

Abstract
Biliary atresia (BA) is thought to be associated with infections by viruses such as Reoviridae and is characterized histologically by fibrosclerosing cholangitis with proinflammatory cytokine-mediated inflammation. Interleukin (IL)-32 affects the continuous inflammation by increasing the production of proinflammatory cytokines. In this study, the role of IL-32 in the cholangitis of BA was examined. Immunohistochemistry for IL-32 and caspase 1 was performed using 21 samples of extrahepatic bile ducts resected from BA patients. Moreover, using cultured human biliary epithelial cells (BECs), the expression of IL-32 and its induction on stimulation with a Toll-like receptor [(TLR)-3 ligand (poly(I:C)] and proinflammatory cytokines was examined. BECs composing extrahepatic bile ducts showing cholangitis expressed IL-32 in BA, but not in controls. Caspase 1 was expressed constantly on BECs of both BA and control subjects. Furthermore, poly(I:C) and proinflammatory cytokines [(IL-1β, interferon (IFN)-γ and tumour necrosis factor (TNF)-α] induced IL-32 expression strongly in cultured BECs, accompanying the constant expression of TLR-3 and caspase 1. Our results imply that the expression of IL-32 in BECs was found in the damaged bile ducts of BA and induced by biliary innate immunity via TLR-3 and proinflammatory cytokines. These findings suggest that IL-32 is involved initially in the pathogenic mechanisms of cholangitis in BA and also plays an important role in the amplification and continuance of periductal inflammatory reactions. It is therefore tempting to speculate that inhibitors of IL-32 could be useful for attenuating cholangitis in BA.
Keywords: biliary atresia, biliary epithelial cells, IL-32, innate immunity, TLR
Introduction
The obliterative lesion of biliary atresia (BA) is characterized by a progressive sclerosing cholangitis accompanying severe inflammation, fibrosis and epithelial injuries; this characteristic feature is known as fibrosclerosing cholangitis. Little is known about the aetiology and pathogenesis of BA, but infections by viruses such as Reoviridae (reovirus and rotavirus) having a double-stranded RNA (dsRNA) have been implicated, although conflicting results have also been reported 1–8. Our recent study has demonstrated that biliary epithelial cells (BECs) possess an innate immune system consisting of Toll-like receptors (TLR), especially TLR-3, which is an innate immune-recognition receptor recognizing dsRNA, including dsRNA viruses as pathogen-associated molecular patterns (PAMPs) 9,10. Furthermore, the biliary innate immune response to artificial dsRNA was also shown to be associated with the induction of biliary apoptosis via the tumour necrosis factor-related apoptosis-inducing ligand (TRAIL) and, differing from the innate immune response to TLR-4 ligand [lipopolysaccharide (LPS)], lack of subsequent tolerance to dsRNA using cultured human biliary epithelial cells 9–11.
Interleukin (IL)-32 is a recently described cytokine produced by T lymphocytes, natural killer (NK) cells, monocytes and some epithelial cells 12,13. Primarily, IL-32 was discovered in the synovial fluid of patients with rheumatoid arthritis and first reported as a transcript in IL-2 activated NK and T cells 14,15. There are six isoforms (α, β, γ, δ, ε and ξ) caused by alternative mRNA splicing, resulting in proteins with a molecular weight ranging from 14·9 to 26·7 kD. IL-32α is the most abundant transcript. IL-32 exhibits several properties typical of proinflammatory cytokines 16. For example, it stimulates the secretion of proinflammatory cytokines and chemokines such as IL-1α, tumour necrosis factor (TNF)-α, IL-6, IL-8 and vascular endothelial growth factor (VEGF) through the activation of nuclear factor-κB (NF-κB) and p38 mitogen-activated protein kinases (MAPKs) 15–18. In contrast, the production of IL-32 is induced or enhanced by the presence of proinflammatory cytokines, including IL-1β, IFN-γ and TNF-α via the activation of caspase 1 17,19,20. IL-32 has been implicated in inflammatory disorders such as rheumatoid arthritis, inflammatory bowel diseases, chronic obstructive pulmonary diseases, atopic dermatitis and allergic rhinitis 14,19–22.
Although human hepatocytes and hepatoma cells express IL-32 in hepatitis C virus (HCV)-associated chronic hepatitis and this expression is regulated by proinflammatory stimuli 23, the pathophysiological role of IL-32 in innate immune-related biliary diseases, including BA, remains unclear. We therefore investigated the IL-32 expression in the inflamed bile ducts of BA patients and the effect of innate immune stimulation by ligands of TLR-3 and cytokines on IL-32 expression in cultured human BECs. Our results provide evidence that biliary epithelial cells are sufficient sources of IL-32 for the biliary inflammation at sites of BA, and IL-32 may therefore play a role in the pathophysiology of BA.
Materials and methods
Patients and tissue preparations
A total of 21 patients with BA (surgical specimens; average age 1·7 months; age range 0·7–12 months; nine male/12 female) and age-matched control patients consisting of one neonatal hepatitis (giant cell hepatitis; wedge biopsy; 3 months; male) and six non-hepatobiliary diseases (congenital heart anomalies; autopsied specimens; average age 2·5 months; three male/three female) were examined. Resected common bile ducts and wedge liver biopsy specimens were obtained from patients with BA using the Kasai procedure. These specimens had been fixed in 10% neutral-buffered formalin and embedded in paraffin; 4 μm-thick sections were prepared for histological observation and immunohistochemistry.
