

Abstract
Intravenous immunoglobulin (IVIg) is used to treat autoimmune and systemic inflammatory diseases caused by derailment of humoral and cellular immunity. In this study we investigated whether IVIg treatment can modulate regulatory T cells (Tregs) in humans in vivo. Blood was collected from IVIg-treated patients with immunodeficiency or autoimmune disease who were treated with low-dose (n = 12) or high-dose (n = 15) IVIg before, immediately after and at 7 days after treatment. Percentages and activation status of circulating CD4+CD25+forkhead box protein 3 (FoxP3+) Tregs and of conventional CD4+FoxP3− T-helper cells (Tconv) were measured. The suppressive capacity of Tregs purified from blood collected at the time-points indicated was determined in an ex-vivo assay. High-dose, but not low-dose, IVIg treatment enhanced the activation status of circulating Tregs, as shown by increased FoxP3 and human leucocyte antigen D-related (HLA-DR) expression, while numbers of circulating Tregs remained unchanged. The enhanced activation was sustained for at least 7 days after infusion, and the suppressive capacity of purified Tregs was increased from 41 to 70% at day 7 after IVIg treatment. The activation status of Tconv was not affected by IVIg. We conclude that high-dose IVIg treatment activates Tregs selectively and enhances their suppressive function in humans in vivo. This effect may be one of the mechanisms by which IVIg restores imbalanced immune homeostasis in patients with autoimmune and systemic inflammatory disorders.
Keywords: human studies, intravenous immunoglobulin, IVIg, regulatory T cells
Introduction
Intravenous immunoglobulin (IVIg) was introduced initially as a replacement therapy for patients with immune deficiencies, but high-dose IVIg is now used widely for the treatment of autoimmune and systemic inflammatory diseases caused by autoantibodies and/or derailment of the cellular immune system 1,2. Moreover, IVIg has shown efficacy in prevention and treatment of organ allograft rejection and graft-versus-host disease (GVHD) 3–6. Several possible mechanisms of action that explain the beneficial effects of high-dose IVIg in autoantibody- and immune complex-mediated diseases have been unravelled during recent decades. These mechanisms include inhibition of the binding of immune complexes or cell-bound immunoglobulin (Ig)G to activating Fcγ receptors, saturation of the neonatal FcR resulting in an enhanced clearance of autoantibodies and up-regulation of the inhibitory FcγRIIb 1,2,7. However, the mechanisms by which high-dose IVIg is effective in diseases which are caused mainly by cell-mediated immune responses, such as Kawasaki disease, dermatomyositis, GVHD and acute transplant rejection, have not yet been elucidated fully. Recent in-vitro and mice studies suggest that one of the mechanisms by which IVIg suppresses cellular immunity is by activating CD4+CD25+forkhead box protein 3 (FoxP3+) regulatory T cells (Tregs) 8,9.
Tregs are critical regulators of cell-mediated immune responses and suppress pathogenic immune responses in autoimmune diseases, transplantation and GVHD 10. Current immunosuppressive drugs used to treat or to prevent these diseases exert generalized inhibition of the immune system, thereby disabling protective immunity against pathogens and malignancies. Therapeutic modalities that stimulate Treg-mediated immune regulation without affecting global immune functions are attractive. Recently, we found that IVIg can activate both human and mouse CD4+CD25+FoxP3+ Tregs in vitro and increase their ability to suppress allogeneic T cell proliferation 11. By triggering functional activation of Tregs, IVIg prevented graft rejection in a fully mismatched skin transplant model. In line with our data, it has been shown that IVIg can prevent mice from developing experimental autoimmune encephalomyelitis 12 and herpes simplex virus-induced encephalitis 13 by enhancing the suppressive capacity and stimulating the expansion of Tregs. In addition, IVIg was found to enhance the suppressive capacity of human Tregs in vitro 14.
These data suggest that IVIg treatment can stimulate Treg expansion and function, and may therefore be an attractive tool to re-establish Treg-mediated immune regulation. However, the data on the effects of IVIg on Tregs are restricted to in-vitro and mice experiments and it is unknown whether IVIg treatment actually affects Treg expansion and function in patients. Therefore, in the present study we analysed systematically the effects of either low- or high-dose IVIg treatment on the percentages, activation status and suppressive capacity of circulating Tregs in autoimmune and immunodeficient patients. We observed increased activation of Tregs after high-dose IVIg treatment, which was not observed for conventional T cells (Tconv). Additionally, Tregs isolated after high-dose IVIg treatment showed enhanced suppressive capacity ex vivo.
