1 hexanes/EtOAc to afford 39 as a white solid (110 mg, 93%). mp: 182.6 - 183.8 °C; 1H NMR (300 MHz, CDCl3) δ 7.39 (d, 2H, J = 6.6 Hz), 7.32 (d, 2H, J = 6.6 Hz), 7.26 (m, 2H), 6.89 (d, 1H, J = 8.7 Hz), 4.29 (m, 2H), 4.06 (dd, 1H, J = 10.5, 6.0 Hz), 3.88 (s, 3H), 3.81 (d, 1H, J = 13.2 Hz), 3.75 (dd, 1H, J = 10.8, 3.0 Hz), 3.60 (s, 3H), 3.49 (s, 3H), 3.40 (d, 1H, J = 13.2 Hz), 3.15 (m, 2H), 2.62 (m, 1H), 2.42 (m, 1H), 1.97 (m, 1H), 1.84 (m, 1H), 1.49 (s, 12H), 1.37 (t, 3H, J = 7.2 Hz); 13C NMR (75 MHz, CDCl3) δ 173.9 (C), 172.9 (C), 157.8 (C), 139.4 (2 × C), 136.8 (C), 133.1 (C), 129.5 (C), 129.3 (2 × CH), 128.0 (2 × CH), 126.8 (CH), 110.4 (CH), 107.8 (CH), 106.5 (C), 83.9 (2 × C), 60.7 (CH2), 58.3 (CH3), 56.3 (CH), 53.2 (CH), 52.7 (CH2), 51.2 (CH3), 29.6 (CH2), 29.6 (CH3), 27.9 (CH2), 25.1 (4 × CH3), 21.8 (CH2), 14.3 (CH3); HRMS (ESI-TOF) m/z: (M + H)+ Calcd for C33H44BN2O7 591.3236, Found 591.3265; Anal. Calcd for C33H43BN2O7: C, 67.12; H, 7.34; N, 4.74. Found: C, 66.91; H, 7.53; N, 4.60.
(1R,1'R,3S,3'S)-diethyl-2,2'-dibenzyl-6,6'-dimethoxy-1,1'-bis(3-methoxy-3-oxopropyl)-9,9'-dimethyl-2,2',3,3',4,4',9,9'-octahydro-1H,1'H-[5,5'-bipyrido[3,4-b]indole]-3,3'-dicarboxylate (38a) and its atropodiastereomer (1S,1'S,3R,3'R)-diethyl 2,2'-dibenzyl-6,6'-dimethoxy-1,1'-bis(3-methoxy-3-oxopropyl)-9,9'-dimethyl-2,2',3,3',4,4',9,9'-octahydro-1H,1'H-[5,5'-bipyrido[3,4-b]indole]-3,3'-dicarboxylate (38b) [Entry 9, Table 3]
General procedure for entries 1 – 8 is the same as described below.
To a resealable Schlenk tube possessing a Teflon screw valve were added 36a (11.8 mg, 0.02 mmol), 39 (17.6 mg, 0.03 mmol), Pd(OAc)2 (0.44 mg, 0.002 mmol), S-Phos (1.6 mg, 0.004 mmol), and K3PO4 (8.4 mg, 0.04 mmol). The Schlenk tube was capped with a rubber septum and then evacuated and backfilled with argon (this sequence was carried out a total of three times). Freshly degassed THF (1 mL) and water (0.1 mL) were added via syringe through the septum. The septum was then replaced with a Teflon screw valve under a positive argon pressure and the Schlenk tube was sealed. The reaction mixture was heated to 50 °C and stirred at that temperature for 48 h. At the end of this period, the reaction mixture was cooled to rt and was passed through a short pad of celite. The celite pad was further washed with EtOAc (10 mL) and the combined filtrate was washed with water (5 mL) and brine (5 mL). The organic extracts were dried (Na2SO4) and concentrated under reduced pressure. The crude residue thus obtained was purified by flash chromatography on a silica gel column, eluted with 1: 1 hexanes/EtOAc to afford 38a as an off-white solid (6.8 mg, 37%), 38b (3.3 mg, 18%) as a light yellow solid and recovered 35 (20%).
The 1H NMR spectra's for 38a and 38b were identical to that reported in the communication.27b
Intermolecular oxidative coupling of the model substrate, β-carboline 35
Entry 1: To a stirred solution of the β-carboline 35 (150 mg, 0.32 mmol) in dry CH2Cl2 (25 mL) at 0 °C was added solid PIFA (84 mg, 0.194 mmol) and boron trifluoride diethyl etherate dropwise (114.5 mg, 0.807 mmol), after which the reaction mixture was allowed to warm to rt and stirred for 2 h. Analysis of the reaction mixture by TLC [silica gel, CHCl3/MeOH (v/v, 9 : 1)] indicated the presence of the starting material 35, and atropdiastereomers 38a and 38b, with a considerable amount of colored impurities formed at the baseline. No further purification for the separation of the diastereomers was attempted.