Immunohistochemistry and immunocytochemistry
For immunocytochemistry using cultured BECs, formalin-fixed, paraffin-embedded sections of cell blocks were prepared according to the protocol reported by Mayall et al. 24. The deparaffinized and rehydrated sections were heated in 10 mM citrate buffer for 20 min in a microwave oven. Following the blocking of endogenous peroxidase, these sections were incubated at 4°C overnight with antibody against the C-terminus of IL-32 [rabbit polyclonal immunoglobulin (Ig)G, 1 μg/ml; Lifespan, Seattle, WA, USA], TLR-3 (rabbit polyclonal IgG, 1 μg/ml; Santa Cruz, Santa Cruz, CA, USA) and caspase 1 (rabbit monoclonal IgG, diluted 1:1000; Abcam, Tokyo, Japan) and then at room temperature for 1 h with anti-rabbit immunoglobulins conjugated to a peroxidase-labelled dextran polymer (Simple Staining Kit; Nichirei, Tokyo, Japan). After a benzidine reaction, sections were counterstained lightly with haematoxylin. As a negative control, normal rabbit IgG was used as the primary antibody; no staining was obtained.
For semiquantitative evaluation of the immunohistochemistry, intrahepatic bile ducts and extrahepatic common bile ducts were chosen in each section for assessment and IL-32 immunoreactivity in these bile ducts was graded semiquantitatively as follows: score 0, absence of expression; score 1, low constitutive expression; score 2, intermediate expression; and score 3, high expression.
In addition, simultaneous detection of IL-32 and cytokeratin (CK)19 was performed using double immunohistochemical staining. After IL-32 immunostaining, CK19 antibody (mouse monoclonal IgG1kappa, 0·45 μg/ml; Dako Japan, Tokyo, Japan) was applied overnight at 4°C, followed by immunoglobulins conjugated with alkaline phosphatase labelled-dextran polymer (Nichirei). Colour development of IL-32 and CK19 was achieved with diaminobenzidine (brown) and Vector blue (Vector Laboratories, Burlingame, CA, USA), respectively.
Cultured human BECs and stimulation with PAMPs and proinflammatory cytokines
A cultured cell line of human intrahepatic BECs was established from the explant liver of a 24-year-old male with BA who had already received the Kasai procedure during the newborn period, and cultured as reported previously 25. The cultured BECs were incubated with a culture medium composed of Dulbecco's modified Eagle's medium (DMEM)/F-12 (Invitrogen, Tokyo, Japan), 5% newborn calf serum (Invitrogen), 0·18 mM adenine (Sigma, St Louis, MO, USA), hydrocortisone (0·4 μg/ml), cholera toxin (10 ng/ml), tri-iodo-thyronine (1·3 μg/l), ITS+ (Becton Dickinson, Franklin Lakes, NJ, USA), 25 mM sodium bicarbonate (Sigma), 1% antibiotics anti-mycotic, human epidermal growth factor (20 ng/ml) (Invitrogen) and human hepatocyte growth factor (10 ng/ml) (Invitrogen). The cells were grown as monolayers in a humidified incubator with 5% CO2 at 37°C. More than 95% of the cells were confirmed to be biliary epithelial cells by the expression of a biliary-type cytokeratin (CK19). The cultured BECs were used between passages 4 and 9. Informed consent for human research was obtained from the patient prior to surgery. This study was approved by the Kanazawa University Ethics Committee. Moreover, as control cultured cells, a commercially available cell line derived from human hepatocellular carcinoma, HepG2, was obtained from the Health Science Research Resources Bank (Osaka, Japan).
These cultured cells were stimulated with a TLR-3 ligand, polyinosinic–polycytidylic acid [poly(I:C), a synthetic analogue of viral dsRNA, 25 μg/ml; Invivogen, San Diego, CA, USA] and recombinant cytokines [IL-1β, IFN-γ, TNF-α, transforming growth factor (TGF)-β1 and IL-10, l000 U/ml; PeproTech, London and IL-32, 1000 U/ml; R&D Systems, Minneapolis, MN, USA] for 3 h (molecular analysis) and 48 h (protein analysis by immunocytochemistry and Western blotting analysis).
Isolation of RNA, reverse transcription–polymerase chain reaction (RT–PCR) and real-time PCR
For evaluation of mRNA of IL-32, caspase 1, TLR-3, IL-1β and IL-6 in cultured BECs, isolation of RNA from BECs and reverse transcription were performed using the RNeasy Total RNA System (Qiagen, Hilden, Germany) and ReverTra Ace (Toyobo, Osaka, Japan). First, to examine the presence of target molecules and the validity of the newly designed primers, conventional PCR was performed. Specific primers for IL-32, caspase 1, TLR-3 and glyceraldehyde 3 phosphate dehydrogenase (GAPDH, positive control) were designed: IL-32 forward: 5′-AGCTGGAGGACGACTTCAAA-3′, reverse: 5′-TTGAGGATTGGGGTTCAGAG-3′ [predicted size, 258 base pairs (bp)]; TLR-3 forward: 5′-CCATTCCAGCCTCTTCGTAA-3′, reverse: 5′-GGATGTTGGTATGGGTCTCG-3′ (predicted size, 505 bp); caspase 1 forward: 5′-CCACAATGGGCTCTGTTTTT-3′, reverse: 5′-CATCTGGCTGCTCAAATGAA-3′ (predicted size, 117); IL-1β forward: 5′-CCAGGGACAGGATATGGAGCA-3′, reverse: 5′-TTCAACACGCAGGACAGGTACAG-3′ (predicted size, 129 bp); IL-6 forward: 5′-AGTGAGGAACAAGCCAGAGC-3′, reverse: 5′-AAGCTGCGCAGAATGAGAT-3′ (predicted size, 189 bp); and GAPDH forward: 5′-GGCCTCCAAGGAGTAAGACC-3′, reverse: 5′-AGGGGTCTACATGGCAACTG-3′ (predicted size, 147 bp). The reaction profile consisted of initial denaturation at 94°C for 3 min followed by 25–40 cycles with 30 s of denaturation at 94°C, 30 s of annealing of primers at 55°C and a 60-s extension at 72°C. Next, to carry out relative quantification, real-time quantitative PCR was performed according to a standard protocol using the Brilliant II SYBR Green QPCR Reagents and Mx300P QPCR system (Stratagene Japan, Tokyo, Japan). Relative gene expression was calculated using the comparative cycle threshold method and adjusted based on expression of the housekeeping gene (GAPDH). Results were obtained from three independent experiments and shown as relative mRNA expression compared with the level without any treatments. Negative controls were obtained by replacing the reverse transcriptase or cDNA samples with RNase and DNase free water.