Material and methods
Patients
Twenty-seven patients (21 female/six male) who were treated with IVIg were included in this study. To assess whether IVIg had a dose-dependent effect on Treg activation, we subdivided the patients into two groups: those who received ‘low-dose’ IVIg and those who received ‘high-dose’ IVIg. Because patients with immunodeficiency started initially with a supplementary dose of 0·4–0·6 g/kg, we defined ‘low-dose’ IVIg as a dose ≤0·6 g/kg and ‘high-dose’ IVIg as a dose >0·6 g/kg. The median dose of IVIg received by low-dose patients was 0·39 g/kg (range 0·25–0·59) and that of high-dose patients was 0·98 g/kg (range 0·65–1·71). High-dose IVIg-treated patients showed a significantly higher rise in plasma IgG levels (+17·3 mg/ml) compared to low-dose IVIg-treated patients (+8·2 mg/ml) immediately after treatment (P < 0·001), as quantified by immunoturbometric analysis (Roche Diagnostics GmbH, Mannheim, Germany). At day 7, plasma IgG levels dropped again in both groups (data not shown). The indications for IVIg treatment in these patients are depicted in Table 1a, b. In the high-dose treatment group (Table 2), five patients received IVIg for licensed indications, including common variable immunodeficiency (n = 2), hypogammaglobulinaemia (n = 1) and idiopathic thrombocytopenic purpura (n = 2). One patient with Good's syndrome received high-dose IVIg as supplementary therapy for hypogammaglobulinaemia. Other patients included in Table 2 received high-dose IVIg as anti-inflammatory therapy ‘off-label’, as they did not respond to conventional immunosuppressive treatment. The IVIg preparations used for treatment were Nanogam® (n = 13; Sanquin, Amsterdam, the Netherlands), Kiovig® (n = 9; Baxter, Deerfield, IL, USA), Flebogamma® (n = 4; Grifols, Barcolona, Spain) and Octagam® (n = 1; Octapharma, Lachen, Switzerland). With the exception of three patients, all patients had been treated previously with IVIg, with an average of 28 days (range 21–35 days) before this study. Twenty-one patients were receiving IVIg monotherapy and six patients received additional corticosteroid treatment. All patients showed clinical improvement after treatment.
Table 1a.
Patient characteristics in low-dose intravenous immunoglobulin (IVIg)-treated patients
| IVIg treatment indication | No. of patients | Age in years, median (range) | IVIg dose in g/kg, median (range) |
|---|---|---|---|
| Common variable immunodeficiency | 5 | 41 (20–57) | 0·38 (0·30–0·48) |
| Hypogammaglobulinaemia | 6 | 60 (45–77) | 0·39 (0·25–0·59) |
| Agammaglobulinaemia | 1 | 30 | 0·25 |
Table 1b.
Patient characteristics in high-dose intravenous immunoglobulin (IVIg)-treated patients
| IVIg treatment indication | No. of patients | Age in years, median (range) | IVIg dose in g/kg, median (range) |
|---|---|---|---|
| Polymyositis† | 3 | 41 (33–56) | 1·00 (0·67–1·67) |
| Common variable immunodeficiency | 2 | 40 (37–43) | 0·68 (0·65–0·70) |
| Immune thrombocytopenic purpera | 2 | 68 (65–71) | 1·00 |
| Acquired von Willebrand syndrome† | 2 | 52 (40–63) | 0·95 (0·93–0·98) |
| Hypogammaglobulinaemia | 1 | 40 | 0·66 |
| Good's syndrome/hypogammaglobulinaemia | 1 | 69 | 0·86 |
| Polyserositis e.c.i. | 1 | 24 | 0·78 |
| Polychondritis† | 1 | 68 | 0·69 |
| Dermatomyositis† | 1 | 46 | 1·71 |
| Systemic vasculitis e.c.i.† | 1 | 64 | 1·4 |
IVIg treatment was indicated based on unresponsiveness to conventional treatment.
e.c.i., e causa ignota.
Ethics statement
The medical ethics committee of the Erasmus University Medical Centre approved the study, and written informed consent was obtained from all patients participating in the study.
Sample collection and preparation
Heparin-decoagulated blood samples were collected from healthy blood bank donors and from patients immediately before IVIg infusion immediately after IVIg infusion (4–24 h after the start of the infusion) and 7 days after infusion. Due to practical issues, we were unable to obtain blood samples from one patient immediately after IVIg treatment and from two patients 7 days after treatment. Plasma and peripheral blood mononuclear cells (PBMCs) were isolated from whole blood by density gradient sedimentation using Ficoll-Paque (GE Healthcare, Uppsala, Sweden). Plasma was collected from the gradient and centrifuged at 1157 g for 10 min at 4°C to remove thrombocytes and debris. For storage of PBMCs, the cells were cryopreserved in RPMI-1640 (Gibco BRL Life Technologies, Breda, the Netherlands) supplemented with 20% heat-inactivated fetal bovine serum (Hyclone, Logan, UT, USA) and 10% dimethylsulphoxide (DMSO; Sigma-Aldrich, St Louis, MO, USA). Until further analysis, the PBMC samples were stored at −135°C and plasma at −80°C. To minimize the possible effects of interassay variation, measurements in plasma and on PBMC obtained at different time-points from the same patient were performed on the same day.
Antibodies and flow cytometry
For the dentification of T cell subsets, PBMCs were stained with anti-CD3-AmCyan, anti-CD4-allophycocyanin (APC)-H7 and CD8-Pacific blue (all from BD Biosciences, San Jose, CA, USA). Percentages and activation status of Tregs were determined by surface labelling with anti-CD4-APC-H7, anti-CD25-phycoerythrin (PE)-cyanin 7 (Cy7) (both from BD Biosciences), anti-CD127-PE (Beckman Coulter, Immunotech, Marseille, France), anti-CD38-Pacific Blue (ExBio antibodies, Praha, Czech Republic), anti-human leucocyte antigen D-related (HLA-DR)-peridinin chlorophyll (PerCP) (eBioscience, San Diego, CA, USA) monoclonal antibody (mAb) and proper isotype controls. For the detection of memory (CD45RO+) Tregs, we additionally labelled the cell surface using CD45RO-fluorescein isothiocyanate (FITC) mAb (Beckman Coulter). Cells (1 × 106) were incubated with the mAb in 50 μl phosphate-buffered saline (PBS) (Lonza, Verviers, Belgium) for 30 min on ice and protected from light. Subsequently, intracellular FoxP3+ staining was performed using anti- FoxP3–APC mAb (or rat IgG2a isotype control mAb) purchased from eBioscience, according to the manufacturer's instructions. To study proliferation, intracellular labelling with anti-Ki-67-FITC (or mIgG1 isotype control mAb) from BD Biosciences was performed. After staining, the cells were washed and resuspended in 100 μl PBS for measurement by flow cytometry (FACS Canto, BD Biosciences, San Jose, CA, USA). A minimum of 1·5 × 105 mononuclear cells (MNC) were acquired. Analyses were performed by fluorescence activated cell sorter (FACS) Diva software (BD Bioscience). Viable MNC were gated based on forward-/side-scatter. For the calculation of percentages of cells expressing a certain marker, or for calculation of the geometric mean of fluorescence intensities (MFI), fluorescence of isotype-matched control mAb was subtracted from fluorescence of specific mAb.
