

Abstract
2-Chlorodeoxyadenosine (cladribine, CdA) is an immunosuppressive drug that is licensed to treat hairy cell leukaemia, and has been shown recently to have beneficial effects in patients with multiple sclerosis (MS). The therapeutic effects of CdA have been suggested to be mediated partly through its potent toxicity towards lymphocytes. However, the effects of CdA on other immune cells are poorly understood. In the present study, we investigated the effects of CdA on the induction of apoptosis in human monocytes, monocyte-derived immature (ImDC) and mature (mDC) dendritic cells. Treatment of monocytes with CdA strongly induced apoptosis after 24 h, while apoptosis induction in DC was evident after 72 h. Furthermore, CdA treatment strongly induced caspase-3 and caspase-9 in monocytes, whereas activation of caspases was undetected in DC. The mitochondrial membrane potential in DC was reduced significantly after CdA treatment. DNA hypodiploid assessment showed fragmented nuclei in DC after CdA treatment together with activation of p53 protein. These results revealed that CdA induces caspase-independent apoptosis in DC and suggest cell type specific effects of CdA. This mechanism may contribute to the effect of CdA in autoimmune diseases.
Keywords: caspases, cladribine, DNA fragmentation, mitochondrial potential, phosphatidylserine
Introduction
Dendritic cells (DC) are professional antigen-presenting cells that have a unique ability to prime antigen-specific T cells. After capturing the antigen, DC migrate to the draining lymph nodes and become mature under the influence of several inflammatory stimuli. Upon maturation DC display certain phenotypical changes, such as up-regulation of the co-stimulatory molecules CD86 and CD40 and an increased release of proinflammatory cytokines 1. Mature DC can process and present antigens efficiently, while their antigen capturing ability is reduced.
In recent years, the role of DC in regulating autoimmune disorders such as arthritis and multiple sclerosis (MS) has been highlighted 2. MS is an autoimmune disease of young adults characterized by inflammatory demyelinating lesions in the central nervous system (CNS) resulting from infiltration of immune cells from the periphery 3. Among these are self-reactive T cells that are believed to be responsible for the neuronal damage 4. Previous studies have shown that secondary progressive MS patients have an increased frequency of CD80 expressing blood DC compared to relapsing–remitting MS or healthy subjects 5. An increased number of plasmacytoid DC has been observed in the cerebrospinal fluid (CSF) of untreated MS patients during the relapse phase, which decreased substantially in the remission phase 6. Although monocyte-derived DC (MoDC) share certain characteristics of circulating blood DC, they closely resemble the inflammatory DC 7,8. Interestingly, MoDC from MS patients release high levels of cytokines such as tumour necrosis factor (TNF)-α and interleukin (IL)-6 and express increased levels of CD1a, which is involved in the presentation of lipid antigens to T cells 9,10. These findings hint at the potential role of DC in MS pathogenesis, and the drugs which can regulate their function might affect the progression of the disease.
Several approved drugs for MS have been shown to influence DC responses. Glatiramer acetate induced production of the anti-inflammatory mediator IL-10 in DC and reduced IL-12p70 synthesis in lipopolysaccharide (LPS)-activated DC 11. In Natalizumab-treated MS patients, del Pilar Martin and colleagues have found a reduced number of DC and CD4+ T cells in cerebral vascular spaces 12. Moreover, interferon (IFN)-β treatment induces apoptosis in bone marrow-derived mature DC 13.
Recently, the immunosuppressive drug cladribine (2-chlorodeoxyadenosine, CdA) has been shown to be effective in MS 14,15. The principle effect of CdA, the induction of apoptosis in lymphocytes, has been studied widely and a similar effect was also reported for monocytes 16,17. CdA is phosphorylated intracellularly into CdAMP by a rate-limiting enzyme deoxycytidine kinase (DCK) and subsequently into CdA-5′-triphosphate (CdATP) by the action of other enzymes 18. CdATP is incorporated into the DNA, causing cell cycle arrest and thereby inducing apoptosis. The apoptosis is shown to be mediated via activation of different caspases such as caspase-3, -9 or -8; however, caspase-independent apoptosis has been also documented 19. In a recent study, we have shown that CdA can also induce apoptosis in microglia 20. Apart from the induction of apoptosis other immunomodulatory effects of CdA, such as reduction in CSF levels of IL-8 and CSF/serum levels of RANTES (regulated upon activation normal T cell expressed and secreted), have also been discussed 21.
However, the direct influence of CdA in vitro on DC has not been addressed so far. Therefore, we studied the effects of CdA on human MoDC cultures. Our findings demonstrate that CdA induces caspase-dependent apoptosis in monocytes and caspase-independent apoptosis in DC.
Materials and methods
Monocyte cell cultures
Human peripheral blood mononuclear cells (PBMC) were isolated from blood of healthy donors received from the blood bank of the Hannover Medical School. PBMC were separated using a Biocoll separating solution (Biochrom, Berlin, Germany), density (1·077 g/ml) and were washed twice with phosphate-buffered saline (PBS) containing 0·5% bovine serum albumin (BSA) and 2 mM ethylenediamine tetraacetic acid (EDTA) (Sigma, Deissenhofen, Germany). Monocytes were then purified by a positive selection method using human CD14 magnetic affinity cell sorting (MACS) microbeads, as described by the manufacturer (Miltenyi Biotech, Bergisch Gladbach, Germany). Monocytes were cultured in RPMI-1640 medium (Invitrogen, Karlsruhe, Germany) supplemented with 10% fetal bovine serum (FBS) (Biochrom), 1% penicillin/streptomycin (Gibco, Karlsruhe, Germany), 1% HEPES buffer (Sigma) and 1% L-alanyl-L-glutamine (Invitrogen) at 37°C in a humidified atmosphere containing 5% CO2. After 20 min non-adherent cells were removed by changing the medium and adherent cells were used further. Monocytes isolated by this method had a purity of >95% as assessed by flow cytometry with a fluorescein isothiocyanate (FITC)-conjugated CD14 antibody (eBioscience, Hatfield, UK). For testing CdA effects on blood DC, isolated human PBMC were left untreated or treated with CdA (1 and 10 μM) for 24 h. Blood DC and monocytes were then characterized by staining with anti-human CD141 allophycocyanin (APC) (clone AD5-14H12; Miltenyi Biotech) and CD14 phycoerythrin (PE) (clone 134620; R&D Systems Inc., Wiesbaden-Nordenstadt, Germany), respectively.