Western blotting
Cell lysates of poly(I:C)-stimulated or unstimulated cultured cell lines (10 μg protein/lane) and the culture medium were subjected to sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE). Recombinant IL-32 protein (0·1 μg; R&D) was used as a positive control. Separated proteins were transferred onto a nitrocellulose membrane; the membrane was blocked in 5% bovine serum albumin, then probed for 1 h with a primary antibody against human IL-32 (0·1 μg/ml). After washing, the membrane was incubated for 1 h with the Simple Staining Kit, and visualized with the benzidine reaction. The band density was evaluated quantitatively using NIH images.
Statistical analysis
Data were analysed using the paired t-test or Welch's t-test; P < 0·05 was considered statistically significant.
Results
Expression of IL-32, caspase 1 and TLR-3 in extrahepatic bile ducts of BA
Immunohistochemistry revealed the expression of IL-32 in BECs, infiltrating inflammatory cells and endothelial cells at various intensities. In particular, damaged common bile ducts showing cholangitis in BA expressed IL-32 strongly, accompanying many IL-32-positive inflammatory cells and vessels (Fig. 1a–c). As shown in Fig. 1f, double immunohistochemistry highlighted that CK19-postive bile ducts clearly expressed IL-32. However, non-damaged biliary epithelium found at the margin of resected common bile ducts did not express IL-32 (Fig. 1g,h). In wedge liver biopsies, hepatocytes were also positive for IL-32 in addition to small bile ducts (interlobular bile ducts), but the intensity was lower than that in damaged common bile ducts (Fig. 1i,j). Moreover, congestive bile in intrahepatic bile ducts was also strongly positive for IL-32 (Fig. 1j). In contrast, BECs in common bile ducts and intrahepatic bile ducts of age-matched controls expressed only weakly or lacked IL-32 (Fig. 2a,d). The semiquantitative analysis for immunoreaction confirmed that the expression of IL-32 in damaged common bile ducts of BA was up-regulated significantly, compared with those in non-damaged/normal bile ducts of BA and age-matched controls (Fig. 3). Caspase 1 and TLR-3 were expressed constantly in BECs of extrahepatic bile ducts in both the BA and control patients (Fig. 2b,c).
Fig. 1.
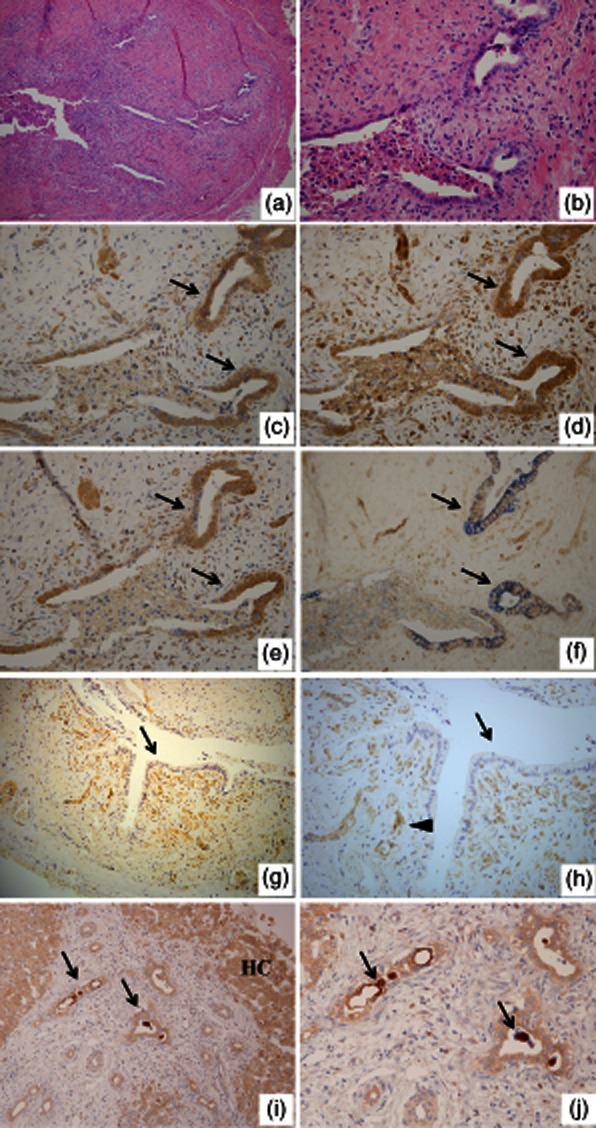
Histology and immunohistochemistry for interleukin (IL)-32, Toll-like receptor (TLR)-3 and caspase 1 in biliary atresia (BA). (a,b) Transverse sections of biliary remnants. Damaged extrahepatic bile ducts line inconsistently by desquamated columnar epithelium and surrounding fibroplasia with an inflammatory cell infiltrate; (b) a higher magnification of (a). Haematoxylin and eosin (H&E) staining. Original magnification (a) ×100 and (b) ×400. Immunohistochemistry for IL-32 (c), TLR-3 (d) and caspase 1 (e). The strong expression of IL-32, TLR-3 and caspase 1 was found in biliary epithelial cells (arrows) of damage bile ducts. Original magnification ×400. (f) Double immunohistochemistry for CK19 and IL-32 highlighted the CK19-positive bile ducts (blue) clearly expressed IL-32 (brown) (arrows). Original magnification ×400. (g,h) Immunohistochemistry for IL-32. Undamaged extrahepatic bile duct located at the resected margin in BA. IL-32-positive neovascular structures (arrowhead) were found, but undamaged biliary epithelium lacked IL-32 expression (arrows); (h) is higher magnification of (g). Original magnification (g) ×200 and (h) ×400. (i,j) Immunohistochemistry for IL-32 using wedge liver specimens of BA. Interlobular bile ducts (arrows in i) and hepatocytes (HC in i) expressed IL-32. Moreover, condensed bile in dilated bile ducts was also strongly positive for IL-32 (arrows in j); (j) is a higher magnification of (i). Original magnification (e) ×200 and (f) ×400.