Suppression assay
To determine the effect of IVIg treatment on the suppressive capacity of Tregs ex vivo, we collected additional blood from seven patients in the study who received high-dose IVIg. To purify Tregs and T responder cells (Tresp), we first purified non-touched CD3+ cells from thawed PBMCs from each time-point by performing immunomagnetic depletion of non-T cells using PE-conjugated antibodies against BDCA1 (Miltenyi Biotec, Bergisch Gladbach, Germany), CD14, CD56 (both from BD Bioscience) CD19, CD56, CD123 (all from Beckman Coulter) as well as CD15- and CD235-microbeads (both from Miltenyi Biotec). The purified CD3+ cells were then stained with anti-CD3-AmCyan, anti-CD4-APC-H7, anti-CD25-APC, 7-aminoactinomycin D (7-AAD) (all from BD Bioscience) and anti-CD127-PE mAb (Beckman Coulter Coulter). Subsequently, Tregs were obtained by flow cytometric sorting 7-AAD–CD3+CD4+CD127–CD25+ cells using FACS Aria (BD Biosciences). Purity of the Tregs (defined as CD4+CD25+CD127–), as determined by flow cytometry, was 98±1%. Tresp were obtained by sorting both 7-AAD-CD3+CD4– and 7-AAD–CD3+CD4+CD127+CD25– cells, which resulted in a purity of 99±0·4% (defined as CD3+CD127+CD25– cells), as determined by flow cytometry. Cells were recovered after cell sorting by incubation in culture medium [RPMI-1640 supplemented with penicillin (100 U/ml), streptomycin (100 μg/ml) and 10% fetal bovine serum (Hyclone, Logan, UT, USA)] at 37°C for at least 3 h prior to co-culture.
To compare their suppressive capacity, Tregs purified from the patient blood collected before IVIg treatment, immediately after treatment and at day 7 after treatment were co-cultured with Tresp (5 × 104) obtained before treatment that were stimulated with 1 μg/ml phytohaemagglutinin (PHA) in the presence of 40 Gy-irradiated autologous PBMC (5 × 104) at Treg : Tresp ratios of 1:4 in culture medium in round-bottomed 96-well plates (Greiner, Alphen a/d Rijn, the Netherlands). After 5 days, supernatants were collected and the cumulative interferon (IFN)-γ secretion by Tresp was quantified by enzyme-linked immunosorbent assay (ELISA) (Ready-SET-Go®; eBioscience) according to the manufacturer's instructions. Each condition was tested at least in triplicate, from which means were calculated. These means were used to calculate the percentage of suppression using the formula: (IFN-γ levelTresp–IFN-γ levelTresp+Treg)/IFN-γ levelTresp × 100%.
Statistical analyses
Differences in measured variables between time-points before and after IVIg treatment were analysed pairwise using the Wilcoxon signed-rank test. For analysis of the Treg suppression assay we used the paired Student's t-test, as the differences in IFN-γ production by Tresp collected at different time-points before and after IVIg treatment were distributed normally. Two-sided P-values < 0·05 were considered significant. Statistical analysis was performed using GraphPad Prism version 5·0 (GraphPad Software, San Diego, CA, USA). In accordance with the statistical test used, all data presented in dot-plots show median values, but data on the suppressive capacity of Treg (Fig. 2b) are depicted as means ± standard error of the mean (s.e.m.).
Fig. 2.