Generation of dendritic cells
DC were generated by previously described protocols, with some modifications 22. Briefly, immature dendritic cells (ImDC) were obtained by culturing monocytes with 50 ng/ml granulocyte–macrophage colony-stimulating factor (GM-CSF) and 1000 U/ml IL-4 for 5 days. Every 3 days fresh RPMI medium containing GM-CSF and IL-4 was added to the cultures. On day 6, half the cells were kept in the same medium and the other half were incubated with medium containing 10 ng/ml TNF-α and 25% monocyte-conditioned medium (MCM) to obtain mature dendritic cells (mDC). Prepared MoDC had a purity of >90% as assessed by flow cytometry with a PE-conjugated CD1a antibody (eBioscience). After 2 days fresh medium with the respective cytokines was added to the unstimulated (ImDC) and TNF-α stimulated (mDC) cells. Thereafter, cells were treated with different concentrations of CdA for a defined time-period. For some experiments LPS was used for activation of DC, and the influence of CdA on DC maturation (16 h) was examined using immunostaining. All cytokines used to produce DC were obtained from Peprotech, Hamburg, Germany.
Immunostaining
Immunostaining for different cell surface proteins was performed for characterization of monocytes and MoDC. Monocytes were stained immediately after isolation, whereas the MoDC were stained on the 8th day of culture. Semi-adherent DC were detached from the culture flasks by gently pipetting the medium onto the cells. Cells were washed once in PBS and placed in 5-ml fluorescence activated cell sorter (FACS) tubes at a density of 2–2·5 × 105 cells/100 μl PBS, and human Fc receptor blocker (Biolegend, Fell, Germany) was added for 15 min at 4°C. Cells were then labelled with different fluorescence antibodies against human CD14 (FITC, clone 61D3; eBioscience), CD11c (APC, clone 3·9; Biolegend), CD1a (PE, clone HI149; eBioscience), human leucocyte antigen D-related (HLA-DR) (APC, clone L243; Biolegend), CD86 (FITC, clone 2331; BD Biosciences, Heidelberg, Germany) and also with corresponding isotype control immunoglobulin (Ig)G. Nucleic acid dye 7-amino-actinomycin D (7-AAD) was added to the samples to exclude dead cells from analysis. Cells were analysed by flow cytometry on a FACScalibur™ Becton-Dickinson flow cytometer using CellQuest™ software. The percentage of positive cells for different markers and total mean fluorescence intensity (MFI) were calculated for each sample. The MFI of untreated samples was normalized to 100% and percentage change in different surface markers was calculated. For each sample a minimum 10 000 events were collected and analysed.
Western blot analyses
Cells were washed with cold PBS and lysed in lysis buffer [42 mM Tris-HCL, 1·3% sodium dodecyl sulphate (SDS), 6·5% glycerin and 100 μM sodium orthovanadate and 2% protease and phosphatase inhibitor]. Before electrophoresis, Laemmli buffer (5% mercaptoethanol, 10% glycerol, 2% SDS, 65 mM Tris-HCL and bromophenol blue) was added to the samples. For caspase-3, -9, DCK and phospho-p53 immunoblotting, 15–20 μg of protein from each sample were used for SDS-polyacrylamide gel electrophoresis (PAGE) on a 12% gel. Proteins were then transferred onto a polyvinylidene difluoride (PVDF) membrane (Millipore, Schwalbach/Ts., Germany) by the wet-blotting method. The membrane was blocked for 30 min at room temperature using a 5% skimmed milk solution (Santa Cruz Biotechnology, Heidelberg, Germany). The membrane was then incubated with the respective primary antibody in 1% milk solution overnight at 4°C. The following primary antibodies were used: rabbit anti-cleaved caspase-3 (1 μg/ml; Abcam, Cambridge, UK), rabbit anti-cleaved caspase-9 (Asp330) (1 μg/ml; Cell Signaling, Danvers, MA, USA), rabbit anti-DCK (1:1000; Abcam), rabbit anti-phospho-p53 (Cell Signaling) and mouse anti-actin (1:3000; Santa Cruz Biotechnology). After extensive washing (three times for 15 min each in Tris-buffered saline (TBS) containing 0·1% Tween 20), proteins were detected with horseradish peroxidase (HRP)-coupled goat anti-rabbit IgG (1:3000; R&D Systems) or HRP-coupled goat anti-mouse IgG (1:5000; R&D Systems) using chemiluminescence reagents (Thermo Fisher Scientific, Bonn, Germany). All Western blot experiments were carried out at least three times.
Annexin V staining for apoptotic cells
Apoptosis was quantified by FITC-conjugated annexin V staining of externalized phosphatidylserine (PS), a reliable marker for early apoptosis 23. 7-AAD was added to quantify dead cells. This staining distinguishes between early apoptotic (annexin V+), late apoptotic or necrotic (annexin V+/7-AAD+) and necrotic cells (7-AAD+) cells. At the end of the incubation period, cells (adhering and detached) were collected from culture dishes with slow pipette blows in PBS. Cells were centrifuged at 370 g for 6 min, washed twice with PBS and resuspended in binding buffer provided in the assay kit (FITC annexin V apoptosis detection kit; Becton Dickinson GmbH). Cells were transferred into 5-ml FACS tubes at a density of 1·5 × 105 cells (for monocytes and MoDC) and 2 × 106 cells per tube (for PBMC) and were left unstained or stained with annexin V and 7-AAD for 15 min in the dark at room temperature. After staining, cells were washed once in binding buffer and 7-AAD was added to the samples before acquiring on flow cytometry. Cells were analysed by flow cytometry on a FACScalibur Becton-Dickinson flow cytometer using CellQuest™ software. For each sample a minimum 10 000 events were collected and analysed.
Mitochondrial transmembrane potential measurement
Mitochondrial transmembrane potential (ΔΨM) was measured using the Cell Meter™ orange assay kit, as described by the manufacturer (AAT Inc., Sunnyvale, CA, USA). Briefly, DC were incubated with different concentrations of CdA (0·1–10 μM) for 24 h and 72 h. Two μl of MitoLite™ fluorescence dye (provided with the assay kit) was added to the cells and cells were incubated at 37°C, 5% CO2 for an additional 20 min. The incubation was stopped by placing the cells on ice for 10 min and cells were collected into 5-ml tubes. After washing with PBS, cells were resuspended in assay buffer and analysed by flow cytometry. In live cells, the fluorescence intensity of MitoLite™ orange is increased, whereas it is low in apoptotic cells with collapsed mitochondria. Fluorescence intensity was read in the orange–red channel (excitation 488 nm) of the flow cytometer and was analysed using CellQuest™ software. For each sample a minimum 10 000 events were collected and analysed.