Fig. 2.
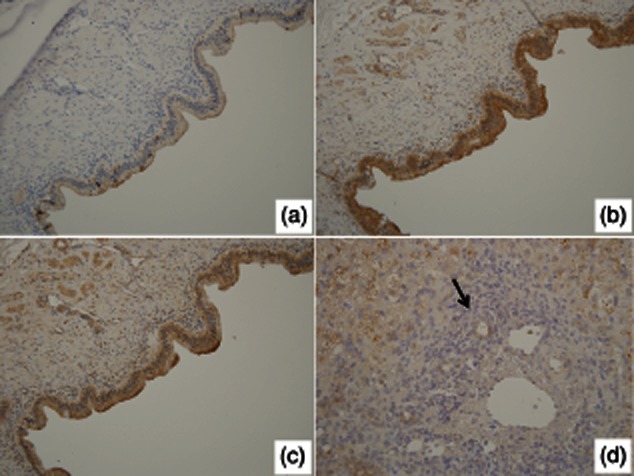
Immunohistochemistry for interleukin (IL)-32 (a,d), Toll-like receptor (TLR)-3 (b) and caspase 1 (c) in age-matched controls. (a–c) Biliary epithelial cells in common bile ducts of non-hepatobiliary diseases (congenital heart anomalies) expressed TLR-3 (b) and caspase 1 (c), but lacking or faintly expressed IL-32 (a) was faint or negative. Original magnification ×200. (d) Interlobular bile duct in neonatal hepatitis was negative for IL-32 (arrow). Original magnification ×400.
Fig. 3.
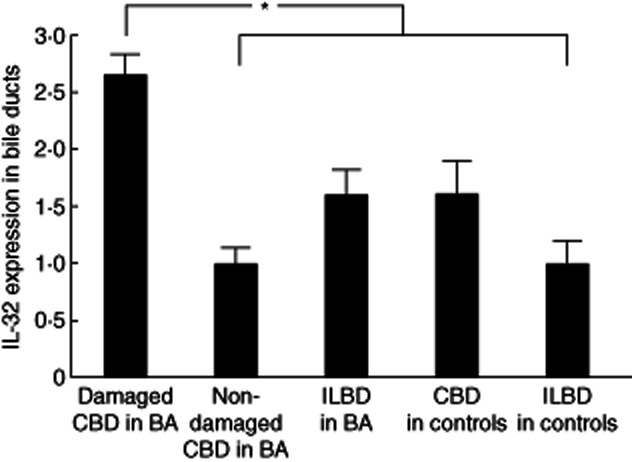
Semiquantitative analysis of immunohistochemistry for interleukin (IL)-32. The expression of IL-32 in damaged common bile ducts (CBD) of biliary atresia (BA) was up-regulated significantly compared with those of non-damaged CBD and interlobular bile ducts (ILBD) in BA, and of CBD and ILBD in age-matched controls. *P<0·05.
Induction of IL-32 expression by PAMPs and cytokines in cultured BECs
To examine the presence of target molecules and the validity of the newly designed primers, RT–PCR at 40 cycles was performed and an amplification of all molecules could be detected as a single band from cultured BECs at the expected size. Moreover, the BECs constantly expressed the mRNA of TLR-3 and caspase 1, which is necessary for the recognition of poly(I:C) and the production of functional IL-32 protein, respectively. The real-time PCR analysis revealed that TLR-3 ligand, poly(I:C) and proinflammatory cytokines (IL-1β, IFN-γ and TNF-α), but not regulatory cytokines (TGF-β1 and IL-10), enhanced the mRNA expression of IL-32, the increases being statistically significant (Fig. 4a). In contrast, stimulation with IL-32 did not up-regulate significantly the expression of BEC-producing cytokines (IL-1β, IL-6 and IL-32), TLR-3 and caspase 1 in cultured BECs (Fig. 4b). Although the control cell line, HepG2, also expressed IL-32 mRNA, up-regulation of IL-32 was not significant by stimulation with poly(I:C) or IL-32 (Fig. 4c).