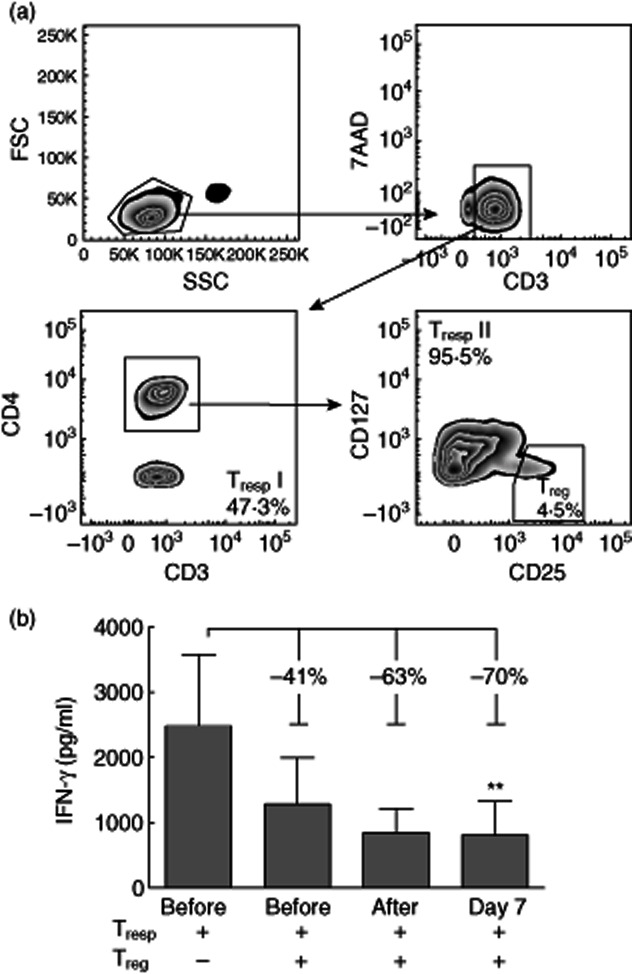
Enhanced suppressive activity of regulatory T cells (Tregs) purified from blood after intravenous immunoglobulin (IVIg) treatment. (a) Regulatory T cells (Tregs) were obtained by flow cytometric sorting on 7-aminoactinomycin D (7-AAD)–CD3+CD4+CD127–CD25+ cells (Tregs) from peripheral blood mononuclear cells (PBMC) collected from seven high-dose IVIg-treated patients before treatment, immediately after treatment and at day 7 after treatment, and T responder cells (Tresp) were obtained by sorting both 7-AAD–CD3+CD4– (Tresp I) and 7-AAD–CD3+CD4+CD127+CD25– (Tresp II) cells from autologous PBMCs collected before IVIg-treatment. (b) Tregs purified from the patients before, after and at day 7 after treatment were co-cultured with Tresp (5 × 104) obtained before treatment that were stimulated with 1 μg/ml phytohaemagglutinin (PHA) in the presence of 40 Gy-irradiated autologous PBMCs (5 × 104) at a Treg : Tresp ratio of 1:4 in culture medium in round-bottomed 96-well plates. After 5 days, supernatants were collected and the cumulative interferon (IFN)-γ production by Tresp was quantified by enzyme-linked immunosorbent assay. Each condition was tested at least in triplicate from which means were calculated. From each patient, these means were used to calculate the percentage of suppression using the formula: (IFN-γ levelTresp–IFN-γ levelTresp–Treg)/IFN-γ levelTresp × 100%. Bar graphs represent mean ± standard error of the mean. **P < 0·01, compared to Tresp alone.
Results
IVIg enhances selectively the activation status of Tregs in vivo
IVIg treatment had no effect on the numbers of circulating CD4+ and CD8+ T cells (Fig. 1a). To study the effects of IVIg treatment on Tregs, we measured the percentages and immunophenotype of circulating Tregs. The gating strategy used to determine CD4+CD25+FoxP3+ cells is shown in Fig. 1b. At baseline, we observed comparable percentages of circulating Tregs in patients who were treated with low-dose IVIg (n = 12) or high-dose IVIg (n = 15) and in healthy controls (n = 10) (Fig. 1c). In addition, baseline activation status of the Tregs in the low- and high-dose patients were comparable to healthy controls, as assessed by expression levels of FoxP3 (Fig. 1d) and HLA-DR (Fig. 1e).
Fig. 1.
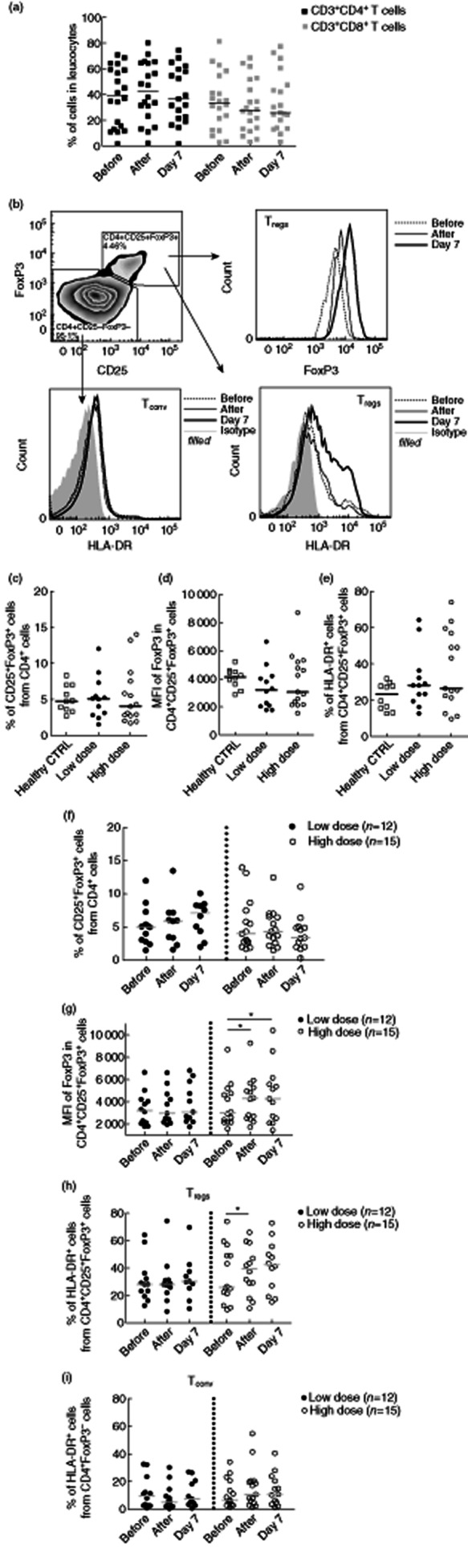
Intravenous immunoglobulin (IVIg) treatment selectively activates circulating regulatory T cells (Tregs), but not conventional T helper cells (Tconv). (a) Percentages of CD3+CD4+ and CD3+CD8+ in peripheral blood mononuclear cells (PBMCs) did not alter after IVIg treatment. (b) Density plot shows the gating strategy of Tregs and Tconv after gating on CD4+ cells. Histograms show intracellular forkhead box protein 3 (FoxP3) expression in Tregs and surface expression of human leucocyte antigen D-related (HLA-DR) on Tregs and Tconv obtained before, immediately after and at day 7 after IVIg treatment of a representative patient. (c) Baseline percentages of CD25+FoxP3+ Tregs within circulating CD4+ T cells, (d) mean fluorescence intensity (MFI) of FoxP3 in Tregs and (e) percentages of Tregs expressing HLA-DR from healthy blood donors (n = 10) and patients treated with low-dose IVIg (n = 12) or high-dose IVIg (n = 15). (f) Percentages of CD25+FoxP3+ Tregs within the CD4+ population before, immediately after and 7 days after IVIg treatment in patients who received low-dose (n = 12) or high-dose IVIg (n = 15). (g) MFI of FoxP3 in Tregs, (h) percentages of Tregs and (i) percentages of Tconv expressing HLA-DR before, immediately after and 7 days after IVIg treatment in patients who received low-dose (n = 12) or high-dose IVIg treatment (n = 15). Horizontal lines represent median. *P < 0·05.