Measurement of DNA damage
DNA damage was assessed by propidium iodide (PI) staining of fragmented nuclei (DNA release assay), as described previously 24. Briefly, after the respective incubation time, cells were washed in PBS and fixed in 4% paraformaldehyde (PFA) solution. Cells were then incubated in permeabilization solution (0·1% sodium citrate, 0·1% Triton X-100) containing 25 μg/ml PI for 2 h at 4°C. Cells were analysed by flow cytometry using CellQuest™ software.
TNF-α enzyme-linked immunosorbent assay
Cell culture supernatants were collected and centrifuged at 400 g for 10 min at 4°C. Supernatants were used to measure TNF-α release using enzyme-linked immunosorbent assay as described by the manufacturer (eBioscience).
Statistical analyses
All experiments were performed at least three times and mean ± standard error of the mean was calculated. Values were compared using one-way analysis of variance (anova) with the post-hoc Student–Newman–Keuls test (multiple comparisons); for analysis of phospho-p53 protein levels, the non-parametric Mann–Whitney test was performed using Graphpad Prism version 5·0 software. P values < 0·05 were considered statistically significant (*P < 0·05; **P < 0·01; ***P < 0·001).
Results
CdA induces apoptosis in monocytes, MoDC and blood DC
CdA is known to induce apoptosis in monocytes and T lymphocytes, but its effects on DC are still unknown. Hence, our initial effort was to determine if CdA-treated DC exhibit the hallmarks of cells undergoing apoptosis. Human monocytes were differentiated into DC and were characterized using immunostaining (Supplementary Fig. S1). As reported in other studies, we observed down-regulation of CD14 and up-regulation of CD1a on the surface of MoDC 10,22,25. An early event in apoptosis is the externalization of PS from the inner leaflet of the plasma membrane, which can be detected readily, and quantified by annexin V staining. We observed that treatment of monocytes with CdA (1 and 10 μM) for 24 h led to a significant increase in the percentage of cells undergoing apoptosis. An increase of 40–46% of annexin V+ cells was observed in monocyte cultures treated with CdA compared to untreated controls. There was no notable increase in annexin V+/7-AAD+ cells, which is a characteristic of cells undergoing delayed apoptosis or necrosis 26. In contrast, treatment of MoDC (ImDC and mDC) with lower concentrations of CdA (1 μM) did not show any increase in annexin V+ cells. At a higher concentration (10 μM) of CdA, only a minimal (8%) increase in annexin V+ cells was observed (Fig. 1a,b). However, at this time-point camptothecin (campto, 10 μM), a drug known to induce apoptosis, increased the number of annexin V+ cells. Interestingly, longer treatment (72 h) with CdA (0·1, 1 and 10 μM) induced a significant increase in annexin V+ MoDC. In addition, there was an increase in annexin V+/7-AAD+ cells. These effects were concentration-dependent, with a maximum at 10 μM CdA (Fig. 1c,d). We studied further the effect of CdA on circulating blood DC by PBMC staining. DC are a heterogeneous group of cells and commonly express the CD11c and HLA-DR markers; however, these markers are also present on other cell types and thus require a complex antibody-staining combination to characterize DC in PBMC. Therefore, we used CD141 as a more specific marker expressed on the myeloid DC subset 27. The assessment of circulating blood DC (CD141high/CD14–) together with 7-AAD and annexin V staining in flow cytometry showed an induction of apoptosis after treatment of PBMC with CdA (1 and 10 μM) early after 24 h, comparable to the effects of CdA that we observed on monocytes (Supplementary Fig. S2). However, it remains unclear as to whether this earlier apoptotic effect of CdA on circulating blood DC in comparison to MoDC is a direct effect of the drug, because in our experimental setting an influence of cell–cell interactions with monocytes and/or lymphocytes on kinetics of apoptosis cannot be excluded. Because of the very low cell number of circulating blood DC (∼0·1%), in-vitro experiments with an isolated cell population of blood DC were not practicable.
Fig. 1.

Effect of 2-chlorodeoxyadenosine (CdA) on phosphatidylserine (PS) exposure in monocytes (Mo) and monocyte-derived dendritic cells (MoDC). Cells were treated with CdA (0·1, 1 and 10 μM) for 24 and 72 h and were assayed for apoptosis using annexin V-fluorescein isothiocyanate (FITC)/7-aminoactinomycin D (7-AAD) staining followed by flow cytometry. (a,c) Dot-plots showing the percentages of annexin V−/7-AAD− cells, annexin V+/7-AAD− cells and annexin V+/7-AAD+ cells. Camptothecin (campto, 10 μM) was used as a positive control. (b,d) Percentages of annexin V+ and 7-AAD− cells. Data are represented as mean ± standard error of the mean (n = 5). *P < 0·05; **P < 0·01; ***P < 0·001 versus untreated control.
CdA treatment disrupts the mitochondrial transmembrane potential in monocytes and MoDC
Mitochondria are the key regulators of cell death mechanisms, and CdA-induced apoptosis in leukaemic cells has been shown to be dependent upon the disruption of ΔΨM 28,29. Therefore, we investigated the ΔΨM of CdA-treated DC using the fluorescent dye MitoLite™. There was a loss of ΔΨM in monocytes but not in DC treated with increasing concentrations of CdA (0·1, 1 and 10 μM) for 24 h (Fig. 2a). The loss of ΔΨM in DC occurred only after 72 h of CdA treatment (Fig. 2b). These results further confirm the above findings that induction of apoptosis is delayed in DC.
Fig. 2.
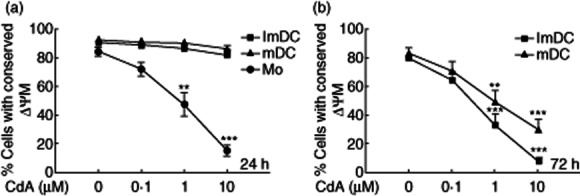
Effect of 2-chlorodeoxyadenosine (CdA) on mitochondrial transmembrane potential (ΔΨM) in monocytes (Mo) and monocyte-derived dendritic cells (MoDC). Cells were treated with CdA (0·1, 1 and 10 μM) for 24 and 72 h and were stained with MitoLite™ fluorescence dye and analysed by flow cytometry. Normal cells with conserved ΔΨM show high fluorescence while apoptotic cells with lost ΔΨM show low fluorescence, as measured in the orange–red channel of the flow cytometer. (a,b) The line graph shows the CdA-induced reduction in percentage of cells with conserved ΔΨM after 24 and 72 h, respectively. Data are represented as mean ± standard error of the mean (n = 4). **P < 0·01; ***P < 0·001) versus untreated control.
