

Abstract
Eotaxins induce the trafficking of eosinophils to the sites of inflammation via CC chemokine receptor 3 (CCR3). In this study, we investigated eotaxin-3/CC chemokine ligand 26 (CCL26) expression in the inflamed mucosa of patients with inflammatory bowel disease (IBD), and characterized the molecular mechanisms responsible for eotaxin-3 expression in human colonic myofibroblasts. Eotaxin-3 mRNA and protein expression was evaluated by real time-polymerase chain reaction (PCR) and enzyme-linked immunosorbent assay (ELISA), respectively. Eotaxin-3 mRNA expression was elevated significantly in the active lesions of ulcerative colitis (UC) patients. Significant elevations were also observed in the active lesions of Crohn's disease (CD) patients, but this was significantly lower than that detected in the active UC lesions. There were no significant increases in the inactive lesions of UC or CD patients. Colonic myofibroblasts were identified as a major source of eotaxin-3 in the colonic mucosa, and interleukin (IL)-4 and IL-13 enhanced eotaxin-3 mRNA and protein expression significantly in these cells. There was a significant positive correlation between mucosal eotaxin-3 and IL-4 mRNA expression in the active lesions of IBD patients. The IL-4- and IL-13-induced eotaxin-3 mRNA expression was regulated by the signal transducer and activator of transcription-6 (STAT-6) and suppressor of cytokine signalling (SOCS)1-mediated pathways. Interferon (IFN)-γ acts as a negative regulator on the IL-4- and IL-13-induced eotaxin-3 expression via STAT-1 activation. Eotaxin-3 expression was elevated specifically in the active lesions of IBD, in particular UC. Eotaxin-3 derived from colonic myofibroblasts may play an important role in the pathophysiology of UC.
Keywords: eosinophil, IBD, SOCS, STAT
Introduction
Inflammatory bowel diseases (IBDs), ulcerative colitis (UC) and Crohn's disease (CD) are chronic intestinal disorders of unknown aetiology 1. The most widely accepted hypothesis on the pathogenesis of IBD is that the mucosal immune system has an aberrant response towards luminal antigens such as dietary factors and/or commensal bacteria in genetically susceptible individuals 1–6. This may be supported by recent findings that the genes encoding innate immune responses are also responsible for determining susceptibility to IBD 7,8. In addition, IBD is often characterized by an imbalance between the effector and the regulatory activities of intestinal immunity, with a preponderance of proinflammatory cytokines 9.
Eosinophils participate in a number of biological processes such as wound healing, defence against parasites and allergic inflammation 10. Eosinophils are characterized by specific cytoplasmic granules containing four major proteins: eosinophil cationic protein (ECP), eosinophil peroxidase (EPO), eosinophil protein X (EPX) and major basic protein (MBP) 11. Recent studies suggest that eosinophils also play a role in tissue repair through fibroblast stimulation by releasing ECP and transforming growth factor (TGF)-β 10.
A growing number of studies support a functional role for eosinophils in IBD 11–14. Eosinophil numbers were increased in active disease compared with normal control subjects 10. Other studies have documented the increased faecal content of ECP in patients with UC, and a high content of ECP and EPX in the stool of patients with UC and CD 12,15,16. Eosinophil granule proteins are increased in the whole-gut lavage fluid from patients with CD and UC 17. Lampinen et al. reported that eosinophils are activated in the remission phase rather than the active phase of UC patients, suggesting a role of these cells in tissue repair. Eosinophils are increased and activated during inflammation in IBD, but a clear role for them in the pathogenesis of IBD has not yet been elucidated.
The eotaxin subfamily of CC chemokines consists of eotaxin-1/CCL11, eotaxin-2/CCL24 and eotaxin-3/CCL26 18. All eotaxins induce the trafficking of eosinophils to the sites of inflammation via CC chemokine receptor 3 (CCR3), which is also expressed by several different cell types, including basophils, dendritic cells, smooth muscle cells, epithelial cells and fibroblasts 19. The sequence similarity between the three eotaxins is limited (<40%), but their functional properties are very similar 20. Eotaxin-1 and -2 are expressed by both haematopoietic and non-haematopoietic cells, but eotaxin-3 expression has been reported to be limited to non-haematopoietic cells 18. Interleukin (IL)-4 is the main inducer for eotaxin-3 expression, whereas eotaxin-1 is up-regulated by IL-4 and the proinflammatory cytokine tumour necrosis factor (TNF)-α 21. Eotaxin-3 is expressed in vascular endothelial cells and human dermal fibroblasts after IL-4 and IL-13 stimulation 21, and this is dependent upon the IL-4-/IL-13-specific transcription factor, signal transducers and activator of transcription (STAT)-6 20,21. Eotaxin-3 is expressed on the surface of IL-4-stimulated endothelial cells and promotes eosinophil transmigration 22.