IVIg treatment had no effect on the percentages of circulating Tregs in both low- and high-dose IVIg-treated patients (Fig. 1f). Although low-dose patients showed a slight increase of Treg percentages, this increase was not significant. Additionally, Ki-67 staining in Tregs showed no increased proliferation of Tregs after IVIg treatment in both groups (data not shown). However, in patients treated with high-dose IVIg, we observed an increased expression level of FoxP3 in CD4+CD25+FoxP3+ Tregs immediately after IVIg treatment, with an average increase of 29% (Fig. 1b,g, P = 0·04). This increased FoxP3 expression level was sustained on day 7 after infusion (+35%, P = 0·02).
Next, we analysed the expression of the activation marker HLA-DR on CD4+CD25+FoxP3+ Treg, gated as shown in Fig. 1b. We found that the proportions of HLA-DR expressing Treg increased gradually after IVIg treatment, again only in high-dose-treated patients, and was 37% higher immediately after (P = 0·02) and 39% at day 7 after treatment (P = 0·1) (Fig. 1b,h). Prednisone treatment did not influence the expression of FoxP3 and HLA-DR, as all six patients who were co-treated with prednisone (low-dose n = 3, high-dose n = 3) showed the same patterns as patients without prednisone treatment (data not shown). In addition, the same trends of Treg activation were found in patients with autoimmune disease and immunodeficient patients who were treated with high-dose IVIg (Supplementary Fig. S1).
To determine whether IVIg also affects the activation status of Tconv, we measured HLA-DR expression on CD4+FoxP3– Tconv (Fig. 1b,i). Compellingly, HLA-DR expression remained low on Tconv after IVIg treatment, even in high-dose IVIg-treated patients, which may suggest that IVIg treatment selectively activates CD4+CD25+FoxP3+ Tregs, but not CD4+FoxP3– Tconv in humans in vivo.
To study the effect of IVIg on the differentiation stage of Tregs, we measured CD45RO expression on Tregs. We found no effect of IVIg treatment on numbers of CD45RO+ Tregs in peripheral blood in both groups, suggesting that IVIg did not drive conversion from naive into memory Tregs within the time-frame of the study (data not shown).
IVIg increases the suppressive capacity of Tregs ex vivo
To investigate whether or not IVIg treatment stimulates the suppressive capacity of Tregs, we isolated CD3+CD4+CD25+CD127– Tregs and autologous CD3+CD25–CD127+ Tresp from PBMCs of seven patients who received high-dose IVIg that were collected before, after and at day 7 after IVIg treatment. The gating strategy for flow cytometric purification of these cells is depicted in Fig. 2a. Tresp isolated from blood collected before IVIg infusion were stimulated with PHA and co-cultured with Tregs purified from blood samples collected before, after and at day 7 after treatment in a Treg : Tresp ratio of 1:4. After 5 days of culture, cumulative IFN-γ production by Tresp was determined. Tregs that were isolated before treatment showed suppression of IFN-γproduction of 41%, while Tregs isolated at day 7 demonstrated a suppression of 70% (P = 0·001, Fig. 2b). Thus, in accordance with the observed increased Treg activation status at day 7 after IVIg treatment, Tregs isolated at 7 days also showed a higher suppressive capacity. These data suggest that IVIg treatment may stimulate the suppressive function of circulating Tregs for up to at least 1 week after treatment.
Discussion
Multiple ongoing studies are being performed to induce Treg expansion or stimulate Treg function in order to control T cell-mediated disorders. Interestingly, the current study suggests that high-dose IVIg treatment may be useful to enhance Treg activation and suppressive function in vivo. We show that IVIg treatment enhances both the activation status and suppressive function of Tregs in patients. Upon high-dose IVIg treatment, a gradual rise in FoxP3 and HLA-DR expression in circulating CD4+CD25+FoxP3+ Tregs was found, with the highest expression on day 7 after infusion. This finding indicates that IVIg treatment activates Tregs for at least 1 week, when IVIg levels already declined. Treg activation was clearly dependent upon the IVIg dose, as patients who received low-dose IVIg (≤0·6 g/kg) did not show significant effects. Activation of Treg was not restricted to patients with autoimmune disease, as immunodeficient patients who received high-dose IVIg also showed a rise in FoxP3 and HLA-DR expression on Tregs. Thus, our data support the generally accepted idea that a high-dose treatment regimen is required in order to gain anti-inflammatory activity 7.