Entry 2: To a stirred solution of the β-carboline 35 (100 mg, 0.215 mmol) in dry CH2Cl2 (2.0 mL) at 0 °C was added solid PIFA (95 mg, 0.219 mmol) and boron trifluoride diethyl etherate dropwise (122.2 mg, 0.861 mmol), after which the reaction mixture was allowed to warm to rt and stirred for 8 h. Analysis of the reaction mixture by TLC [silica gel, CHCl3/MeOH (v/v, 9 : 1)] indicated complete conversion of the starting material 35. The reaction mixture was diluted CH2Cl2 (25 mL) and cooled to 0 °C, after which it was brought to pH = 8 with a cold aq solution of saturated NaHCO3. The aq layer which resulted was extracted with CH2Cl2 (3 × 15 mL), and the combined organic layers were washed with a saturated aq solution of NaHSO3 (2 × 15 mL) and dried (Na2SO4). The solvent was removed under reduced pressure to afford a dark oil. The crude diastereomeric mixture was purified by flash chromatography on silica gel (hexanes/ethyl acetate) to provide a combined yield of 38a + 38b: 21 mg (11%), with a diastereomeric ratio of 3 : 2 in favor of 38a.
Entry 3: To a stirred solution of the β-carboline 35 (100 mg, 0.215 mmol) in dry CH2Cl2 (2.0 mL) at −40 °C was added solid PIFA (95 mg, 0.219 mmol) and boron trifluoride diethyl etherate (122.2 mg, 0.861 mmol) dropwise, after which the reaction mixture was allowed to warm to rt and stirred for 1.5 h. Analysis of the reaction mixture by TLC [silica gel, CHCl3/MeOH (v/v, 9 : 1)] indicated complete conversion of the starting material 35. Fewer baseline impurities were observed as a result of lowering the reaction temperature in this case. The workup and purification procedure were the same as entry 2: combined yield of 38a + 38b: 39 mg (20%) with a diastereomeric ratio of 3 : 2 in favor of 38a.
Entry 4: To a stirred solution of the β-carboline 35 (55 mg, 0.118 mmol) in dry CH2Cl2 (1.0 mL) under an inert atmosphere at −40 °C was added a solution of PIFA (40 mg, 0.094 mmol) and boron trifluoride diethyl etherate (50.41 mg, 0.355 mmol) dropwise in CH2Cl2 (2.0 mL), which had been precooled to −40 °C via a double ended needle transfer. The reaction mixture which resulted was stirred at −40 °C for 0.5 h. Analysis of the reaction mixture by TLC [silica gel, CHCl3/MeOH (v/v, 9 : 1)] indicated complete conversion of the starting material 35 and a much cleaner reaction. The workup and purification procedure were the same as entry 2: combined yield of 38a + 38b: 33 mg (30%) with a diastereomeric ratio of 4 : 1 in favor of M-38a via analysis of the integration values of the 1H NMR spectrum.
Entry 5: To a stirred solution of the β-carboline 35 (63 mg, 0.135 mmol) in dry CH2Cl2 (1.0 mL) under an inert atmosphere at −78 °C was added a solution of PIFA (47 mg, 0.108 mmol) and boron trifluoride diethyl etherate (57.5 mg, 0.405 mmol) dropwise in CH2Cl2 (2.0 mL) precooled to −78 °C via a double ended transfer needle. The reaction mixture which resulted was stirred at −78 °C for 0.5 h. Analysis of the reaction mixture by TLC [silica gel, CHCl3/ MeOH (v/v, 9 : 1)] indicated the presence of unreacted starting material 35 along with the diastereomers 38a and 38b. The workup and purification procedure were the same as entry 2: combined yield of 38a + 38b: 31.4 mg (25%, based on recovered starting material) with a diastereomeric ratio of 4 : 1 in favor of 38a.
Entry 6: To a stirred solution of the β-carboline 35 (50 mg, 0.107 mmol) in dry CH2Cl2 (1.0 mL) under an inert atmosphere at −40 °C was added a solution of PIDA (27.7 mg, 0.086 mmol) and boron trifluoride diethyl etherate (45.8 mg, 0.322 mmol) dropwise in CH2Cl2 (2.0 mL) precooled to −40 °C via a double ended transfer needle. The reaction mixture which resulted was stirred at −40 °C for 2.5 h. Analysis of the reaction mixture by TLC [silica gel, CHCl3/ MeOH (v/v, 9 : 1)] indicated very little formation of the diastereomers 38a and 38b with a considerable amount of starting material 35 remaining. The workup and purification procedure were the same as entry 2. No further purification for the separation of the diastereomers (38a and 38b) was attempted.
Entry 7: To a stirred solution of the β-carboline 35 (50 mg, 0.107 mmol) in dry MeCN (2.0 mL) under an inert atmosphere at rt was added solid thallium(III) trifluoroacetate (46.7 mg, 0.086 mmol) and boron trifluoride diethyl etherate (45.8 mg, 0.322 mmol) dropwise and the solution which resulted allowed to stir at rt for 15 min. Analysis of the reaction mixture by TLC [silica gel, CHCl3/MeOH (v/v, 9 : 1)] indicated the absence of starting material 35 and formation of traces of atropdiastereomers 38a and 38b. No further purification for the separation of the diastereomers was attempted.
Note: Thallium compounds are toxic. Do not breath, ingest or get on skin: Use Caution.