The mechanisms responsible for the accumulation of eosinophils in IBD lesions remain unclear, but it is likely that various steps involving several chemotactic factors operate in order to recruit eosinophils from the peripheral blood into the tissues. The purpose of this study was to investigate mucosal eotaxin expression in IBD patients. Moreover, we examined the molecular mechanisms underlying eotaxin induction in the inflamed mucosa of IBD.
Materials and methods
Reagents
Recombinant human cytokines were purchased from R&D Systems (Minneapolis, MN, USA). All other reagents were purchased from Sigma Chemical Co. (St Louis, MO, USA). Human eotaxin enzyme-linked immunosorbent assay (ELISA) kits were purchased from R&D Systems. STAT-1-specific, STAT-6-specific, suppressor of cytokine signalling (SOCS)-specific and control siRNAs were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA).
Tissue samples
The diagnosis of IBD was based on conventional clinical and endoscopic criteria. Biopsied specimens from 42 patients with UC (21 active and 21 inactive patients) and 33 patients with CD (16 active and 17 inactive patients) were used, with informed consent. The disease activity of the samples was determined by endoscopic and histological findings. Normal colorectal tissues (n = 16) were obtained by surgical resection of colon cancer at distal tumour sites. The ethics committee of the Shiga University of Medical Science approved this project.
Culture of human colonic myofibroblasts
Primary colonic myofibroblast cultures were prepared according to the method reported by Mahida et al. 23. The cellular characteristics and culture conditions have also been described in our previous report 24. Samples of the human adult colonic mucosa were obtained from surgical specimens (>5 cm from the tumour margin) from patients undergoing a partial colectomy for carcinoma, with their informed consent. All studies were performed on passages 3–6 of myofibroblasts isolated from six resection specimens. The human colon cancer cell lines HT-29 and Caco-2 were obtained from the American Type Culture Collection (Manassas, VA, USA) 25.
ELISA for the quantification of antigenic eotaxin-3
Eotaxin-3 protein in the samples was quantified by an ELISA kit, purchased from R&D Systems.
Immunohistochemical staining for eotaxin-3 protein
The cells were grown on a culture slide system (BD, Franklin Lakes, NJ, USA). The cells were fixed and reacted with anti-eotaxin-3 antibodies (R&D Systems) and cyanin 2 (Cy2)-labelled secondary antibody (Rockland Immunochemicals, Gilbertsville, PA, USA). Nuclei were visualized using mounting medium with 4-6-diamidino-2-phenylindole (DAPI) (Vector Laboratories, Burlingame, CA, USA). A digital confocal laser scanning microscope (Nikon, Tokyo, Japan) was used for analysis.
Real-time polymerase chain reaction
The expression of mRNA in the samples was assessed by real-time PCR analyses. The oligonucleotide primers used in this study were human eotaxin-3 (sense: GACCTGGGTGCGAAGCTATG, anti-sense: TGGGAGGAAACACCCTCTCC), human IL-4 (sense: TGCCTCCAAGAACACAACTG, anti-sense: GTTTCAGGAATCGGATCAGC), human IL-13 (sense: GTGGCCCAGTTTGTAAAGGA, anti-sense: CAGCACAGGCTGAGGTCTAA) and β-actin (sense: TGACCCAGATCATGTTTGAGACCT, anti-sense: CCACGTCACACTTCATGATGATGGAG). Real-time PCR was performed using a LightCycler 480 system (Roche Applied Science, Tokyo, Japan). The PCR was performed using a SYBR Premix Ex Taq (Takara, Shiga, Japan). The data were normalized versus β-actin for human eotaxin-3.
Western blot analyses
The stimulated cells were lysed in a sodium dodecyl sulphide (SDS) sample buffer containing orthovanadate. Western blots were then performed according to a method described previously 26. Detection was performed using the enhanced chemiluminescence Western blotting system (GE Healthcare, Little Chalfont, UK).
STATs and SOCSs mRNA interference (RNAi) experiments
The siRNA for STAT-1, STAT-6, SOCS1, SOCS3, SOCS5 and a control siRNA were used. Human colonic subepithelial myofibroblasts (SEMFs) were cultured for 4 days in complete medium that did not contain antibiotics. The cells were then seeded onto a six-well plate 1 day prior to the transfection, and cultured to 60–70% confluence on the following day. For the RNAi experiments, Lipofectamine™ RNAi MAX Reagent (Invitrogen, Carlsbad, CA, USA) was used.