Our ex-vivo functional assays showed that IVIg treatment may not only stimulate the activation status, but may also enhance the suppressive function of circulating Tregs. We observed an increased suppressive function of Tregs that was highest on day 7 after IVIg treatment, which is in line with the time-dependent increase of the activation status after treatment. Supporting our data, previous studies have shown that Tregs with a higher HLA-DR 11 and FoxP3 15,16 expression exert higher suppressive capacity. Further studies are warranted to confirm this finding, as the suppressive function was studied in a small number of patients. Moreover, it will be extremely interesting to monitor the activation status and suppressive function of Tregs after high-dose IVIg therapy over a longer time-period to study the durability of this effect.
In order to assess the suppressive capacity of Tregs, we did not use proliferation as the classical read-out, as interleukin (IL)-2 produced by Tresp in the co-culture system may influence proliferation of Tregs, hence affecting overall [3H]-thymidine incorporation 17. Therefore, we analysed IFN-γ secretion by Tresp, which is independent of the extent of Tregs hyporesponsiveness and may therefore be more unbiased.
While a few previous studies have shown an increase of circulating Treg numbers in patients treated with IVIg 18–21, we show an increase in both the activation status and functional suppressive capacity of Treg upon high-dose IVIg therapy in humans in vivo. Although the effects are moderate this finding may be of importance, as in several autoimmune diseases the numbers of Tregs are normal, yet these cells display a functional defect 22–24. A possible explanation for the fact that we did not find an increase in the circulating Treg numbers after IVIg treatment is that our patients did not have a deficit of Treg numbers compared to healthy subjects (Fig. 1c), while the previous studies included patients with acute inflammatory diseases which had decreased numbers of circulating Tregs before IVIg treatment 18–20.
Compellingly, high-dose IVIg treatment selectively activated CD4+CD25+FoxP3+ Tregs, but not CD4+FoxP3– Tconv. This observation may be interesting for the clinic, as conventional immunosuppressive drugs used for treating autoimmune diseases and allograft rejection, in particular calcineurin inhibitors, do not only suppress Tconv, but also Tregs 25–32. By selective activation of CD4+CD25+FoxP3+ Tregs, but not Tconv, IVIg may be superior to classical immunosuppressive drugs. Similar observations were also found in vitro and in mice models 11,12,14. In addition, IVIg selectivity for Tregs has also been suggested by Maddur and colleagues 33, who showed that IVIg inhibited T helper type 17 (Th17) cell differentiation and amplification, while it increased the numbers of Tregs among memory T cells.
The mechanism by which IVIg selectively activates Tregs is still unclear. Our previous data 11, together with the study by Ephrem et al. 12, suggest that the activation may be mediated by direct binding of IVIg to an unknown ‘IVIg receptor’ present on Tregs but not on Tconv. Because we observed that human Tregs do not express classical Fcγ receptors 11, this receptor must be a non-classical IgG-binding molecule. Alternatively, IVIg may bind to specific surface receptors by its F(ab′)2 fragment, as suggested by Maddur et al. 33, or IVIg may contain Treg epitopes that can activate Tregs by presentation on major histocompatibility complex (MHC) II+ antigen-presenting cells towards Tregs, as proposed by De Groot and colleagues 34,35.
The current study focused upon patients treated with IVIg for different indications. Although the underlying diseases are variable, the positive effect of high-dose IVIg on Treg activation is seen consistently in patients treated with high-dose IVIg. These results suggest that the effects of IVIg on Treg activation are not restricted to a certain disease, but to high-dose treatment. It would, however, be interesting to confirm the dose-dependent effect of IVIg in patients with the same treatment indication in the future.
In summary, we have demonstrated that high-dose IVIg therapy in humans in vivo can activate Tregs selectively and enhance their suppressive capacity. As the effects in our patient cohort were moderate, we propose that Treg stimulation may be one of the mechanisms by which IVIg controls inflammation in patients with autoimmune and systemic inflammatory disorders, but other mechanisms are probably also required to restore imbalanced immune homeostasis. The data presented in this study underline the multi-faceted character of the immunomodulatory mechanisms of IVIg.
Acknowledgments
The authors thank the Netherlands Organization for Scientific Research (NWO 017.007.055) for granting a Mosaic grant to A.S.W. Tjon and Biotest Pharma (Dreieich, Germany) for granting an unrestricted grant to J. Kwekkeboom, M. van der Ent (Department of Internal Medicine) and P. Matthijssen (Department of Hematology) for their contribution to collection of the patient blood samples, Dr N. Litjens (Laboratory of Transplant Immunology, Department of Internal Medicine) for her technical advice on the Treg suppression assays, S. Mancham and H. Jaadar for their technical support, M. van der Heide and Y. Liu for flow cytometric sorting and Dr B.E. Hansen for statistical advices (all from the Department of Gastroenterology and Hepatology).
Disclosure
The authors declare no financial or commercial conflicts of interest.
Supporting information
Additional Supporting Information may be found in the online version of this article at the publisher's website:
Fig. S1. Regulatory T cell (Treg) activation status after high-dose intravenous immunoglobulin (IVIg) treatment. Mean fluorescence intensity (MFI) of forkhead box protein 3 (FoxP3) expression in Tregs from (a) autoimmune disease patients and (b) immunodeficient patients and percentages of human leucocyte antigen D-related (HLA-DR) expression on Tregs in (c) autoimmune disease patients and (d) immunodeficient patients that are obtained before, immediately after and at day 7 after IVIg infusion. Horizontal lines represent median; n.s.: non-significant.