Entry 8: To a stirred solution of the β-carboline 35 (50 mg, 0.108 mmol) in dry acetonitrile (2.0 mL) under an inert atmosphere at −40 °C was added a solution of thallium(III) trifluoroacetate (46.7 mg, 0.086 mmol) and boron trifluoride diethyl etherate dropwise (45.8 mg, 0.322 mmol) in MeCN (2.0 mL), which was precooled to −40 °C, via a double ended transfer needle. The reaction mixture which resulted was stirred at −40 °C for 20 min. Analysis of the reaction mixture by TLC [silica gel, CHCl3/MeOH (v/v, 9: 1)] indicated a cleaner reaction had occurred with complete conversion of the starting material 35 and formation of the two atropdiastereomers (38a and 38b). The cold reaction mixture was diluted with CH2Cl2 (25 mL) and the solvent was removed under reduced pressure to give a brown residue. The residue was dissolved in fresh CH2Cl2 (25 mL) and cooled to 0 °C after which it was brought to pH = 8 with a cold aq solution of saturated NaHCO3. The aq layer which resulted was extracted with CH2Cl2 (3 × 15 mL) and the combined organic layers were washed with brine (2 × 15 mL) and dried (Na2SO4). The solvent was removed under reduced pressure to afford a dark brown oil. The crude diastereomeric mixture was purified by flash chromatography on silica gel (hexanes/ethyl acetate) to provide a combined yield of 38a + 38b: 37.9 mg (38%) with a diastereomeric ratio of 2 : 3 in favor of 38b.
Entry 9: To a stirred solution of the β-carboline 35 (50 mg, 0.108 mmol) in dry acetonitrile (2.0 mL) under an inert atmosphere at −78 °C was added a solution of thallium(III) trifluoroacetate (29.2 mg, 0.054 mmol) and boron trifluoride diethyl etherate (38.2 mg. 0.269 mmol) in MeCN (2.0 mL) precooled to −78 °C via a double ended transfer needle. The reaction mixture which resulted was stirred at −78 °C for 40 min. Analysis of the reaction mixture by TLC [silica gel, CHCl3/MeOH (v/v, 9 : 1)] indicated formation of the two atropdiastereomers (38a and 38b) and complete conversion of the starting material 35 with a considerable amount of impurities formed at the baseline. The workup and purification procedure were the same as entry 8: combined yield of 38a + 38b: 29.9 mg (30%) with a diastereomeric ratio of 3 : 7 in favor of 38b.
Entry 10: To a stirred solution of the β-carboline 35 (200 mg, 0.430 mmol) in dry acetonitrile (10 mL) under an inert atmosphere at −40 °C was added a solution of thallium(III) acetate (115.0 mg, 0.301 mmol) and boron trifluoride diethyl etherate (183.0 mg, 1.29 mmol) in MeCN (10 mL) precooled to −40 °C via a double ended transfer needle. The reaction mixture which resulted was stirred at −40 °C for 1.25 h. Analysis of the reaction mixture by TLC [silica gel, CHCl3/MeOH (v/v, 9 : 1)] indicated the presence of starting material 37 and formation of the two atropdiastereomers (38a and 38b). The workup and purification procedure were the same as entry 8: combined yield of 38a + 38b: 227 mg (67%) with a diastereomeric ratio of 3 : 7 in favor of 38b; recovered starting material 35 (28 mg, 14%). Recrystallization of 38b from ethanol gave light brown crystals. X-ray analysis of 38b established the axial chirality (at the C9-C9') as P(S).
Supplementary Material
ACKNOWLEDGEMENTS
We wish to acknowledge NIMH (in part) and the Lynde and Harry Bradley Foundation for support of this work. We thank Dr. Jeffrey Deschamps for X-ray crystallographic studies (supported by NIDA-NRL Interagency Agreement Number Y1-DA1101). This paper is dedicated to Dr. Alfred Bader for his interest in organic chemistry worldwide.
Footnotes
Supporting Information 1H and 13C NMR spectra, and single crystal X-ray crystallographic data (CIF) for compounds 16 and 36a. This material is available free of charge via the Internet at http://pubs.acs.org
REFERENCES