CdA induces caspase-3 and caspase-9 activation in human monocytes but not in MoDC
Activation of caspases is a central phenomenon in the regulation of apoptosis in different cells 30. Therefore, we investigated whether or not treatment of human monocytes and MoDC with CdA resulted in the activation of apoptotic caspases. We found that monocytes treated with CdA (1 and 10 μM) for 24 h strongly induced caspase-3 and -9 activation, whereas similar treatment did not activate these caspases in MoDC (Fig. 3a). These findings suggest that while CdA has strong effects on the activation of caspases in monocytes, the DC appear to be unaffected. This observed difference is not due to the inability of DC to activate pro-caspases, as treatment of DC with the apoptosis-inducing drug camptothecin resulted in the activation of caspase-3. However, the signal for caspase-9 was not detected even in camptothecin-treated DC. To substantiate further if DC exhibit a delayed kinetics of caspase activation, we checked caspase-3 activation after 48 and 72 h of CdA treatment. Irrespective of the time of CdA treatment, no signal for caspase-3 was detected in DC (Fig. 3b).
Fig. 3.
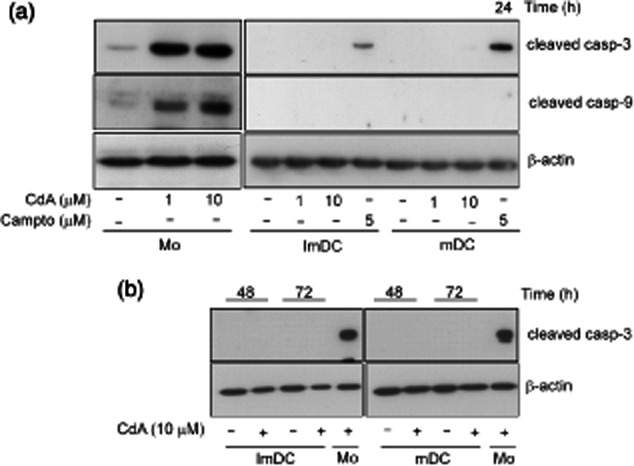
Effect of 2-chlorodeoxyadenosine (CdA) on the activation of caspase-3 and -9 in monocytes (Mo) and monocyte-derived dendritic cells (MoDC). Cells were treated with CdA (1 and 10 μM) or camptothecin (campto; 5 μM) as positive control for 24–72 h and whole-cell lysates were resolved by sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE), transferred to polyvinylidine difluoride (PVDF) membrane and probed with anti-caspases and β-actin antibodies. (a) Activation of caspase-3 and -9 at 24 h. (b) Activation of caspase-3 at 48 and 72 h. Mo = CdA-treated monocytes were used as a positive control. Blots are representative of four independent experiments.
CdA induces p53 activation and DNA fragmentation in MoDC
p53 is involved critically in the cellular response to stress or apoptotic signals 31. Monocytes and MoDC were treated with CdA (10 μM) for 6–24 h and activation of p53 was evaluated by detection of its phosphorylated form by Western blotting. Further supporting the above findings, we observed activation of p53 as early as 6 h after CdA treatment in monocytes, while it was evident at 24 h in DC (Fig. 4a,b).
Fig. 4.
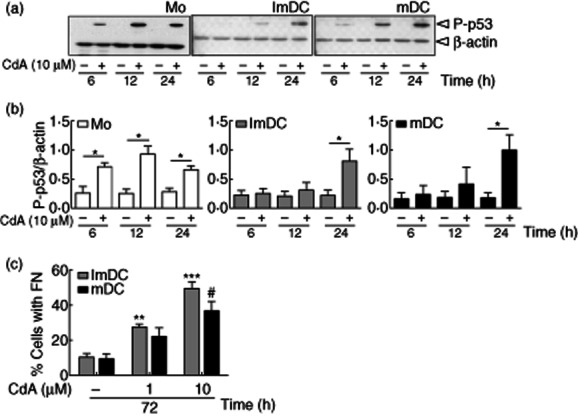
Effect of 2-chlorodeoxyadenosine (CdA) on p53 activation and DNA fragmentation in monocyte-derived dendritic cells (MoDC). (a) Cells were treated with CdA (10 μM) for 6–24 h and whole-cell lysates were resolved by sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE), transferred to polyvinylidine difluoride (PVDF) membrane and probed with anti-phospho-p53 and β-actin antibodies. Blots are representative of four independent experiments. (b) Densitograph analysis is shown and data are represented as mean ± standard error of the mean (n = 4). (c) Cells were treated with CdA (1 and 10 μM) for 72 h, permeabilized, stained with propidium iodide (PI) and analysed by flow cytometry. Data are represented as percentage of cells with fragmented nuclei (FN). *#P < 0·05; **P < 0·01; ***P < 0·001 versus untreated control.
A characteristic feature of apoptotic cells is the fragmentation of DNA at the internucleosomal sections 32. The fragmented DNA confers a hypodiploid state and this can be detected readily on FACS by using PI, a nucleic acid stain. The FACS analysis showed that approximately 50% of ImDC and 36% of mDC that were treated with 10 μM CdA for 72 h displayed a subdiploid peak (Fig. 4c). These observations support the fact that CdA induces apoptosis but not necrosis in DC, as the subdiploid peak is lacking in the cells subjected to necrosis 33,34. These results signify the role of CdA in inducing DNA damage and delayed activation of downstream signalling molecules in DC in comparison to monocytes.
Expression of DCK in monocytes and MoDC
In order to mediate its effects, CdA has to be phosphorylated within the cell and this step is catalyzed by the rate-limiting DCK 35. To test if the observed delay in induction of apoptosis in MoDC is a result of differential expression of DCK, we compared the protein levels of DCK in untreated monocytes and DC. We observed no significant differences in the expression of DCK in monocytes and DC (Fig. 5a,b). Hence, it is evident from these results that the delayed apoptotic effects of CdA in DC in comparison to monocytes are not due to absence or low DCK expression.
Fig. 5.