References
- 1.Kazatchkine MD, Kaveri SV. Immunomodulation of autoimmune and inflammatory diseases with intravenous immune globulin. N Engl J Med. 2001;345:747–755. doi: 10.1056/NEJMra993360. [DOI] [PubMed] [Google Scholar]
- 2.Negi VS, Elluru S, Siberil S, et al. Intravenous immunoglobulin: an update on the clinical use and mechanisms of action. J Clin Immunol. 2007;27:233–245. doi: 10.1007/s10875-007-9088-9. [DOI] [PubMed] [Google Scholar]
- 3.Kwekkeboom J, Tha-In T, Tra WM, et al. Hepatitis B immunoglobulins inhibit dendritic cells and T cells and protect against acute rejection after liver transplantation. Am J Transplant. 2005;5:2393–2402. doi: 10.1111/j.1600-6143.2005.01029.x. [DOI] [PubMed] [Google Scholar]
- 4.Bucuvalas JC, Anand R Studies of Pediatric Liver Transplantation Research G. Treatment with immunoglobulin improves outcome for pediatric liver transplant recipients. Liver Transpl. 2009;15:1564–1569. doi: 10.1002/lt.21843. [DOI] [PubMed] [Google Scholar]
- 5.Casadei DH, del C Rial M, Opelz G, et al. A randomized and prospective study comparing treatment with high-dose intravenous immunoglobulin with monoclonal antibodies for rescue of kidney grafts with steroid-resistant rejection. Transplantation. 2001;71:53–58. doi: 10.1097/00007890-200101150-00009. [DOI] [PubMed] [Google Scholar]
- 6.Sokos DR, Berger M, Lazarus HM. Intravenous immunoglobulin: appropriate indications and uses in hematopoietic stem cell transplantation. Biol Blood Marrow Transplant. 2002;8:117–130. doi: 10.1053/bbmt.2002.v8.pm11939601. [DOI] [PubMed] [Google Scholar]
- 7.Nimmerjahn F, Ravetch JV. Anti-inflammatory actions of intravenous immunoglobulin. Annu Rev Immunol. 2008;26:513–533. doi: 10.1146/annurev.immunol.26.021607.090232. [DOI] [PubMed] [Google Scholar]
- 8.Tha-In T, Bayry J, Metselaar HJ, Kaveri SV, Kwekkeboom J. Modulation of the cellular immune system by intravenous immunoglobulin. Trends Immunol. 2008;29:608–615. doi: 10.1016/j.it.2008.08.004. [DOI] [PubMed] [Google Scholar]
- 9.Kwekkeboom J. Modulation of dendritic cells and regulatory T cells by naturally occurring antibodies. Adv Exp Med Biol. 2012;750:133–144. doi: 10.1007/978-1-4614-3461-0_10. [DOI] [PubMed] [Google Scholar]
- 10.Sakaguchi S, Sakaguchi N, Shimizu J, et al. Immunologic tolerance maintained by CD25+ CD4+ regulatory T cells: their common role in controlling autoimmunity, tumor immunity, and transplantation tolerance. Immunol Rev. 2001;182:18–32. doi: 10.1034/j.1600-065x.2001.1820102.x. [DOI] [PubMed] [Google Scholar]
- 11.Tha-In T, Metselaar HJ, Bushell AR, Kwekkeboom J, Wood KJ. Intravenous immunoglobulins promote skin allograft acceptance by triggering functional activation of CD4+Foxp3+ T cells. Transplantation. 2010;89:1446–1455. doi: 10.1097/TP.0b013e3181dd6bf1. [DOI] [PubMed] [Google Scholar]
- 12.Ephrem A, Chamat S, Miquel C, et al. Expansion of CD4+CD25+ regulatory T cells by intravenous immunoglobulin: a critical factor in controlling experimental autoimmune encephalomyelitis. Blood. 2008;111:715–722. doi: 10.1182/blood-2007-03-079947. [DOI] [PubMed] [Google Scholar]
- 13.Ramakrishna C, Newo AN, Shen YW, Cantin E. Passively administered pooled human immunoglobulins exert IL-10 dependent anti-inflammatory effects that protect against fatal HSV encephalitis. PLoS Pathog. 2011;7:e1002071. doi: 10.1371/journal.ppat.1002071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kessel A, Ammuri H, Peri R, et al. Intravenous immunoglobulin therapy affects T regulatory cells by increasing their suppressive function. J Immunol. 2007;179:5571–5575. doi: 10.4049/jimmunol.179.8.5571. [DOI] [PubMed] [Google Scholar]
- 15.Wan YY, Flavell RA. Regulatory T-cell functions are subverted and converted owing to attenuated Foxp3 expression. Nature. 2007;445:766–770. doi: 10.1038/nature05479. [DOI] [PubMed] [Google Scholar]
- 16.Itose I, Kanto T, Kakita N, et al. Enhanced ability of regulatory T cells in chronic hepatitis C patients with persistently normal alanine aminotransferase levels than those with active hepatitis. J Viral Hepat. 2009;16:844–852. doi: 10.1111/j.1365-2893.2009.01131.x. [DOI] [PubMed] [Google Scholar]
- 17.McMurchy AN, Levings MK. Suppression assays with human T regulatory cells: a technical guide. Eur J Immunol. 2012;42:27–34. doi: 10.1002/eji.201141651. [DOI] [PubMed] [Google Scholar]
- 18.Chi LJ, Wang HB, Zhang Y, Wang WZ. Abnormality of circulating CD4(+)CD25(+) regulatory T cell in patients with Guillain–Barre syndrome. J Neuroimmunol. 2007;192:206–214. doi: 10.1016/j.jneuroim.2007.09.034. [DOI] [PubMed] [Google Scholar]