- 1).(a) Lounasmaa M, Hanhinen P, Westersund M. The Sarpagine Group of Indole Alkaloids. In: Cordell G, editor. The Alkaloids. Vol. 52. Academic Press; San Diego, CA: 1999. pp. 103–195. [Google Scholar]; (b) Koskinen A, Lounasmaa M. The Sarpagine-Ajmaline Group of Indole Alkaloids. In: Herz W, Grisebach H, Kirby GW, editors. Progress in the Chemistry of Organic Natural Products. Vol. 43. Springer-Verlag; New York: 1983. pp. 267–346. [Google Scholar]; (c) Taylor WI. The Ajmaline-Sarpagine Alkaloids. In: Manske RHF, editor. The Alkaloid: Chemistry and Physiology. Vol. 11. Academic Press; New York: 1968. pp. 41–72. [Google Scholar]; (d) Taylor WI. The Ajmaline-Sarpagine Alkaloids. In: Manske RHF, editor. The Alkaloid: Chemistry and Physiology. Vol. 8. Academic Press; New York: 1965. pp. 785–814. [Google Scholar]; (e) Hamaker LK, Cook JM. The Synthesis of Macroline Related Sarpagine Alkaloids. In: Pelletier SW, editor. Alkaloids: Chemical and Biological Perspectives. Vol 9. Elsevier. Science; New York: 1995. pp. 23–84. [Google Scholar]
- 2).(a) Gröger DI. Alkaloids derived from Tryptophan. In: Mothes K, Schutte HR, Luckner M, editors. Biochemistry of Alkaloids. VCH Publishers; Florida: 1985. pp. 272–313. references therein. [Google Scholar]; (b) Cordell GA, Quinn-Beattie ML, Farnsworth NR. Phytother. Res. 2001;15:183. doi: 10.1002/ptr.890. [DOI] [PubMed] [Google Scholar]
- 3).(a) Talapatra SK, Chaudhury NA. Sci. Cult. 1958;24:243. [Google Scholar]; (b) Wright CW, Allen D, Cai Y, Phillipson JD, Said IM, Kirby GC, Warhurst DC. Phytother. Res. 1992;6:121. [Google Scholar]; (c) Keawpradup N, Eno-Amooquaye E, Burke PJ, Houghton PJ. Planta Med. 1999;65:311. doi: 10.1055/s-1999-13992. [DOI] [PubMed] [Google Scholar]; (d) Keawpradup N, Kirby GC, Steele JC, Houghton PJ. Planta Med. 1999;65:690. doi: 10.1055/s-1999-14043. [DOI] [PubMed] [Google Scholar]; (e) Feng Y, Gao H, Zeng G. Acta Pharm. Sin. 1986;21:1. [PubMed] [Google Scholar]
- 4).(a) Pfitzner A, Stöckigt J. Tetrahedron Lett. 1983;24:5197. [Google Scholar]; (b) Ruppert M, Ma X, Stöckigt J. Current Org. Chem. 2005;9:1431. [Google Scholar]; (c) Koskinen A, Lounasmaa M. Planta Med. 1982;45:248. doi: 10.1055/s-2007-971385. [DOI] [PubMed] [Google Scholar]
- 5).Lounasmaa M, Hanhinen P. The Ajmaline Group of Indole Alkaloids. In: Cordell G, editor. The Alkaloids. Vol. 55. Academic Press; San Diego, CA: 2001. pp. 1–87. [DOI] [PubMed] [Google Scholar]
- 6).(a) Wetzel S, Bon RS, Kumar K, Waldmann H. Angew. Chem. Int. Ed. 2011;50:10800. doi: 10.1002/anie.201007004. [DOI] [PubMed] [Google Scholar]; (b) Nören-Müller A, Reis-Corrêa JI, Prinz H, Rosenbaum C, Saxena K, Schwalbe H, Vestweber D, Cagna G, Schunk S, Schwarz O, Schiewe H, Waldmann H. Proc. Natl. Acad. Sci. USA. 2006;103:10606. doi: 10.1073/pnas.0601490103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7).(a) Nören-Müller A, Wilk W, Saxena K, Schwalbe H, Kaiser M, Waldmann H. Angew. Chem. Int. Ed. 2008;47:5973. doi: 10.1002/anie.200801566. [DOI] [PubMed] [Google Scholar]; (b) Wilk W, Nören-Müller A, Kaiser M, Waldmann H. Chem. Eur. J. 2009;15:11976. doi: 10.1002/chem.200901797. [DOI] [PubMed] [Google Scholar]
- 8).(a) Bialy L, Waldmann H. Angew. Chem. Int. Ed. 2005;44:3814. doi: 10.1002/anie.200461517. [DOI] [PubMed] [Google Scholar]; (b) Dye C, Scheele S, Dolin P, Pathania V, Raviglione MC. J. Am. Med. Assoc. 1999;282:677. doi: 10.1001/jama.282.7.677. [DOI] [PubMed] [Google Scholar]
- 9).Huang KC, Williams WM. The Pharmacology of Chinese Herbs. 2nd ed. CRC Press; Boca Raton, FL: 1998. Antihypertensive Herbs; pp. 86–89. [Google Scholar]
- 10).Lin M, Yang B, Yu D-Q. Acta Pharm. Sin. 1986;21:114. [PubMed] [Google Scholar]
- 11).(a) Orazi OO, Corral RA, Stoichevich ME. Can. J. Chemistry. 1966;44:1523. [Google Scholar]; (b) Lin M, Yu D, Liu X, Fu F, Zheng Q, He C, Bao G, Yu C. Acta Pharm. Sin. 1985;20:198. [PubMed] [Google Scholar]