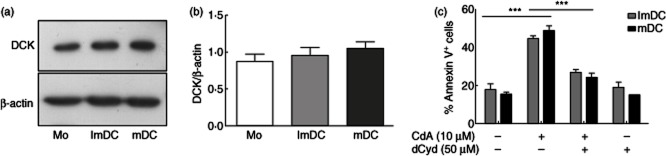
Analysis of protein expression of the enzyme deoxycytidine kinase (DCK) in monocytes (Mo) and monocyte-derived dendritic cells (MoDC). (a) Untreated cell lysates were resolved by sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE), transferred to polyvinylidine difluoride (PVDF) membrane and probed with anti-DCK or β-actin antibodies. Blots are representative of four independent experiments. (b) Densitometric analysis of four independent experiments. (c) Cells were treated with deoxycytidine (dCyd, 50 μM) prior to the addition of 10 μM 2-chlorodeoxyadenosine (CdA) for 72 h and annexin V staining was performed. Data are represented as mean ± standard error of the mean (n = 3). ***P < 0·001.
Further, we tested if the CdA-induced apoptotic effects in DC were dependent upon its phosphorylation via DCK. DC were treated with deoxycytidine (50 μM), a preferential substrate for DCK, prior to the addition of CdA for 72 h and apoptosis was measured by annexin V staining. CdA-induced apoptosis was inhibited significantly by the ablation of DCK required for its phosphorylation (Fig. 5c). These findings suggest that although there is a delay in the induction of apoptosis in DC, this effect of CdA is still mediated by its phosphorylation and requires the activity of DCK.
Effects of CdA on LPS-induced maturation and TNF-α release in ImDC
To investigate the effects of CdA on DC maturation, cells were treated with CdA (10 μM) for 16 h in the presence or absence of LPS (100 ng/ml). LPS is a Toll-like receptor (TLR)-4 agonist that is used widely to induce DC maturation and triggers a proinflammatory response in DC 36. Flow cytometric analysis showed that LPS led significantly to increased surface expression of maturation markers CD86 and HLA-DR in DC; however, the addition of CdA did not show any effect (Fig. 6a,b). In addition, CdA has no effect on LPS-induced release of TNF-α in DC (Fig. 6c). Taken together, these results suggest that CdA induces apoptosis in DC but does not interfere with other cell functions like their maturation and release of proinflammatory cytokine such as TNF-α.
Fig. 6.
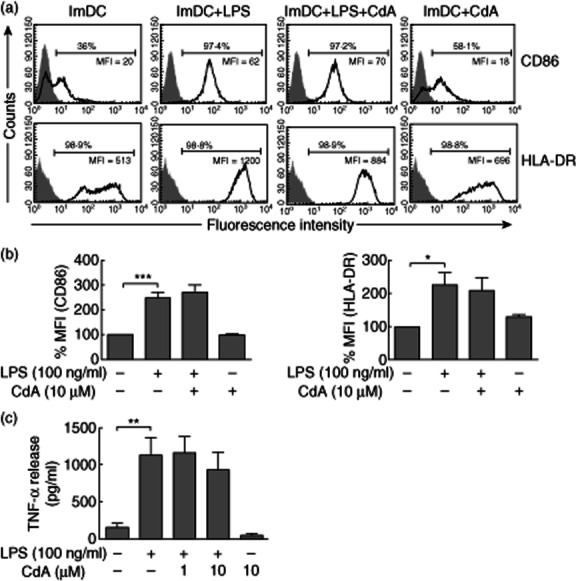
Effect of 2-chlorodeoxyadenosine (CdA) on lipopolysaccharide (LPS)-induced dendritic cell (DC) maturation and tumour necrosis factor (TNF)-α release. Immature (Im)DC were treated with 10 μM CdA in the absence or presence of LPS (100 ng/ml) and immunostaining for maturation markers [CD86 and human leucocyte antigen D-related (HLA-DR)] was performed and analysed by flow cytometry. (a) Histograms are representative for four independent experiments. Data were normalized to isotype controls and percentage of positive cells and mean fluorescence intensity (MFI) are presented. Isotype controls (shaded) were included for each staining. (b) Data are presented as the percentage change in the MFI. (c) The release of TNF-α (pg/ml) was measured in the culture medium. Data are represented as mean ± standard error of the mean (n = 4). *P < 0·05; **P < 0·01; ***P < 0·001 versus untreated ImDC control.
Discussion
Immunosuppressive drugs are of significant therapeutic importance in treating several autoimmune diseases. Due to its known apoptotic effects on T lymphocytes 37 cladribine (CdA) was recommended as a therapeutic option for multiple sclerosis (MS), a T cell-mediated autoimmune disease of the CNS 15,38. DC also play a key role in priming autospecific T cells, and hence can be another key target for the action of CdA to mediate its immunosuppressive effects. However, the effects of CdA on DC are yet to be characterized. In this study, using certain key parameters of apoptosis such as PS externalization, caspase activation and DNA fragmentation, we compared the effects of CdA on monocytes and MoDC. The CdA concentrations (0·1–10 μM) used in this study are highly relevant to the physiological values in human subjects. For instance, plasma and cellular concentrations of CdA have been demonstrated to be ∼0·1 and 10 μM, respectively, in treated leukaemic patients 39,40.
We demonstrate that CdA induces apoptosis in monocytes after short incubation periods, which is in line with previous findings 17. Interestingly, once the monocytes are differentiated into DC they are partially resistant to the action of the drug and require longer incubation periods to induce apoptosis. Our study focused mainly on MoDC for two reasons. First, obtaining sufficient numbers of human blood DC, which are found in extremely low frequencies in blood, was always a challenge. Secondly, MoDC closely resemble the inflammatory DC in terms of their phenotype and function 7. Nevertheless, we also attempted to study the effects of CdA on circulating blood DC by treating PBMC with the drug. The effects observed on blood DC followed similar kinetics, such as monocytes, and contrasted with that observed in MoDC (Supplementary Fig. S2). We cannot confirm at this point if the observed early apoptosis in blood DC is a direct effect of the drug or a bystander effect of other cells in the culture. Long-term, but not short-term, treatment of MoDC with CdA strongly reduced ΔΨM, and this phenomenon paralleled the kinetics of PS externalization. We believe that externalization of PS in DC might be the consequence of the loss of ΔΨM. A similar phenomenon has been reported in dexamethasone-treated thymocytes, where a reduction in ΔΨM was a prerequisite for PS exposure 41.