- 19.Furuno K, Yuge T, Kusuhara K, et al. CD25+CD4+ regulatory T cells in patients with Kawasaki disease. J Pediatr. 2004;145:385–390. doi: 10.1016/j.jpeds.2004.05.048. [DOI] [PubMed] [Google Scholar]
- 20.Olivito B, Taddio A, Simonini G, et al. Defective FOXP3 expression in patients with acute Kawasaki disease and restoration by intravenous immunoglobulin therapy. Clin Exp Rheumatol. 2010;28(1 Suppl. 57):93–97. [PubMed] [Google Scholar]
- 21.Bayry J, Mouthon L, Kaveri SV. Intravenous immunoglobulin expands regulatory T cells in autoimmune rheumatic disease. J Rheumatol. 2012;39:450–451. doi: 10.3899/jrheum.111123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Costantino CM, Baecher-Allan C, Hafler DA. Multiple sclerosis and regulatory T cells. J Clin Immunol. 2008;28:697–706. doi: 10.1007/s10875-008-9236-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lan RY, Ansari AA, Lian ZX, Gershwin ME. Regulatory T cells: development, function and role in autoimmunity. Autoimmun Rev. 2005;4:351–363. doi: 10.1016/j.autrev.2005.01.007. [DOI] [PubMed] [Google Scholar]
- 24.Miyara M, Gorochov G, Ehrenstein M, Musset L, Sakaguchi S, Amoura Z. Human FoxP3+ regulatory T cells in systemic autoimmune diseases. Autoimmun Rev. 2011;10:744–755. doi: 10.1016/j.autrev.2011.05.004. [DOI] [PubMed] [Google Scholar]
- 25.Segundo DS, Ruiz JC, Izquierdo M, et al. Calcineurin inhibitors, but not rapamycin, reduce percentages of CD4+CD25+FOXP3+ regulatory T cells in renal transplant recipients. Transplantation. 2006;82:550–557. doi: 10.1097/01.tp.0000229473.95202.50. [DOI] [PubMed] [Google Scholar]
- 26.Demirkiran A, Hendrikx TK, Baan CC, van der Laan LJ. Impact of immunosuppressive drugs on CD4+CD25+FOXP3+ regulatory T cells: does in vitro evidence translate to the clinical setting? Transplantation. 2008;85:783–789. doi: 10.1097/TP.0b013e318166910b. [DOI] [PubMed] [Google Scholar]
- 27.Demirkiran A, Sewgobind VD, van der Weijde J, et al. Conversion from calcineurin inhibitor to mycophenolate mofetil-based immunosuppression changes the frequency and phenotype of CD4+FOXP3+ regulatory T cells. Transplantation. 2009;87:1062–1068. doi: 10.1097/TP.0b013e31819d2032. [DOI] [PubMed] [Google Scholar]
- 28.Baan CC, van der Mast BJ, Klepper M, et al. Differential effect of calcineurin inhibitors, anti-CD25 antibodies and rapamycin on the induction of FOXP3 in human T cells. Transplantation. 2005;80:110–117. doi: 10.1097/01.tp.0000164142.98167.4b. [DOI] [PubMed] [Google Scholar]
- 29.Coenen JJ, Koenen HJ, van Rijssen E, Hilbrands LB, Joosten I. Rapamycin, and not cyclosporin A, preserves the highly suppressive CD27+ subset of human CD4+CD25+ regulatory T cells. Blood. 2006;107:1018–1023. doi: 10.1182/blood-2005-07-3032. [DOI] [PubMed] [Google Scholar]
- 30.Zeiser R, Nguyen VH, Beilhack A, et al. Inhibition of CD4+CD25+ regulatory T-cell function by calcineurin-dependent interleukin-2 production. Blood. 2006;108:390–399. doi: 10.1182/blood-2006-01-0329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gao W, Lu Y, El Essawy B, Oukka M, Kuchroo VK, Strom TB. Contrasting effects of cyclosporine and rapamycin in de novo generation of alloantigen-specific regulatory T cells. Am J Transplant. 2007;7:1722–1732. doi: 10.1111/j.1600-6143.2007.01842.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Miroux C, Morales O, Carpentier A, et al. Inhibitory effects of cyclosporine on human regulatory T cells in vitro. Transplant Proc. 2009;41:3371–3374. doi: 10.1016/j.transproceed.2009.08.043. [DOI] [PubMed] [Google Scholar]
- 33.Maddur MS, Vani J, Hegde P, Lacroix-Desmazes S, Kaveri SV, Bayry J. Inhibition of differentiation, amplification, and function of human TH17 cells by intravenous immunoglobulin. J Allergy Clin Immunol. 2011;127:823–30 e1-7. doi: 10.1016/j.jaci.2010.12.1102. [DOI] [PubMed] [Google Scholar]
- 34.Cousens LP, Najafian N, Mingozzi F, et al. In vitro and in vivo studies of IgG-derived Treg epitopes (Tregitopes): a promising new tool for tolerance induction and treatment of autoimmunity. J Clin Immunol. 2012;33:43–49. doi: 10.1007/s10875-012-9762-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.De Groot AS, Moise L, McMurry JA, et al. Activation of natural regulatory T cells by IgG Fc-derived peptide ‘Tregitopes’. Blood. 2008;112:3303–3311. doi: 10.1182/blood-2008-02-138073. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.