- 12).(a) Kam T-S, Choo Y-M. Bisindole Alkaloids. In: Cordell GA, editor. The Alkaloids. Vol. 63. Academic Press; San Diego: 2006. pp. 182–345. [DOI] [PubMed] [Google Scholar]; (b) Cordell GA, Saxton JE. Indole Alkaloids. In: Manske RHF, Rodrigo RGA, editors. The Alkaloids. Vol. 20. Academic Press; New York: 1981. pp. 189–204. [Google Scholar]
- 13).Arbain D, Dachriyanus, Firmansyah, Sargent MV, Skelton BW, White AH. J. Chem. Soc., Perkin Trans. 1998;1:2537. [Google Scholar]
- 14).Mao L, Yu D-Q, Liu X, Fu F-Y, Zheng Q-T, He C-H, Hong BG, Xu C-F. Acta Pharm. Sin. 1985;20:203. [Google Scholar]
- 15).(a) Lin X, Zheng Q, Zhang Y. Jiegou Huaxue. 1987;6:89. [Google Scholar]; (b) Lin M, Yang BQ, Yu DQ, Lin XY, Zhang YJ. Acta Pharm. Sin. 1987;22:833. [PubMed] [Google Scholar]
- 16).(a) Tian B-H, Zeng G-Y. Acta Pharm. Sin. 1988;9:58. [Google Scholar]; (b) Zeng G-Y, Tian B-H. Acta Pharm. Sin. 1991;12:471. [Google Scholar]
- 17).Plat M, Lemay R, Le Men J, Janot M-M, Djerassi C, Budzikiewicz H. Bull. Soc. Chim. Fr. 1965:2497. [PubMed] [Google Scholar]
- 18).(a) Abaul J, Philogene E, Bourgeois P, Merault G, Poupat C, Ahond A, Potier P. J. Nat. Prod. 1986;49:829. [Google Scholar]; (b) Martínez JA, Valero R, Sosa ME, Manchúa M. Rev. Cubana Quim. 1992;6:48. [Google Scholar]; (c) Valencia A. E, Valenzuela V. E, Barros B. E, Zarraga O. M, Madinaveitia M. A, Gonzalez AG, Bermejo B. YJ. Bol. Soc. Chil. Quim. 1996;41:347. [Google Scholar]
- 19).Kiang AK, Wan ASC. J. Chem. Soc. 1960:1394. [Google Scholar]
- 20).Chen W-M, Yan Y-P, Wang Y-P, Liang X-T. Acta Pharm. Sin. 1985;20:906. [PubMed] [Google Scholar]
- 21).Banerji A, Chakrabarty M. Phytochemistry. 1974;13:2309. [Google Scholar]
- 22).(a) Arnold W, Berlage F, Bernauer H, Schmid H, Karrer P. Helv. Chim. Acta. 1958;41:1505. [Google Scholar]; (b) Karrer P, Schmid H. Helv. Chim. Acta. 1946;29:1853. [Google Scholar]; (c) Khan ZM, Hesse M, Schmid H. Helv. Chim. Acta. 1965;48:1957. doi: 10.1002/hlca.19670500403. [DOI] [PubMed] [Google Scholar]
- 23).Wang L, Zhang Y, He H-P, Zhang Q, Li S-F, Hao X-J. Planta Med. 2011;77:754. doi: 10.1055/s-0030-1250569. [DOI] [PubMed] [Google Scholar]
- 24).Inaba M, Nagashima K. Jpn. J. Cancer. Res. (Gann) 1986;77:197. [PubMed] [Google Scholar]
- 25).(a) Zaima K, Koga I, Iwasawa N, Hosoya T, Hirasawa Y, Kaneda T, Ismail IS, Lajis NH, Morita H. J. Nat. Med. 2013;67:9. doi: 10.1007/s11418-012-0638-y. [DOI] [PubMed] [Google Scholar]; (b) Svoboda GH, Blake DA. In: The Catharanthus Alkaloids. Taylor WI, Farnsworth NR, editors. Marcel Dekker; New York: 1974. p. 45. [Google Scholar]
- 26).Tan S-J, Lim K-H, Subramaniam G, Kam T-S. Phytochemistry. 2013;85:194. doi: 10.1016/j.phytochem.2012.08.016. [DOI] [PubMed] [Google Scholar]
- 27).(a) Edwankar CR, Edwankar RV, Deschamps JR, Cook JM. Angew. Chem. 2012;124:11932. doi: 10.1002/anie.201206015. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Edwankar CR, Edwankar RV, Deschamps JR, Cook JM. Angew. Chem. Int. Ed. 2012;51:11762. doi: 10.1002/anie.201206015. This article was featured in Synfacts 2013, 9, 0015. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Edwankar CR. Ph.D. Thesis. University of Wisconsin-Milwaukee; Milwaukee, WI: 2011. [Google Scholar]
- 28).(a) Edwankar CR, Edwankar RV, Rallapalli SK, Cook JM. Nat. Prod. Commun. 2008;3:1839. [Google Scholar]; (b) Lewis SE. Tetrahedron. 2006;62:8655. [Google Scholar]
- 29).(a) Wang W, Xiong C, Yang J, Hruby VJ. Tetrahedron Lett. 2001;42:7717. [Google Scholar]; (b) Yin W. Ph.D. Thesis. University of Wisconsin-Milwaukee; Milwaukee, WI: 2007. [Google Scholar]
- 30).Blaser G, Sanderson JM, Bastanov AS, Howard JAK. Tetrahedron Lett. 2008;49:2795. [Google Scholar]
- 31).Tamaka M, Hikawa H, Yokoyama Y. Tetrahedron. 2011;67:5897. [Google Scholar]
- 32).(a) Zhao S, Liao X, Wang T, Flippen-Anderson J, Cook JM. J. Org. Chem. 2003;68:6279. doi: 10.1021/jo030055u. [DOI] [PubMed] [Google Scholar]; (b) Liao X. Ph.D. Thesis. University of Wisconsin-Milwaukee; Milwaukee, WI: 2007. [Google Scholar]