Statistical analysis
The statistical significance of the differences was determined by Student's t-test. Differences resulting in P-values less than 0·05 were considered to be statistically significant.
Results
Eotaxin-3 expression in IBD mucosa
Eotaxin-3 mRNA expression was analysed by real-time PCR in biopsied samples from IBD patients. As shown in Fig. 1, eotaxin-3 mRNA was detected very weakly in the samples from the normal mucosa. A remarkable and significant increase of eotaxin-3 mRNA expression was observed in the samples from the active lesions of UC patients. Significant elevation was also observed in the samples from the active lesions of CD patients, but this was significantly lower than that detected in the active lesions of UC patients. There was no significant increase in the inactive lesions of UC and CD patients.
Fig. 1.
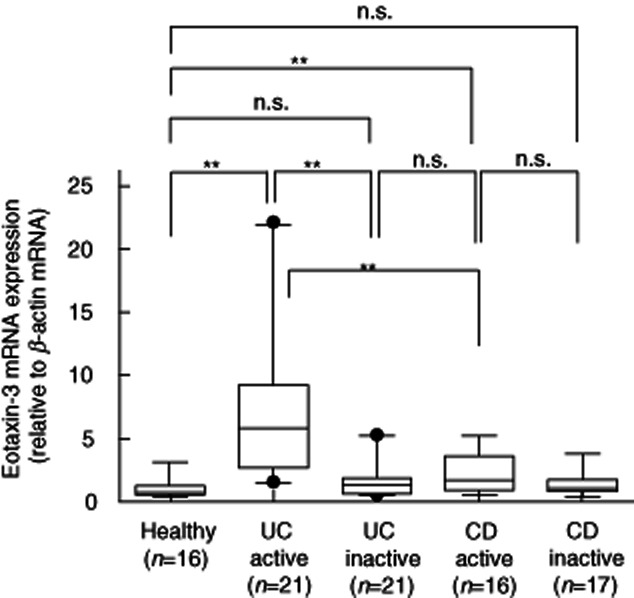
Eotaxin-3 mRNA expression in the inflammatory bowel disease (IBD) mucosa. Total RNA was extracted from biopsied samples, and eotaxin-3 mRNA expression was evaluated by real-time polymerase chain reaction (PCR) analyses. The eotaxin-3 mRNA expression was expressed relative to the β-actin mRNA expression. **P < 0·01; n.s.: not significant.
Regulation of eotaxin-3 expression in colonic myofibroblasts and IBD mucosa
Based on the in-vivo expression of eotaxin-3 in the inflamed IBD mucosa, we examined eotaxin-3 expression in isolated human colonic myofibroblasts. Colonic myofibroblasts were stimulated with various cytokines and lipopolysaccharide (LPS) for 24 h, and eotaxin-3 mRNA expression was then determined by real-time PCR (Fig. 2a). Very weak eotaxin-3 mRNA expression was detected in the unstimulated colonic myofibroblasts, and IL-4 and IL-13 stimulation enhanced eotaxin-3 mRNA expression significantly. Immunohistochemical analyses showed cytoplasmic expression of eotaxin-3 protein in the IL-4- and IL-13-stimulated colonic myofibroblasts (Fig. 2b).
Fig. 2.