These findings led us to speculate if MoDC have differential kinetics of drug metabolism. In order to mediate its effects, CdA has to be phosphorylated into biologically active CdATP within the cell. This process is catalyzed by the enzyme DCK; this is the rate-limiting step for CdA activity 18. It has been reported previously that cells lacking this enzyme are resistant to CdA-induced apoptosis 42. Protein expression analysis revealed similar expression levels of DCK in monocytes and DC. This excludes the possibility that absence or low levels of DCK in DC might delay the induction of apoptosis. In addition, pharmacological depletion of the DCK by using a preferential substrate, deoxycytidine, protected DC from CdA-induced apoptosis. These findings suggest that although apoptosis is delayed in CdA-treated DC, it is still dependent upon DCK-mediated phosphorylation of the drug.
In many cell types, CdA-induced apoptosis is known to be triggered through the activation of cysteine proteases known as caspases 43. Several studies have demonstrated caspase-dependent apoptosis in CdA-treated lymphocytes. Recently we have also shown caspase-dependent apoptosis in CdA-treated microglia 20. However, we observed that CdA treatment of monocytes strongly activates caspase-3 and -9 after 24 h, whereas neither of the caspases were detected in CdA-treated DC even after longer treatments. The absence of caspase activation suggests a caspase-independent apoptotic mechanism operating in CdA-treated DC. Marzo et al. have demonstrated the involvement of caspase-independent apoptotic pathways in CdA-treated U937 leukaemic cells 19. Furthermore, a study by Nicolo et al. has shown a delayed apoptotic response in mDC that were subjected to ultraviolet B-induced stress 44. This was attributed to the presence of higher levels of the anti-apoptotic protein Bcl-2 in mDC. Intriguingly, we observed lower levels of Bcl-2 in untreated DC than in monocytes (data not shown). Nevertheless, participation of other anti-apoptotic mechanisms resulting in delayed apoptosis cannot be ruled out 45.
It is known widely that p53 is a key protein activated in response to cellular stress. Our results have shown the activation of p53 both in monocytes and DC, but the kinetics of p53 activation is delayed in CdA-treated DC compared to the monocytes. p53 stimulates a wide network of signals that operate through major apoptotic pathways 46. Hence, we believe that p53 acts upstream of apoptotic pathways and the timing of p53 activation might regulate the kinetics of apoptosis. There are reports which demonstrate the kinetics of p53 induction and subsequent DNA fragmentation in CdA-treated cells 47,48. Similar results are reported in our study, where DNA fragmentation occurred in DC after longer treatment with CdA.
An interesting observation was made when the effect of CdA on maturation and function of DC was studied. Although CdA drives DC towards apoptosis, there was no immediate effect of the drug on LPS-induced DC maturation and release of proinflammatory molecules.
CdA toxicity towards lymphocytes has been well documented. In accordance with previous reports we have demonstrated that CdA triggers apoptosis in monocytes. It is noteworthy that DC derived from these monocytes are somewhat robust to CdA action and follow relatively delayed kinetics of apoptosis. CdA-induced apoptosis in DC might prove effective in treating autoimmune diseases such as MS, as DC can infiltrate into the brain and reprime myelin-specific T cells and cause inflammatory damage 49. Therefore, targeting DC would be an ideal step in controlling T cell-related autoimmune diseases. In addition, the ability of CdA to cross the blood–brain barrier suggests a novel therapeutic approach to eliminate DC from MS lesions.
Acknowledgments
This research work was supported partly by Merck Serono GmbH. The sponsor was not involved in data collection or analysis, drafting the manuscript or decision to publish. This work is part of the PhD thesis of V. Singh.
Disclosure
MS has received honoraria for scientific lectures or consultancy from Bayer Healthcare, Biogen Idec, CSL Behring, Grifols, Merck-Serono, Novartis, Sanofi-Aventis, and Teva. His institution received research support from Bayer Healthcare, Biogen Idec, Merck-Serono, Novartis, and Teva.
Supporting information
Additional Supporting Information may be found in the online version of this article at the publisher's web-site:
Fig. S1. Expression of CD14, CD1a, CD11c, CD86 and human leucocyte antigen D-related (HLA-DR) on monocyte-derived dendritic cells (MoDC). Monocytes and ex-vivo differentiated immature (Im)DC and mature (m)DC were stained with respective antibodies and analysed by flow cytometry. Data were normalized to isotype controls and the percentage of positive cells and mean fluorescence intensity (MFI) are presented. Isotype controls (shaded) were included for each staining. Results are representative of three independent experiments.
Fig. S2. Effect of 2-chlorodeoxyadenosine (CdA) on phosphatidylserine (PS) exposure in blood dendritic cells (DC) and monocytes. Human peripheral blood mononuclear cells (PBMC) were isolated and left untreated or treated with CdA (1 and 10 μM) for 24 h. Blood DC and monocytes were identified by immunostaining and were assayed for apoptosis using annexin V-fluorescein isothiocyanate (FITC)/7-aminoactinomycin D (7-AAD) staining followed by flow cytometry. (a) Dot-plots representing gated CD141+ blood DC and CD14+ monocytes. (b) Dot-plots representing percentages of annexin V−/7-AAD−, annexin V+/7-AAD− and annexin V+/7-AAD+ blood DC and monocytes. Results are representative of three independent experiments.