Fig. 4.
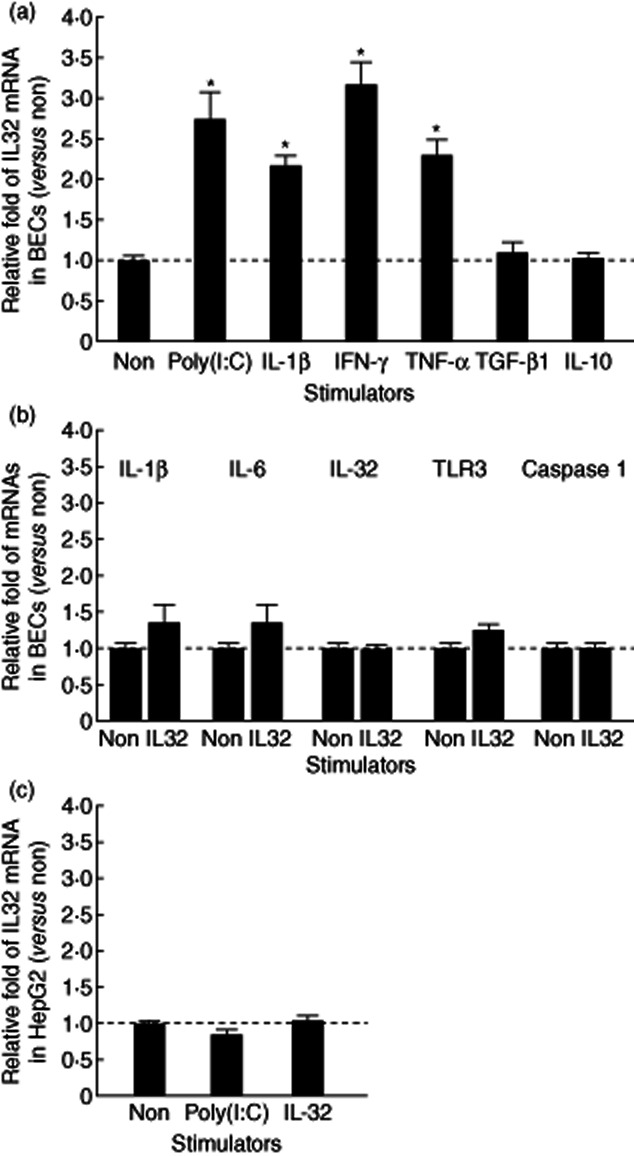
(a) Induction of interleukin (IL)-32 expression by Toll-like receptor (TLR)-3 ligand (poly I:C) and cytokines in cultured biliary epithelial cells (BECs). Quantitative analysis using real-time polymerase chain reaction (PCR) revealed that a TLR-3 ligand, poly(I:C) and proinflammatory cytokines [interleukin (IL)-1β, interferon (IFN)-γ and tumour necrosis factor (TNF)-α], but not regulatory cytokines [transforming growth factor (TGF)-β1 and IL-10], up-regulated significantly the mRNA expression of IL-32. (b) Detection of BEC-producing cytokines (IL-1β, IL-6 and IL-32), TLR-3 and caspase 1 in cultured BECs. The stimulation with IL-32 did not up-regulate the expression of any cytokines, TLR-3 or caspase 1 significantly. (c) Detection of IL-32 in a control cell line, HepG2. Induction of IL-32 expression was not found by stimulation with poly(I:C) or IL-32. Results were obtained from three independent experiments and shown as relative mRNA expression compared with the level without any treatments (Non). Bars indicate the mean ± standard error of the mean. *P < 0·05.
Detection of intracytoplasmic and secreted IL-32protein
To investigate secretion of the IL-32 protein, Western blotting was performed using the cell lysate and culture medium of BECs. IL-32 was detected in the medium as well as lysate from the poly(I:C)-stimulated BECs (Fig. 5a). Semiquantitative analysis using NIH image analysis revealed that the band density was up-regulated in cell lysate and culture medium by stimulation with poly(I:C) (Fig. 5a). Moreover, immunocytochemistry also demonstrated that IL-32 protein was expressed strongly in poly(I:C)-stimulated BECs, compared with non-stimulated BECs (Fig. 5b).
Fig. 5.

Detection of intracytoplasmic and secreted interleukin (IL)-32 protein in cultured biliary epithelial cells (BECs). (a) Western blotting revealed that the culture medium as well as cell lysate of poly(I:C)-treated cultured cells contained IL-32 protein, but the level was faint in untreated cells (Non). As a positive control, recombinant IL-32 (rIL-32, 0·1 μg) was used. Semiquantitative analysis using NIH image analysis confirmed that the density of bands was up-regulated in cell lysate and culture medium by stimulation with poly(I:C). (b) Immunocytochemistry also demonstrated that IL-32 was expressed strongly in the poly(I:C)-stimulated BECs compared with unstimulated BECs (Non). Original magnification ×400.