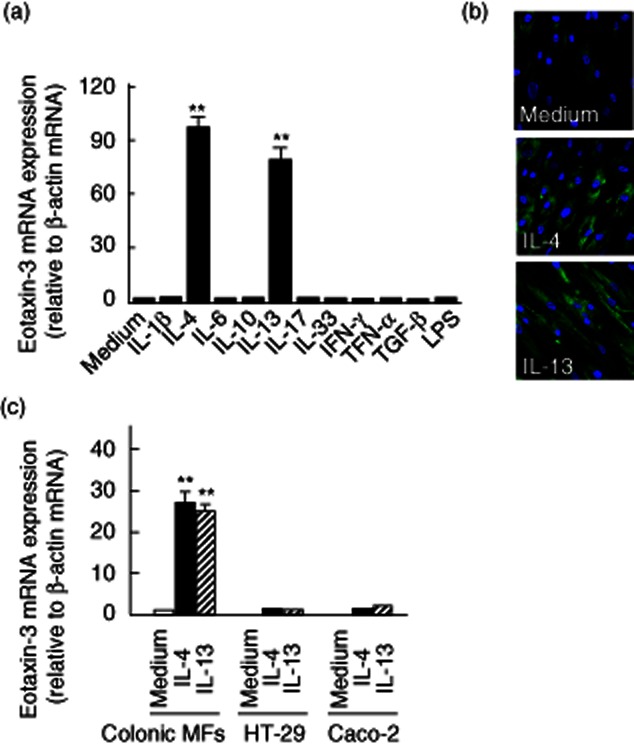
Eotaxin-3 mRNA expression in human colonic myofibroblasts. (a) Colonic myofibroblasts, derived from normal mucosa, were stimulated with cytokines (100 ng/ml) for 24 h. Eotaxin-3 mRNA expression was then analysed by real-time polymerase chain reaction (PCR). (b) Eotaxin-3 protein expression in interleukin (IL)-4 or IL-13 stimulated cells. The cells were grown on culture slides and stimulated with cytokines (100 ng/ml) for 24 h. Eotaxin-3 was stained with anti-eotaxin-3 antibodies. (c) Comparison of eotaxin-3 mRNA expression in intestinal epithelial cell lines (HT-29 and Caco-2 cells) and colonic myofibroblasts. The cells were stimulated with cytokines (100 ng/ml) for 24 h. Eotaxin-3 mRNA expression was expressed relative to the β-actin mRNA expression (mean ± standard deviation from three different experiments). MFs: myofibroblasts. **P < 0·01 versus medium only.
In contrast, in the colonic epithelial cell lines (HT-29 and Caco-2), eotaxin-3 mRNA was not detected in the unstimulated cells. Both IL-4 and IL-13 stimulated eotaxin-3 mRNA expression weakly, but these were somewhat modest compared to the responses of colonic myofibroblasts (Fig. 2c).
These responses were investigated in normal and inflamed mucosa of IBD patients [8 normal mucosa, UC (11 active and 8 inactive lesions) and CD (10 active and eight inactive lesions), total n = 45]. A significant positive correlation was found between eotaxin-3 and IL-4 mRNA expression (Pearson's correlation R = 0.69, P < 0·0001, n = 45) (Fig. 3a). However, there was no correlation between eotaxin-3 and IL-13 mRNA expression (Fig. 3b).
Fig. 3.
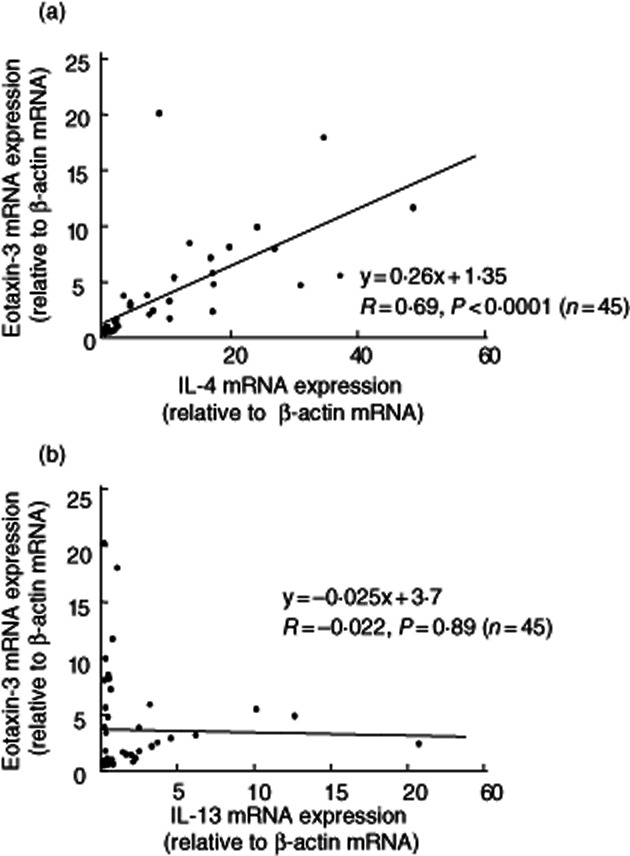
Correlation between mucosal eotaxin3 and interleukin (IL)-4/IL-13 mRNA expression in the mucosa. Eotaxin-3 mRNA expression was expressed relative to the β-actin mRNA expression. (a) A significant positive correlation was observed between eotaxin-3 and IL-4 mRNA levels (Pearson's correlation R = 0·69, P < 0·0001, n = 45). (b) There was no correlation between eotaxin-3 and IL-13 mRNA expression (n = 45).