References
- 1.Dzopalic T, Rajkovic I, Dragicevic A, Colic M. The response of human dendritic cells to co-ligation of pattern-recognition receptors. Immunol Res. 2012;52:20–33. doi: 10.1007/s12026-012-8279-5. [DOI] [PubMed] [Google Scholar]
- 2.Cravens PD, Lipsky PE. Dendritic cells, chemokine receptors and autoimmune inflammatory diseases. Immunol Cell Biol. 2002;80:497–505. doi: 10.1046/j.1440-1711.2002.01118.x. [DOI] [PubMed] [Google Scholar]
- 3.Korn T. Pathophysiology of multiple sclerosis. J Neurol. 2008;255(Suppl. 6):2–6. doi: 10.1007/s00415-008-6001-2. [DOI] [PubMed] [Google Scholar]
- 4.Martino G, Furlan R, Brambilla E, et al. Cytokines and immunity in multiple sclerosis: the dual signal hypothesis. J Neuroimmunol. 2000;109:3–9. doi: 10.1016/s0165-5728(00)00295-2. [DOI] [PubMed] [Google Scholar]
- 5.Karni A, Abraham M, Monsonego A, et al. Innate immunity in multiple sclerosis: myeloid dendritic cells in secondary progressive multiple sclerosis are activated and drive a proinflammatory immune response. J Immunol. 2006;177:4196–4202. doi: 10.4049/jimmunol.177.6.4196. [DOI] [PubMed] [Google Scholar]
- 6.Longhini AL, von Glehn F, Brandao CO, et al. Plasmacytoid dendritic cells are increased in cerebrospinal fluid of untreated patients during multiple sclerosis relapse. J Neuroinflammation. 2011;8:1–4. doi: 10.1186/1742-2094-8-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shortman K, Naik SH. Steady-state and inflammatory dendritic-cell development. Nat Rev Immunol. 2007;7:19–30. doi: 10.1038/nri1996. [DOI] [PubMed] [Google Scholar]
- 8.Osugi Y, Vuckovic S, Hart DN. Myeloid blood CD11c(+) dendritic cells and monocyte-derived dendritic cells differ in their ability to stimulate T lymphocytes. Blood. 2002;100:2858–2866. doi: 10.1182/blood.V100.8.2858. [DOI] [PubMed] [Google Scholar]
- 9.Huang YM, Xiao BG, Ozenci V, et al. Multiple sclerosis is associated with high levels of circulating dendritic cells secreting pro-inflammatory cytokines. J Neuroimmunol. 1999;99:82–90. doi: 10.1016/s0165-5728(99)00106-x. [DOI] [PubMed] [Google Scholar]
- 10.Bartosik-Psujek H, Tabarkiewicz J, Pocinska K, Radej S, Stelmasiak Z, Rolinski J. Immunomodulatory effects of IFN-beta and lovastatin on immunophenotype of monocyte-derived dendritic cells in multiple sclerosis. Arch Immunol Ther Exp (Warsz) 2010;58:313–319. doi: 10.1007/s00005-010-0084-z. [DOI] [PubMed] [Google Scholar]
- 11.Vieira PL, Heystek HC, Wormmeester J, Wierenga EA, Kapsenberg ML. Glatiramer acetate (copolymer-1, copaxone) promotes Th2 cell development and increased IL-10 production through modulation of dendritic cells. J Immunol. 2003;170:4483–4488. doi: 10.4049/jimmunol.170.9.4483. [DOI] [PubMed] [Google Scholar]
- 12.del Pilar Martin M, Cravens PD, Winger R, et al. Decrease in the numbers of dendritic cells and CD4+ T cells in cerebral perivascular spaces due to natalizumab. Arch Neurol. 2008;65:1596–1603. doi: 10.1001/archneur.65.12.noc80051. [DOI] [PubMed] [Google Scholar]
- 13.Yen JH, Ganea D. Interferon beta induces mature dendritic cell apoptosis through caspase-11/caspase-3 activation. Blood. 2009;114:1344–1354. doi: 10.1182/blood-2008-12-196592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Giovannoni G, Comi G, Cook S, et al. A placebo-controlled trial of oral cladribine for relapsing multiple sclerosis. N Engl J Med. 2010;362:416–426. doi: 10.1056/NEJMoa0902533. [DOI] [PubMed] [Google Scholar]
- 15.Giovannoni G, Cook S, Rammohan K, et al. Sustained disease-activity-free status in patients with relapsing–remitting multiple sclerosis treated with cladribine tablets in the CLARITY study: a post-hoc and subgroup analysis. Lancet Neurol. 2011;10:329–337. doi: 10.1016/S1474-4422(11)70023-0. [DOI] [PubMed] [Google Scholar]
- 16.Carson DA, Wasson DB, Taetle R, Yu A. Specific toxicity of 2-chlorodeoxyadenosine toward resting and proliferating human lymphocytes. Blood. 1983;62:737–743. [PubMed] [Google Scholar]
- 17.Carrera CJ, Terai C, Lotz M, et al. Potent toxicity of 2-chlorodeoxyadenosine toward human monocytes in vitro and in vivo. A novel approach to immunosuppressive therapy. J Clin Invest. 1990;86:1480–1488. doi: 10.1172/JCI114865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Beutler E. Cladribine (2-chlorodeoxyadenosine) Lancet. 1992;340:952–956. doi: 10.1016/0140-6736(92)92826-2. [DOI] [PubMed] [Google Scholar]
- 19.Marzo I, Perez-Galan P, Giraldo P, Rubio-Felix D, Anel A, Naval J. Cladribine induces apoptosis in human leukaemia cells by caspase-dependent and -independent pathways acting on mitochondria. Biochem J. 2001;359:537–546. doi: 10.1042/0264-6021:3590537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Singh V, Voss EV, Benardais K, Stangel M. Effects of 2-chlorodeoxyadenosine (Cladribine) on primary rat microglia. J Neuroimmune Pharmacol. 2012;7:939–950. doi: 10.1007/s11481-012-9387-7. [DOI] [PubMed] [Google Scholar]
- 21.Bartosik-Psujek H, Belniak E, Mitosek-Szewczyk K, Dobosz B, Stelmasiak Z. Interleukin-8 and RANTES levels in patients with relapsing-remitting multiple sclerosis (RR-MS) treated with cladribine. Acta Neurol Scand. 2004;109:390–392. doi: 10.1111/j.1600-0404.2004.00259.x. [DOI] [PubMed] [Google Scholar]
- 22.Wiesemann E, Sonmez D, Heidenreich F, Windhagen A. Interferon-beta increases the stimulatory capacity of monocyte-derived dendritic cells to induce IL-13, IL-5 and IL-10 in autologous T-cells. J Neuroimmunol. 2002;123:160–169. doi: 10.1016/s0165-5728(01)00482-9. [DOI] [PubMed] [Google Scholar]
- 23.Koopman G, Reutelingsperger CP, Kuijten GA, Keehnen RM, Pals ST, van Oers MH. Annexin V for flow cytometric detection of phosphatidylserine expression on B cells undergoing apoptosis. Blood. 1994;84:1415–1420. [PubMed] [Google Scholar]