- 33).Ma C, Liu X, Li X, Flippen-Anderson J, Yu S, Cook JM. J. Org. Chem. 2001;66:4525. doi: 10.1021/jo001679s. [DOI] [PubMed] [Google Scholar]
- 34).(a) Larock RC, Yum EK. J. Am. Chem. Soc. 1991;113:6689. [Google Scholar]; (b) Larock RC, Yum EK, Refvik MD. J. Org. Chem. 1998;63:7652. [Google Scholar]
- 35).(a) Yu J, Wearing XZ, Cook JM. J. Org. Chem. 2005;70:3963. doi: 10.1021/jo040282b. [DOI] [PubMed] [Google Scholar]; (b) Zhou H, Liao X, Cook JM. Org. Lett. 2004;6:1187. doi: 10.1021/ol0362212. [DOI] [PubMed] [Google Scholar]
- 36).Ma J, Yin W, Zhou H, Cook JM. Org. Lett. 2007;9:3491. doi: 10.1021/ol071220l. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37).(a) Schöllkopf U, Groth U, Deng C. Angew. Chem. 1981;93:793. [Google Scholar]; (b) Schöllkopf U. Pure. Appl. Chem. 1983;55:1799. [Google Scholar]; (c) Hamaker LK. Ph.D. Thesis. University of Wisconsin-Milwaukee; Milwaukee, WI: 1995. [Google Scholar]
- 38).(a) Kondo Y, Kojima S, Sakamoto T. J. Org. Chem. 1997;62:6507. [Google Scholar]; (b) Lizos D, Tripoli R, Murphy JA. Org. Biomol. Chem. 2003;1:117. doi: 10.1039/b208114h. [DOI] [PubMed] [Google Scholar]
- 39).Snieckus V. Chem. Rev. 1990;90:879. [Google Scholar]
- 40).Robinson B. The Fischer Indole Synthesis. John Wiley & Sons; New York: 1982. [Google Scholar]
- 41).Abramovitch RA, Shapiro DS. J. Chem. Soc., Perkin Trans. 1965;1:4589. [Google Scholar]
- 42).Zhang P, Liu R, Cook JM. Tetrahedron Lett. 1995;36:3103. [Google Scholar]
- 43).Bastiaans HMM, van der Baan JL, Ottenheijm HCJ. J. Org. Chem. 1997;62:3880. [Google Scholar]
- 44).(a) Lorenz M, Van Linn ML, Cook JM. Curr. Org. Synth. 2010;7:189. [Google Scholar]; (b) Cox ED, Cook JM. Chem. Rev. 1995;95:1797. [Google Scholar]; (c) Stöckigt J, Antonchick AP, Wu F-R, Waldmann H. Angew. Chem. Int. Ed. 2011;50:8538. doi: 10.1002/anie.201008071. [DOI] [PubMed] [Google Scholar]
- 45).Wang T, Cook JM. Org. Lett. 2000;2:2057. doi: 10.1021/ol000095+. [DOI] [PubMed] [Google Scholar]
- 46).Yang J, Rallapalli SK, Cook JM. Tetrahedron Lett. 2009;51:815. [Google Scholar]
- 47).Liao X, Zhou H, Yu J, Cook JM. J. Org. Chem. 2006;71:8884. doi: 10.1021/jo061652u. [DOI] [PubMed] [Google Scholar]
- 48).Solé D, Urbaneja X, Bonjoch J. Adv. Synth. Catal. 2004;346:1646. [Google Scholar]
- 49).Yamazaki N, Dokoshi W, Kibayashi C. Org. Lett. 2001;3:193. doi: 10.1021/ol0003228. [DOI] [PubMed] [Google Scholar]
- 50).Zhou H, Han D, Liao X, Cook JM. Tetrahedron Lett. 2005;46:4219. [Google Scholar]
- 51).(a) Bringmann G, Gulder T, Gulder TAM, Breuning M. Chem. Rev. 2011;111:563. doi: 10.1021/cr100155e. [DOI] [PubMed] [Google Scholar]; (b) Bringmann G, Price Mortimer AJ, Keller PA, Gresser MJ, Garner J, Breuning M. Angew. Chem. Int. Ed. 2005;44:5384. doi: 10.1002/anie.200462661. [DOI] [PubMed] [Google Scholar]
- 52).(a) Yeung CS, Dong VM. Chem. Rev. 2011;111:1215. doi: 10.1021/cr100280d. [DOI] [PubMed] [Google Scholar]; (b) Wallace TM. Org. Biomol. Chem. 2006;4:3197. doi: 10.1039/b608470m. [DOI] [PubMed] [Google Scholar]; (c) Cepanec I. Synthesis of Biaryls. 1st ed. Elsevier; San Diego: 2004. [Google Scholar]; (d) Hassan J, Sévignon M, Gozzi C, Schulz E, Lemaire M. Chem. Rev. 2002;102:1359. doi: 10.1021/cr000664r. [DOI] [PubMed] [Google Scholar]
- 53).(a) Ashenhurst JA. Chem. Soc. Rev. 2010;39:540. doi: 10.1039/b907809f. [DOI] [PubMed] [Google Scholar]; (b) Kozlowski MC, Morgan BJ, Linton EC. Chem. Soc. Rev. 2009;38:3193. doi: 10.1039/b821092f. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Bringmann G, Tasler S. Tetrahedron. 2001;57:331. [Google Scholar]
- 54).(a) Monnier F, Taillefer M. Angew. Chem. Int. Ed. 2008;47:3096. doi: 10.1002/anie.200703209. [DOI] [PubMed] [Google Scholar]; (b) Nelson TD, Crouch RD. Organic Reactions. Vol. 63. Wiley Interscience; New York: 2004. Cu, Ni, and Pd Mediated Homocoupling Reactions in Biaryl Syntheses: The Ullmann Reaction; pp. 265–555. [Google Scholar]; (c) Fuson RC, Cleveland EA. Vol. 3. Organic Syntheses; Wiley; New York: 1955. p. 339. Collect. [Google Scholar]