Eotaxin-3 induction by IL-4 and IL-13
The effects of IL-4 and IL-13 were analysed precisely. As shown in Fig. 4a,b, IL-4 and IL-13 induced eotaxin-3 mRNA expression dose- and time-dependently. Similarly, IL-4 and IL-13 induced eotaxin-3 protein secretion in a dose- and time-dependent manner (Fig. 4c,d).
Fig. 4.
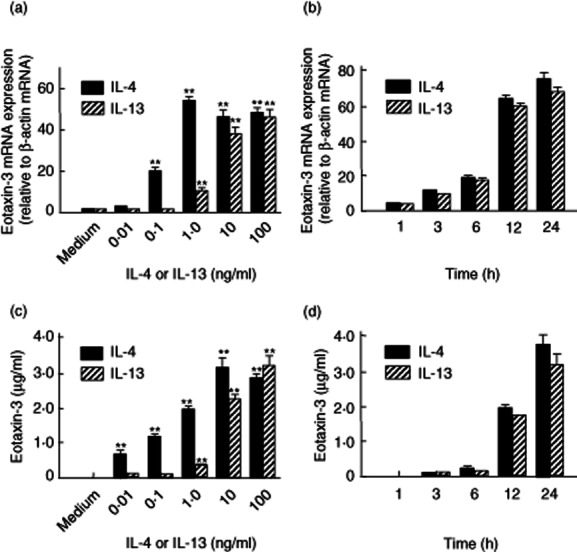
Effects of interleukin (IL)-4 and IL-13 on eotaxin-3 mRNA expression in myofibroblasts derived from normal colonic mucosa. (a) Dose-dependent effects of IL-4 and IL-13 on eotaxin-3 mRNA expression. The cells were incubated for 24 h with increasing concentrations of IL-4 or IL-13. Eotaxin-3 mRNA expression was expressed relative to the β-actin mRNA expression [mean ± standard deviation (s.d.) from three different experiments]. **P < 0·01 versus medium only. (b) Time-dependent effects of IL-4 and IL-13 on eotaxin-3 mRNA expression. The cells were stimulated with IL-4 (100 ng/ml) or IL-13 (100 ng/ml) for the predetermined times. Eotaxin-3 mRNA expression was expressed relative to β-actin mRNA expression (mean ± s.d. from three different experiments). (c) Dose-dependent effects of IL-4 and IL-13 on eotaxin-3 protein secretion. The cells were incubated for 24 h with increasing concentrations of IL-4 or IL-13, and eotaxin-3 levels were determined by enzyme-linked immunosorbent assay (ELISA). All values are expressed as means ± s.d. (n = 3). **P < 0·01 versus medium only. (d) Time-dependent effects of IL-4 and IL-13 on eotaxin-3 protein secretion. The cells were stimulated with IL-4 (100 ng/ml) or IL-13 (100 ng/ml) for predetermined times, and eotaxin-3 levels were determined by ELISA. All values are expressed as mean ± s.d. (n = 3).
Role of STAT-6 activation in IL-4-/IL-13-induced eotaxin-3 expression
It has been reported that IL-4 and IL-13 activate STAT-6, which is a transcription factor required for many biological functions 27. To confirm this response in our system, we evaluated the effects of IL-4 and IL-13 on the activation of STAT-6 in colonic myofibroblasts. As shown in Fig. 5a, IL-4 and IL-13 induced phosphorylation of STAT-6 as early as 15 min after stimulation. In the STAT-6 siRNA-transfected cells, this IL-4- and IL-13-induced eotaxin-3 expression was reduced significantly (Fig. 5b). These findings indicate that IL-4- and IL-13-induced eotaxin-3 expression is dependent upon STAT-6 activation in human colonic myofibroblasts.
Fig. 5.