Discussion
BA is characterized initially by periductal inflammation and fibrosis and the obstruction of common bile ducts, known as fibrosclerosing cholangitis. Recruitment of inflammatory cells results in the release of other proinflammatory cytokines and chemokines, sustaining the cholangitis associated with the biliary innate immune response and promoting chronic cholangitis associated with the subsequent acquired immune response in a later phase 26. IL-32 is a recently described cytokine that is a strong inducer of proinflammatory cytokines whose expression is increased markedly in several inflammatory disorders, including rheumatoid arthritis (RA) and inflammatory bowel disease (IBD), and correlated with the severity of these diseases 14,19. In the present study, human BECs were demonstrated to be the local source of IL-32. Immunohistochemical analysis showed a cytoplasmic distribution of IL-32 in BECs of the damaged common bile ducts in BA cases, although BECs of common bile ducts in age-matched controls were negative or only weakly positive for IL-32, suggesting that IL-32 is associated closely with the histogenesis of periductal inflammation in BA. However, IL-32 production in BECs is not specific to BA alone. In fact, we confirmed the expression of IL-32 in bile ducts of adult biliary diseases such as primary biliary cirrhosis, but its intensity was lower than those in the damaged common bile ducts of BA. Therefore, we speculated that the induction of IL-32 by unique factors such as viral infections in BA was stronger than those in other biliary diseases. Inflammasomes are multi-protein cytoplasmic complexes that mediate the activation of inflammatory caspase-1. For example, caspase-1 cleaves pro-IL-1β to the active form of IL-1β. In this manner, caspase-1 controls the maturation of some of the proinflammatory cytokines, and IL-32 also depends upon caspase 1 activation 17,20. Therefore, the presence of caspase 1 is necessary for the functional expression of IL-32 in BECs. In the present study, BECs constantly expressed caspase 1 in vitro and in vivo, suggesting the expression of functional IL-32 in BECs.
Recent studies have focused upon the role of innate immunity associated with Reoviridae (reovirus and rotavirus) in the pathogenesis of BA. Having a dsRNA genome, Reoviridae in particular are characterized by epithelial tropism 1,3,4,9,10,27,28. The initial sensing of innate immunity is mediated by the recognition of PAMPs through TLRs. IL-32 also appears to play an important role in the host defence against invading micro-organisms 23,29,30; that is, IL-32 is described as a proinflammatory cytokine that enhances host immunity against various microbial pathogens. The present study revealed that stimulation with poly(I:C), a mimic of Reoviridae, enhanced IL-32 expression in cultured BECs, suggesting that the biliary innate immune response directly induces the production of IL-32 in BECs. A control cell line used in this study, HepG2, also expressed IL-32 mRNA, but the up-regulation of IL-32 was not significant by stimulation with poly(I:C). It has already been reported that IL-32 expression is induced in peripheral blood mononuclear cells and monocytes by Mycobacterium tuberculosis 31 but, to our knowledge, this is the first description concerning the production of IL-32 in epithelial cells such as BECs via an innate immune response.
IL-1β, IFN-γ and TNF-α were reported to be inducers of IL-32 expression 16,19. However, the regulatory mechanism of these proinflammatory cytokines remains unclear. In this study, we found that all these proinflammatory cytokines are potent stimulators of IL-32 expression in cultured BECs. In contrast, the aforementioned results suggest that the secretion of IL-32 could stimulate periductal inflammatory and/or immune cells to secrete proinflammatory cytokines and contributes to the deterioration of periductal inflammation. Because these inflammatory cytokines and an innate immunity play important roles in the immune-mediated histogenesis of BA, the inflammatory responses and innate immune response in the affected bile ducts of BA patients may be amplified by constant IL-32-induced secretion of proinflammatory cytokines from BECs and periductal inflammatory cells, suggesting that IL-32 plays a central role in the inflammatory responses involved in BA pathogenesis. However, IL-32 itself could not up-regulate the expression of inflammatory cytokines (IL-1β, IL-6 and IL-32), TLR-3 and caspase 1 in cultured BECs, suggesting that IL-32 produced by BECs was unlikely to be involved in direct reciprocal signalling resulting in up-regulation of inflammatory cytokines and of susceptibility to virus in BECs.
In this study, we demonstrate that stimulation with poly(I:C) induced the transcription of IL-32 mRNA in BECs and also confirmed the presence of the protein in the culture medium as well as cell lysate. Moreover, immunohistochemistry also revealed that a condensed bile in intrahepatic small bile ducts was positive for IL-32. These findings suggest the secretion of IL-32 from IL-32-expressing BECs. Therefore, IL-32 is speculated to be secreted extracellularly in periductal tissue fluids and into bile in BA. As mentioned above, the secreted IL-32 induces the production of proinflammatory cytokines in inflammatory and/or immune cells, resulting in marked amplification of the inflammatory cytokine milieu, and these responses may contribute to the aggravation of BA. Moreover, it was suggested recently that IL-32 acts as a cytoplasmic protein: IL-32 was expressed at high levels in human epidermal keratinocytes after stimulation with IFN-γ and TNF-α, but was not secreted by keratinocytes 21. Moreover, it was also shown that the up-regulation of cytoplasmic IL-32 expression induces apoptosis 21,32. In IBD, the apoptosis of damaged colonic cells by accumulated intracellular IL-32 can be considered a host defence mechanism against invading microorganisms, by which damaged epithelial cells are eliminated efficiently along with invading microorganisms and further invasions of microorganisms can be blocked 19,33. In BA, our previous study found that biliary apoptosis was enhanced in the damaged common bile ducts and associated closely with bile duct loss in BA, which was caused by the production of an apoptosis-inducer, TRAIL, in BECs via the biliary innate immune response to a TLR-3 ligand, poly(I:C) 10. However, this TRAIL-mediated biliary apoptosis is only partially involved in the poly(I:C)-induced mechanism, and other possible mechanisms could also exist 10. Therefore, the IL-32-mediated mechanism is also likely in poly(I:C)-induced biliary apoptosis, and might be associated with enhanced biliary apoptosis in the damaged common bile ducts of BA.