- 55).(a) Cammidge AN, Crepy KVL. Tetrahedron. 2004;60:4377. [Google Scholar]; (b) Castanet A-S, Colobert F, Broutin P-E, Obringer M. Tetrahedron Asymmetry. 2002;13:659. [Google Scholar]; (c) Miyaura N, Suzuki A. Chem. Rev. 1995;95:2457. [Google Scholar]; (d) Ishiyama T, Murata M, Miyaura N. J. Org. Chem. 1995;60:7508. [Google Scholar]; (e) Miyaura N, Yanagi T, Suzuki A. Synth. Commun. 1981;11:513. [Google Scholar]
- 56).(a) Bungard CJ, Morris JC. J. Org. Chem. 2006;71:7354. doi: 10.1021/jo0611364. [DOI] [PubMed] [Google Scholar]; (b) Su W, Urgaonkar S, McLaughlin PA, Verkade JG. J. Am. Chem. Soc. 2004;126:16433. doi: 10.1021/ja0450096. [DOI] [PubMed] [Google Scholar]
- 57).(a) Davies HML, Du Bois J, Yu J-Q. Chem. Soc. Rev. 2011;40:1855. doi: 10.1039/c1cs90010b. [DOI] [PubMed] [Google Scholar]; (b) McMurray L, O'Hara F, Gaunt MJ. Chem. Soc. Rev. 2011;40:1885. doi: 10.1039/c1cs15013h. [DOI] [PubMed] [Google Scholar]; (c) Gutekunst WR, Baran PS. Chem. Soc. Rev. 2011;40:1976. doi: 10.1039/c0cs00182a. [DOI] [PubMed] [Google Scholar]
- 58).(a) Keśeru GM, Nόgrádi M. Natural Products by Oxidative Phenolic Coupling Phytochemistry, Biosynthesis and Synthesis. In: Atta-ur-Rahman, editor. Studies in Natural Products Chemistry. Vol. 20. Elsevier; Amsterdam: 1997. pp. 263–322. [Google Scholar]; (b) Bringmann G, Gunther C, Ochse M, Schupp O, Tasler S. Prog. Chem. Org. Nat. Prod. 2001;82:1. doi: 10.1007/978-3-7091-6227-9_1. [DOI] [PubMed] [Google Scholar]; (c) Taylor WI, Battersby AR. Oxidative Coupling of Phenols. Dekker; New York: 1967. [Google Scholar]
- 59).(a) Toda F, Tanaka K, Iwata S. J. Org. Chem. 1989;54:3007. [Google Scholar]; (b) Lessene G, Feldman KS. Oxidative Aryl-Coupling Reactions in Synthesis. In: Astruc D, editor. Modern Arene Chemistry: Concepts, Synthesis, and Applications. Wiley-VCH; Mörlenbach: 2002. pp. 479–538. [Google Scholar]
- 60).(a) Scholl R, Mansfeld J. Ber. Dtsch. Chem. Ges. 1910;43:1734. [Google Scholar]; (b) Kovacic P, Jones MB. Chem. Rev. 1987;87:357. [Google Scholar]; (c) Scholl R, Seer C. Liebigs Ann. Chem. 1912;394:111. [Google Scholar]
- 61).(a) Watson MD, Fechtenkötter A, Mullen K. Chem. Rev. 2001;101:1267. doi: 10.1021/cr990322p. [DOI] [PubMed] [Google Scholar]; (b) Harvey RG. Polycylcic Aromatic Hydrocarbons. 1st ed. Wiley-VCH; New York: 1997. [Google Scholar]
- 62).Naarmann H, Hanack M, Mattmer R. Synthesis. 1994:477. [Google Scholar]
- 63).Boden N, Bushby RJ, Headdock G, Lozman OR, Wood A. Liq. Cryst. 2001;28:139. [Google Scholar]
- 64).(a) Kubel C, Eckhardt K, Enkelmann V, Wegner G, Müllen K. J. Mater. Chem. 2000;10:879. [Google Scholar]; (b) Simpson CD, Mattersteig G, Martin K, Gherghel L, Bauer RE, Räder HJ, Müllen K. J. Am. Chem. Soc. 2004;126:3139. doi: 10.1021/ja036732j. [DOI] [PubMed] [Google Scholar]
- 65).McKillop A, Turell AG, Young DW, Taylor EC. J. Am. Chem. Soc. 1980;102:6504. [Google Scholar]
- 66).Aylward JB. J. Chem. Soc. B. 1967:1268. [Google Scholar]
- 67).(a) Kramer B, Fröhlich R, Waldvogel SR. Eur. J. Org. Chem. 2003:3549. [Google Scholar]; (b) Kovacic P, Lange RM. J. Org. Chem. 1963;28:968. [Google Scholar]
- 68).Yamaguchi S, Swager TM. J. Am. Chem. Soc. 2001;123:12087. doi: 10.1021/ja016692o. [DOI] [PubMed] [Google Scholar]
- 69).Takada T, Arisawa M, Gyoten M, Hamada R, Tohma H, Kita Y. J. Org. Chem. 1998;63:7698. [Google Scholar]
- 70).Rathore R, Kochi JK. J. Org. Chem. 1995;60:7479. [Google Scholar]
- 71).(a) King BT, Kroulík J, Robertson CR, Rempala P, Hilton CL, Korinek JD, Gortari LM. J. Org. Chem. 2007;72:2279. doi: 10.1021/jo061515x. [DOI] [PubMed] [Google Scholar]; (b) Rempala P, Kroulík J, King BT. J. Am. Chem. Soc. 2004;126:15002. doi: 10.1021/ja046513d. [DOI] [PubMed] [Google Scholar]
- 72).(a) Merkushev EB. Synthesis. 1988:923. [Google Scholar]; (b) Stavber S, Jereb M, Zupan M. Synthesis. 2008:1487. [Google Scholar]; For reviews on iodination of organic compounds see:
- 73).Castanet A-S, Colobert F, Broutin P-E. Tetrahedron Lett. 2002;43:5047. [Google Scholar]
- 74).Moorthy JN, Senapati K, Kumar S. J. Org. Chem. 2009;74:6287. doi: 10.1021/jo9007892. [DOI] [PubMed] [Google Scholar]
- 75).Zhang S, Zhang D, Liebskind LS. J. Org. Chem. 1997;62:2312. doi: 10.1021/jo9700078. [DOI] [PubMed] [Google Scholar]
- 76).(a) Murata M, Watanabe S, Masuda Y. J. Org. Chem. 1997;62:6458. doi: 10.1021/jo971143f. [DOI] [PubMed] [Google Scholar]; (b) Murata M, Oyama T, Watanabe S, Masuda M. J. Org. Chem. 2000;65:164. doi: 10.1021/jo991337q. [DOI] [PubMed] [Google Scholar]
- 77).Billingsley KL, Buchwald SL. J. Org. Chem. 2008;73:5589. doi: 10.1021/jo800727s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78).Baudoin O, Guénard D, Guéritte F. J. Org. Chem. 2000;65:9268. doi: 10.1021/jo005663d. [DOI] [PubMed] [Google Scholar]
- 79).(a) Barder TE, Walker SD, Martinelli JR, Buchwald SL. J. Am. Chem. Soc. 2005;127:4685. doi: 10.1021/ja042491j. [DOI] [PubMed] [Google Scholar]; (b) Martin R, Buchwald SL. Acc. Chem. Res. 2008;41:1461. doi: 10.1021/ar800036s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80).(a) Dohi T, Ito M, Yamaoka N, Morimoto K, Fujioka H, Kita Y. Tetrahedron. 2009;65:10797. [Google Scholar]; (b) Tohma H, Morioka H, Takizawa S, Arisawa M, Kita Y. Tetrahedron. 2001;57:345. [Google Scholar]; (c) Kita Y, Tohma H, Hatanaka K, Takada T, Fujita S, Mitoh S, Sakurai H, Oka S. J. Am. Chem. Soc. 1994;116:3684. [Google Scholar]
- 81).(a) McKillop A, Taylor EC. Recent Advances in Organothallium Chemistry. In: Stone FGA, West R, editors. Advances in Organometallic Chemistry. Vol. 11. Academic Science; New York: 1973. pp. 147–206. [Google Scholar]; (b) Taylor EC, McKillop A. Acc. Chem. Res. 1970;3:338. [Google Scholar]
- 82).Churucca F, SanMartin R, Carril M, Urtiaga MK, Solans X, Tellitu I, Domínguez E. J. Org. Chem. 2005;70:3178. doi: 10.1021/jo0501036. [DOI] [PubMed] [Google Scholar]
- 83).Faggi E, Sebastián RM, Pleixats R, Vallribera A, Shafir A, Rodríguez- Gimeno A, Ramírez de Arellano C. J. Am. Chem. Soc. 2010;132:17980. doi: 10.1021/ja107467c. [DOI] [PubMed] [Google Scholar]
- 84).(a) Adesomojou AA, Davis WA, Rajaraman R, Pelletier JC, Cava MP. J. Org. Chem. 1984;49:3220. [Google Scholar]; (b) Schwartz MA, Rose BF, Vishnuvajjala B. J. Am. Chem. Soc. 1973;95:612. [Google Scholar]
- 85).(a) Magnus P, Schultz J, Gallagher T. J. Am. Chem. Soc. 1985;107:4984. [Google Scholar]; (b) Magnus P, Schultz J, Gallagher T. J. Chem. Soc., Chem. Commun. 1984:1179. [Google Scholar]
- 86).Sawyer JS, Macdonald TL. Tetrahedron Lett. 1988;29:4839. [Google Scholar]
- 87).Taylor EC, Andrade JG, Rall GJH, McKillop A. J. Am. Chem. Soc. 1980;102:6513. [Google Scholar]
- 88).Banerji A, Ray R, Pal SC, Banerji D, Maiti KK. J. Indian Chem. Soc. 1998;75:698. [Google Scholar]
- 89).Tholander J, Bergman J. Tetrahedron. 1999;55:12595. [Google Scholar]
- 90).Keller PA, Yepuri NR, Kelso MJ, Mariani M, Skelton BW, White AH. Tetrahedron. 2008;64:7787. [Google Scholar]
- 91).(a) Lau W, Kochi JK. J. Am. Chem. Soc. 1984;106:7100. [Google Scholar]; (b) Elson IH, Kochi JK. J. Am. Chem. Soc. 1973;95:5060. [Google Scholar]
- 92).A request was made for an authentic sample of natural dispegatrine (1) to the isolation chemists
- 93).(a) Hirasawa Y, Hara M, Nugroho AE, Sugai M, Zaima K, Kawahara N, Goda Y, Awang K, Hadi AHA, Litaudon M, Morita H. J. Org. Chem. 2010;75:4218. doi: 10.1021/jo1006762. [DOI] [PubMed] [Google Scholar]; (b) Tatsuta K, Yamazaki T, Mase T, Yoshimoto T. Tetrahedron Lett. 1998;39:1771. [Google Scholar]; (c) Fukuyama Y, Asakawa Y. J. Chem. Soc., Perkin Trans. 1991;1:2737. [Google Scholar]; For examples of homo- and hetero-atropodiastereomeric natural products that have almost identical 1H NMR and 13C NMR spectra see:
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.