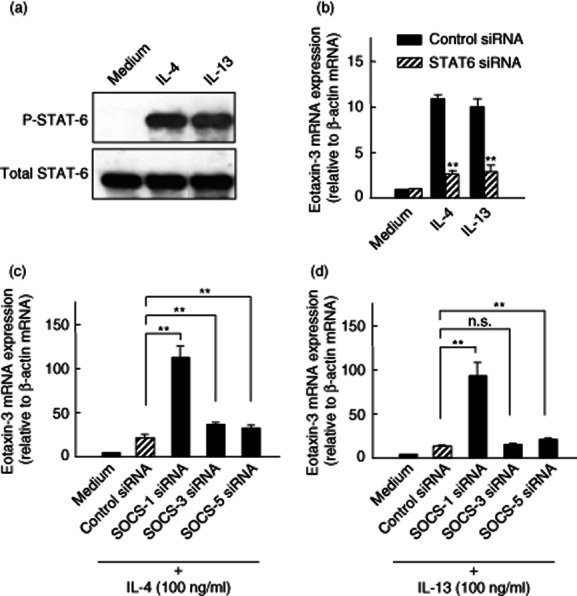
Cellular signalling for interleukin (IL)-4- and IL-13-mediated eotaxin-3 induction. (a) IL-4- and IL-13-induced signal transducer and activator of transcription-6 (STAT6) activation. Colonic myofibroblasts, derived from normal mucosa, were stimulated with cytokines [IL-4 (100 ng/ml) and IL-13 (100 ng/ml)] for 15 min, and the phosphorylated (p-) and total STAT-6 were detected by Western blotting. (b) Effects of STAT-6 silencing on eotaxin-3 expression. The cells were transfected with the siRNA specific for STAT-6 and incubated for 24 h. Eotaxin-3 mRNA expression was expressed relative to β-actin mRNA expression [mean ± standard deviation (s.d.) from three different experiments]. **P < 0·01 versus control siRNA. (c) Effects of suppressor of cytokine signalling (SOCS) silencing on IL-4-induced eotaxin-3 expression. The cells were transfected with siRNAs specific for SOCS1, SOCS3 and SOCS5, and incubated for 24 h with IL-4 (100 ng/ml). Eotaxin-3 mRNA expression was expressed relative to β-actin mRNA expression (mean ± s.d. from three different experiments). (d) Effects of SOCS silencing on IL-13-induced eotaxin-3 expression. The cells were transfected with siRNAs specific for SOCS1, SOCS3 and SOCS5 and incubated for 24 h with IL-13 (100 ng/ml). Eotaxin-3 mRNA expression was expressed relative to β-actin mRNA expression (mean ± s.d. from three different experiments).
SOCS1 plays a central negative role in IL-4-/IL-13-induced eotaxin-3 expression
SOCS1–3 act as negative regulators of cytokine signalling 20. A previous study reported negative regulatory actions of SOCS1 and SOCS3 for the IL-4- and IL-13-induced eotaxin-3 gene expression 20. In addition, SOCS5 has been reported to play a role in eosinophilic inflammation in mice 28. Based on these notions, we evaluated the effects of siRNAs specific for SOCS1, SOCS3 and SOCS5 on IL-4- and IL-13-induced eotaxin-3 mRNA expression in colonic myofibroblasts. As shown in Fig. 5c, silencing SOCS1 induced a significant and marked enhancement of IL-4-induced eotaxin-3 mRNA expression. Silencing SOCS3 and SOCS5 also induced an increase in eotaxin-3 mRNA expression, but these effects were much weaker than the effects of the SOCS1 siRNA. Similar effects of siRNAs specific for SOCS1, SOCS3 and SOCS5 were also observed for IL-13-induced eotaxin-3 mRNA expression (Fig. 5d). Thus, it became clear that SOCS1 is a major negative regulator for the IL-4- and IL-13-induced activation of eotaxin-3 gene expression in human colonic myofibroblasts.
Effects of interferon (IFN)-γ on IL-4-/IL-13-induced eotaxin-3 expression
A T helper type 1 (Th1)/Th2 cytokine imbalance with a predominance of Th1 cytokines has been suggested to be of pathogenic importance in IBD 29. This led us to investigate the effects of IFN-γ, a major Th1 cytokine, on IL-4-/IL-13-induced eotaxin-3 expression in human colonic myofibroblasts. As shown in Fig 6a, IFN-γ suppressed IL-4-induced eotaxin-3 mRNA expression dose-dependently. Similarly, IFN-γ suppressed IL-13-induced eotaxin-3 mRNA expression dose-dependently (Fig. 6b). Thus, the Th1 cytokine IFN-γ had a significant inhibitory effect on Th2 cytokine (IL-4- and IL-13)-induced eotaxin-3 mRNA expression in human colonic myofibroblasts.
Fig. 6.