In conclusion, we have demonstrated that IL-32 expression is enhanced in the damaged common bile ducts of BA patients. Expression of IL-32 in BECs was induced by the innate immune response to dsRNA [poly(I:C)] and proinflammatory cytokines (IL-1β, IFN-γ and TNF-α). This study has identified IL-32 as an important inflammatory cytokine involved in the cholangitis of BA. So far, anti-IL-32 treatment has been studied in only a few diseases, such as rheumatoid arthritis 34,35. The regulation of IL-32 expression may form the basis of a new strategy for the treatment of BA.
Acknowledgments
This work was supported by Grants-in-Aid for Scientific Research from the Ministry of Health, Labour and Welfare of Japan and Grants-in-Aid for Scientific Research (C) from the Ministry of Education, Culture, Sports, Science, and Technology of Japan.
Disclosure
The authors declare no conflicts of interest.
References
- 1.Morecki R, Glaser JH, Cho S, Balistreri WF, Horwitz MS. Biliary atresia and reovirus type 3 infection. N Engl J Med. 1982;307:481–484. doi: 10.1056/NEJM198208193070806. [DOI] [PubMed] [Google Scholar]
- 2.Morecki R, Glaser JH, Johnson AB, Kress Y. Detection of reovirus type 3 in the porta hepatis of an infant with extrahepatic biliary atresia: ultrastructural and immunocytochemical study. Hepatology. 1984;4:1137–1142. doi: 10.1002/hep.1840040608. [DOI] [PubMed] [Google Scholar]
- 3.Tyler KL, Sokol RJ, Oberhaus SM, et al. Detection of reovirus RNA in hepatobiliary tissues from patients with extrahepatic biliary atresia and choledochal cysts. Hepatology. 1998;27:1475–1482. doi: 10.1002/hep.510270603. [DOI] [PubMed] [Google Scholar]
- 4.Riepenhoff-Talty M, Gouvea V, Evans MJ, et al. Detection of group C rotavirus in infants with extrahepatic biliary atresia. J Infect Dis. 1996;174:8–15. doi: 10.1093/infdis/174.1.8. [DOI] [PubMed] [Google Scholar]
- 5.Brown WR, Sokol RJ, Levin MJ, et al. Lack of correlation between infection with reovirus 3 and extrahepatic biliary atresia or neonatal hepatitis. J Pediatr. 1988;113:670–676. doi: 10.1016/s0022-3476(88)80376-7. [DOI] [PubMed] [Google Scholar]
- 6.Bobo L, Ojeh C, Chiu D, Machado A, Colombani P, Schwarz K. Lack of evidence for rotavirus by polymerase chain reaction/enzyme immunoassay of hepatobiliary samples from children with biliary atresia. Pediatr Res. 1997;41:229–234. doi: 10.1203/00006450-199702000-00013. [DOI] [PubMed] [Google Scholar]
- 7.Mack CL. The pathogenesis of biliary atresia: evidence for a virus-induced autoimmune disease. Semin Liver Dis. 2007;27:233–242. doi: 10.1055/s-2007-985068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mack CL, Tucker RM, Lu BR, et al. Cellular and humoral autoimmunity directed at bile duct epithelia in murine biliary atresia. Hepatology. 2006;44:1231–1239. doi: 10.1002/hep.21366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Harada K, Sato Y, Isse K, Ikeda H, Nakanuma Y. Induction of innate immune response and absence of subsequent tolerance to dsRNA in biliary epithelial cells relate to the pathogenesis of biliary atresia. Liver Int. 2008;28:614–621. doi: 10.1111/j.1478-3231.2008.01740.x. [DOI] [PubMed] [Google Scholar]
- 10.Harada K, Sato Y, Itatsu K, et al. Innate immune response to double-stranded RNA in biliary epithelial cells is associated with the pathogenesis of biliary atresia. Hepatology. 2007;46:1146–1154. doi: 10.1002/hep.21797. [DOI] [PubMed] [Google Scholar]
- 11.Harada K, Isse K, Sato Y, Ozaki S, Nakanuma Y. Endotoxin tolerance in human intrahepatic biliary epithelial cells is induced by upregulation of IRAK-M. Liver Int. 2006;26:935–942. doi: 10.1111/j.1478-3231.2006.01325.x. [DOI] [PubMed] [Google Scholar]
- 12.Heinhuis B, Netea MG, van den Berg WB, Dinarello CA, Joosten LA. Interleukin-32: a predominantly intracellular proinflammatory mediator that controls cell activation and cell death. Cytokine. 2012;60:321–327. doi: 10.1016/j.cyto.2012.07.010. [DOI] [PubMed] [Google Scholar]
- 13.Dahl CA, Schall RP, He HL, Cairns JS. Identification of a novel gene expressed in activated natural killer cells and T cells. J Immunol. 1992;148:597–603. [PubMed] [Google Scholar]
- 14.Joosten LA, Netea MG, Kim SH, et al. IL-32, a proinflammatory cytokine in rheumatoid arthritis. Proc Natl Acad Sci USA. 2006;103:3298–3303. doi: 10.1073/pnas.0511233103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kim KH, Shim JH, Seo EH, et al. Interleukin-32 monoclonal antibodies for immunohistochemistry, Western blotting, and ELISA. J Immunol Methods. 2008;333:38–50. doi: 10.1016/j.jim.2007.12.017. [DOI] [PubMed] [Google Scholar]
- 16.Kim SH, Han SY, Azam T, Yoon DY, Dinarello CA. Interleukin-32: a cytokine and inducer of TNFalpha. Immunity. 2005;22:131–142. doi: 10.1016/j.immuni.2004.12.003. [DOI] [PubMed] [Google Scholar]