- 24.Nicoletti I, Migliorati G, Pagliacci MC, Grignani F, Riccardi C. A rapid and simple method for measuring thymocyte apoptosis by propidium iodide staining and flow cytometry. J Immunol Methods. 1991;139:271–279. doi: 10.1016/0022-1759(91)90198-o. [DOI] [PubMed] [Google Scholar]
- 25.Gogolak P, Rethi B, Szatmari I, et al. Differentiation of CD1a- and CD1a+ monocyte-derived dendritic cells is biased by lipid environment and PPARgamma. Blood. 2007;109:643–652. doi: 10.1182/blood-2006-04-016840. [DOI] [PubMed] [Google Scholar]
- 26.Miller E. Apoptosis measurement by annexin V staining. Methods Mol Med. 2004;88:191–202. doi: 10.1385/1-59259-406-9:191. [DOI] [PubMed] [Google Scholar]
- 27.Jongbloed SL, Kassianos AJ, McDonald KJ, et al. Human CD141+ (BDCA-3)+ dendritic cells (DCs) represent a unique myeloid DC subset that cross-presents necrotic cell antigens. J Exp Med. 2010;207:1247–1260. doi: 10.1084/jem.20092140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Genini D, Adachi S, Chao Q, et al. Deoxyadenosine analogs induce programmed cell death in chronic lymphocytic leukemia cells by damaging the DNA and by directly affecting the mitochondria. Blood. 2000;96:3537–3543. [PubMed] [Google Scholar]
- 29.Vaux DL. Apoptogenic factors released from mitochondria. Biochim Biophys Acta. 2011;1813:546–550. doi: 10.1016/j.bbamcr.2010.08.002. [DOI] [PubMed] [Google Scholar]
- 30.Shi Y. Caspase activation: revisiting the induced proximity model. Cell. 2004;117:855–858. doi: 10.1016/j.cell.2004.06.007. [DOI] [PubMed] [Google Scholar]
- 31.Reinhardt HC, Schumacher B. The p53 network: cellular and systemic DNA damage responses in aging and cancer. Trends Genet. 2012;28:128–136. doi: 10.1016/j.tig.2011.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nagata S. Apoptotic DNA fragmentation. Exp Cell Res. 2000;256:12–18. doi: 10.1006/excr.2000.4834. [DOI] [PubMed] [Google Scholar]
- 33.Qiao L, Koutsos M, Tsai LL, et al. Staurosporine inhibits the proliferation, alters the cell cycle distribution and induces apoptosis in HT-29 human colon adenocarcinoma cells. Cancer Lett. 1996;107:83–89. doi: 10.1016/0304-3835(96)04346-7. [DOI] [PubMed] [Google Scholar]
- 34.Riccardi C, Nicoletti I. Analysis of apoptosis by propidium iodide staining and flow cytometry. Nat Protoc. 2006;1:1458–1461. doi: 10.1038/nprot.2006.238. [DOI] [PubMed] [Google Scholar]
- 35.Bontemps F, Meier C, Delacauw A, Balzarini J, Galmarini C, van den Neste E. Study of the efficacy of a pronucleotide of 2-chloro-2′-deoxyadenosine in deoxycytidine kinase-deficient lymphoma cells. Nucleosides Nucleotides Nucleic Acids. 2006;25:997–1000. doi: 10.1080/15257770600889444. [DOI] [PubMed] [Google Scholar]
- 36.Downing I, MacDonald SL, Atkinson AP, Turner ML, Kilpatrick DC. Drug modification of LPS-stimulated human monocyte-derived dendritic cells. Br J Biomed Sci. 2012;69:126–133. [PubMed] [Google Scholar]
- 37.Robak T, Wierzbowska A, Robak E. Recent clinical trials of cladribine in hematological malignancies and autoimmune disorders. Rev Recent Clin Trials. 2006;1:15–34. doi: 10.2174/157488706775246102. [DOI] [PubMed] [Google Scholar]
- 38.Leist TP, Vermersch P. The potential role for cladribine in the treatment of multiple sclerosis: clinical experience and development of an oral tablet formulation. Curr Med Res Opin. 2007;23:2667–2676. doi: 10.1185/030079907x233142. [DOI] [PubMed] [Google Scholar]
- 39.Albertioni F, Lindemalm S, Reichelova V, et al. Pharmacokinetics of cladribine in plasma and its 5′-monophosphate and 5′-triphosphate in leukemic cells of patients with chronic lymphocytic leukemia. Clin Cancer Res. 1998;4:653–658. [PubMed] [Google Scholar]
- 40.Liliemark J. The clinical pharmacokinetics of cladribine. Clin Pharmacokinet. 1997;32:120–131. doi: 10.2165/00003088-199732020-00003. [DOI] [PubMed] [Google Scholar]
- 41.Castedo M, Hirsch T, Susin SA, et al. Sequential acquisition of mitochondrial and plasma membrane alterations during early lymphocyte apoptosis. J Immunol. 1996;157:512–521. [PubMed] [Google Scholar]
- 42.Mansson E, Spasokoukotskaja T, Sallstrom J, Eriksson S, Albertioni F. Molecular and biochemical mechanisms of fludarabine and cladribine resistance in a human promyelocytic cell line. Cancer Res. 1999;59:5956–5963. [PubMed] [Google Scholar]
- 43.Van den Neste E, Cardoen S, Offner F, Bontemps F. Old and new insights into the mechanisms of action of two nucleoside analogs active in lymphoid malignancies: fludarabine and cladribine (review) Int J Oncol. 2005;27:1113–1124. [PubMed] [Google Scholar]
- 44.Nicolo C, Tomassini B, Rippo MR, Testi R. UVB-induced apoptosis of human dendritic cells: contribution by caspase-dependent and caspase-independent pathways. Blood. 2001;97:1803–1808. doi: 10.1182/blood.v97.6.1803. [DOI] [PubMed] [Google Scholar]
- 45.O'Gorman DM, Cotter TG. Molecular signals in anti-apoptotic survival pathways. Leukemia. 2001;15:21–34. doi: 10.1038/sj.leu.2401998. [DOI] [PubMed] [Google Scholar]
- 46.Vogelstein B, Kinzler KW. p53 function and dysfunction. Cell. 1992;70:523–526. doi: 10.1016/0092-8674(92)90421-8. [DOI] [PubMed] [Google Scholar]
- 47.Borner MM, Joncourt F, Hotz MA. Similarity of apoptosis induction by 2-chlorodeoxyadenosine and cisplatin in human mononuclear blood cells. Br J Cancer. 1997;76:1448–1454. doi: 10.1038/bjc.1997.577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Szondy Z. The 2-chlorodeoxyadenosine-induced cell death signalling pathway in human thymocytes is different from that induced by 2-chloroadenosine. Biochem J. 1995;311(Pt 2):585–588. doi: 10.1042/bj3110585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Serafini B, Rosicarelli B, Magliozzi R, et al. Dendritic cells in multiple sclerosis lesions: maturation stage, myelin uptake, and interaction with proliferating T cells. J Neuropathol Exp Neurol. 2006;65:124–141. doi: 10.1097/01.jnen.0000199572.96472.1c. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.