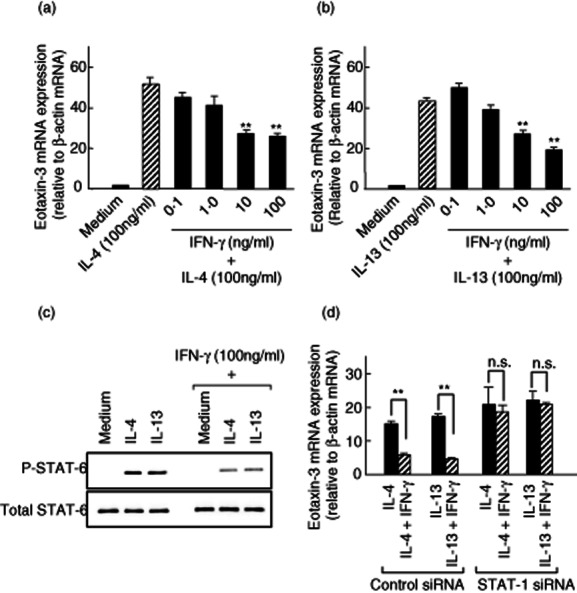
Effects of interferon (IFN)-γ on interleukin (IL)-4 and IL-13-induced eotaxin-3 mRNA expression. (a,b) Colonic myofibroblasts, derived from normal mucosa, were stimulated for 24 h with IL-4 (100 ng/ml) or IL-13 (100 ng/ml) in the presence or absence of increasing concentrations of IFN-γ. Eotaxin-3 mRNA expression was expressed relative to β-actin mRNA expression [mean ± standard deviation (s.d.) from three different experiments]. **P < 0·01 versus IL-4 or IL-13 alone. (c) Effects of IFN-γ on IL-4- and IL13-induced signal transducer and activator of transcription-6 (STAT6) activation. The cells were preincubated with IFN-γ (100 ng/ml) for 3 h, and then stimulated with IL-4 (100 ng/ml) and IL-13 (100 ng/ml) for 15 min. The phosphorylated (p-) and total STAT-6 were detected by Western blotting. (d) Effects of STAT-1 silencing on eotaxin-3 expression. The cells were transfected with the siRNA specific for STAT-1 and incubated for 24 h with IL-4 (100 ng/ml) or IL-13 (100 ng/ml). Eotaxin-3 mRNA expression was expressed relative to β-actin mRNA expression (mean ± s.d. from three different experiments). **P < 0·01.
In order to define the molecular mechanisms underlying the inhibitory effects of IFN-γ, we evaluated the effects of IFN-γ on STAT-6 activation. As shown in Fig. 6c, IFN-γ induced inhibition of STAT-6 activation in colonic myofibroblasts.
Furthermore, we evaluated the effects of a STAT-1-specific siRNA on IFN-γ-induced inhibitory effects on eotaxin-3 mRNA expression, as STAT-1 is a master transcription factor of IFN-γ signalling 30. As shown in Fig. 6d, the STAT-1 siRNA attenuated completely the IFN-γ-induced inhibitory effects on eotaxin-3 mRNA expression.
Discussion
In this study, we have demonstrated that mucosal eotaxin-3 mRNA was elevated significantly in the active lesions of UC patients. This elevation was specific to the active UC patients, as the increase in the active lesions of CD patients was modest and significantly lower than that detected in the active lesions of UC patients. A recent study by Manousou et al. revealed a similar observation of significant elevation in mucosal eotaxin-3 mRNA expression in IBD patients, especially in UC patients 31, but they did not find any differences between active and inactive disease. The reason for this discrepancy concerning disease activity might be associated with the methods used. They performed eotaxin-3 analysis using a reverse transcription (RT)–PCR-based method, but we used a semi-quantitative real-time PCR method, which serves as a much more sensitive and quantitative analysis. In addition, they defined disease activity by the clinical activity index based on clinical symptoms, whereas we determined disease activity according to endoscopic and histological findings. Finally, the majority of patients enrolled in Manousou's study (22 of 32 IBD patients) were clinically active. Thus, to our knowledge, the current study is the first report showing an elevation of mucosal eotaxin-3 expression in the active lesions of UC patients, and to a lesser degree in the active lesions of CD patients, suggesting a role for eotaxin-3 in the pathophysiology of active IBD patients.