- 17.Netea MG, Azam T, Ferwerda G, et al. IL-32 synergizes with nucleotide oligomerization domain (NOD) 1 and NOD2 ligands for IL-1beta and IL-6 production through a caspase 1-dependent mechanism. Proc Natl Acad Sci USA. 2005;102:16309–16314. doi: 10.1073/pnas.0508237102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kobayashi H, Huang J, Ye F, Shyr Y, Blackwell TS, Lin PC. Interleukin-32beta propagates vascular inflammation and exacerbates sepsis in a mouse model. PLoS ONE. 2010;5:e9458. doi: 10.1371/journal.pone.0009458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shioya M, Nishida A, Yagi Y, et al. Epithelial overexpression of interleukin-32alpha in inflammatory bowel disease. Clin Exp Immunol. 2007;149:480–486. doi: 10.1111/j.1365-2249.2007.03439.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jeong HJ, Shin SY, Oh HA, Kim MH, Cho JS, Kim HM. IL-32 up-regulation is associated with inflammatory cytokine production in allergic rhinitis. J Pathol. 2011;224:553–563. doi: 10.1002/path.2899. [DOI] [PubMed] [Google Scholar]
- 21.Meyer N, Zimmermann M, Burgler S, et al. IL-32 is expressed by human primary keratinocytes and modulates keratinocyte apoptosis in atopic dermatitis. J Allergy Clin Immunol. 2010;125:858–65 e10. doi: 10.1016/j.jaci.2010.01.016. [DOI] [PubMed] [Google Scholar]
- 22.Calabrese F, Baraldo S, Bazzan E, et al. IL-32, a novel proinflammatory cytokine in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2008;178:894–901. doi: 10.1164/rccm.200804-646OC. [DOI] [PubMed] [Google Scholar]
- 23.Moschen AR, Fritz T, Clouston AD, et al. Interleukin-32: a new proinflammatory cytokine involved in hepatitis C virus-related liver inflammation and fibrosis. Hepatology. 2011;53:1819–1829. doi: 10.1002/hep.24285. [DOI] [PubMed] [Google Scholar]
- 24.Mayall F, Chang B, Darlington A. A review of 50 consecutive cytology cell block preparations in a large general hospital. J Clin Pathol. 1997;50:985–990. doi: 10.1136/jcp.50.12.985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kamihira T, Shimoda S, Harada K, et al. Distinct costimulation dependent and independent autoreactive T-cell clones in primary biliary cirrhosis. Gastroenterology. 2003;125:1379–1387. doi: 10.1016/j.gastro.2003.07.013. [DOI] [PubMed] [Google Scholar]
- 26.Feldman AG, Mack CL. Biliary atresia: cellular dynamics and immune dysregulation. Semin Pediatr Surg. 2012;21:192–200. doi: 10.1053/j.sempedsurg.2012.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Riepenhoff-Talty M, Schaekel K, Clark HF, et al. Group A rotaviruses produce extrahepatic biliary obstruction in orally inoculated newborn mice. Pediatr Res. 1993;33:394–399. doi: 10.1203/00006450-199304000-00016. [DOI] [PubMed] [Google Scholar]
- 28.Szavay PO, Leonhardt J, Czech-Schmidt G, Petersen C. The role of reovirus type 3 infection in an established murine model for biliary atresia. Eur J Pediatr Surg. 2002;12:248–250. doi: 10.1055/s-2002-34477. [DOI] [PubMed] [Google Scholar]
- 29.Bai X, Ovrutsky AR, Kartalija M, et al. IL-32 expression in the airway epithelial cells of patients with Mycobacterium avium complex lung disease. Int Immunol. 2011;23:679–691. doi: 10.1093/intimm/dxr075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li W, Sun W, Liu L, et al. IL-32: a host proinflammatory factor against influenza viral replication is upregulated by aberrant epigenetic modifications during influenza A virus infection. J Immunol. 2010;185:5056–5065. doi: 10.4049/jimmunol.0902667. [DOI] [PubMed] [Google Scholar]
- 31.Netea MG, Azam T, Lewis EC, et al. Mycobacterium tuberculosis induces interleukin-32 production through a caspase-1/IL-18/interferon-gamma-dependent mechanism. PLoS Med. 2006;3:e277. doi: 10.1371/journal.pmed.0030277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Goda C, Kanaji T, Kanaji S, et al. Involvement of IL-32 in activation-induced cell death in T cells. Int Immunol. 2006;18:233–240. doi: 10.1093/intimm/dxh339. [DOI] [PubMed] [Google Scholar]
- 33.Kim JM, Eckmann L, Savidge TC, Lowe DC, Witthoft T, Kagnoff MF. Apoptosis of human intestinal epithelial cells after bacterial invasion. J Clin Invest. 1998;102:1815–1823. doi: 10.1172/JCI2466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Alsaleh G, Sparsa L, Chatelus E, et al. Innate immunity triggers IL-32 expression by fibroblast-like synoviocytes in rheumatoid arthritis. Arthritis Res Ther. 2010;12:R135. doi: 10.1186/ar3073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Leask A. B cell block: is rituximab a new possible treatment for systemic sclerosis? J Cell Commun Signal. 2010;4:201–202. doi: 10.1007/s12079-010-0107-x. [DOI] [PMC free article] [PubMed] [Google Scholar]