The molecular mechanisms responsible for eotaxin-3 expression in the colonic mucosa have not been identified fully. To investigate the regulatory mechanisms involved in eotaxin-3 expression in the UC mucosa, we used colonic myofibroblasts isolated from the normal human colonic mucosa [23]. Among the various cytokines, Th2 cytokines IL-4 and IL-13 exerted remarkable effects on eotaxin-3 induction. Manousou et al. evaluated eotaxin-3 mRNA expression recently in the colon cancer cell lines HT-29 and Caco-2 cells, and suggested that colonic epithelial cells might be a source of eotaxin-3 in the inflamed mucosa of IBD patients. However, as shown in Fig. 2b, eotaxin-3 mRNA expression in both HT-29 and Caco-2 cells was very weak, even under IL-4 and IL-13 stimulation, compared with the responses of human colonic myofibroblasts. This finding negatively supports the hypothesis that colonic epithelial cells are the source of eotaxin-3 in the mucosa. In contrast, the current observations suggest strongly that colonic myofibroblasts are one of the major sources of eotaxin-3 in the inflamed mucosa of IBD patients. A limitation of the present study is the use of colonic myofibroblasts derived from normal mucosa. To clarify the role of colonic myofibroblasts in the pathophysiology of IBD, in future further studies should be performed using colonic myofibroblasts derived from IBD patients. In addition, eotaxin-3 expression in colonic epithelial cells should be confirmed using non-transformed cells.
Inflammation in UC has been difficult to classify using the Th1/Th2 paradigm 32,33. In patients with UC, the secretion of IL-4 as well as IFN-γ or IL-12 was not increased, and thus the inflammation is clearly not a Th1 or Th2 response 33. The Th2 characteristics of UC inflammation have been explained recently by the increased IL-13 production by natural killer (NK) T cells, which manifest reactivity to antigens presented by epithelial cells 1,34. Heller et al. demonstrated that lamina propria mononuclear cells (LPMCs) isolated from UC patients exhibited a strong capacity to secrete IL-13, and this was much more prominent compared to LPMCs isolated from CD patients 34. However, we could not detect a significant IL-13 mRNA expression in IBD mucosa (data not shown), and there was no correlation between eotaxin-3 mRNA expression and IL-13 mRNA expression. The reason for this discrepancy is unclear, but these findings might be associated with different distributions of IL-4-secreting Th2 cells and IL-13-secreting NK T cells in the inflamed mucosa. In contrast, we found a significant positive correlation between mucosal eotaxin-3 mRNA expression and IL-4 mRNA expression. This was supported by the recent study that circulating Th2 phenotype lymphocytes were increased in UC patients 31. Thus, these findings suggest that, in the inflamed mucosa of UC patients, Th2 cells may drive mucosal eotaxin-3 expression in colonic myofibroblasts via IL-4 secretion. Conversely, in future the role of IL-13 should be investigated further.
The IL-4-/IL-13-induced activation of the eotaxin-3 gene has been reported to be a STAT-6-dependent process mediated by a single STAT-6 binding motif located upstream of the transcription initiation site 20. In human colonic myofibroblasts, IL-4 and IL-13 actually induced rapid phosphorylation of STAT-6, and the eotaxin-3 mRNA expression induced by IL-4 and IL-13 was blocked significantly and almost completely in the STAT-6 siRNA-transfected cells. These findings indicate that the IL-4- and IL-13-induced eotaxin-3 mRNA expression was actually mediated by STAT-6 activation.
Cytokine signalling is regulated strictly by the SOCS family of proteins 33,35. Among them, SOCS1 is relatively specific to IFN-γ/STAT-1 and IL-4/STAT-6, whereas SOCS3 is specific to STAT-3. SOCS1 inhibits cytokine signalling by suppressing Janus kinase (JAK) activity and promoting the degradation of the activated cytokine–receptor complex. SOCS3 has been shown to have strong expression in the colon of CD and UC patients and in most mouse colitis models 33. These facts led us to explore the role of STAT proteins in IL-4- and IL-13-induced eotaxin-3 expression in colonic myofibroblasts. As shown in Fig. 4c, IL-4-induced eotaxin-3 mRNA expression was enhanced by silencing SOCS1, SOCS3 and SOCS5 expression. In particular, a marked enhancement was observed in the SOCS1-specific siRNA transfected cells. Similar responses were also observed for IL-13-induced eotaxin-3 expression. These findings suggest that SOCS1 plays a central role in suppressing IL-4 and IL-13 signalling to induce eotaxin-3 expression in colonic myofibroblasts. In contrast, an important role for SOCS3 in IL-4- and IL-13-induced eotaxin-3 expression has been reported in other cell types 20, but its role in colonic myofibroblasts was considered to be minimal.
IFN-γ is known to antagonize the actions of IL-4 by repressing the expression of a number of IL-4-induced genes, whereas IL-4 has been shown to antagonize IFN-γ 33. In colonic myofibroblasts, IFN-γ suppressed IL-4- and IL-13-induced eotaxin-3 expression dose-dependently, and this was accompanied by the suppression of STAT-6 activation. Moreover, this inhibitory effect on eotaxin-3 induction was blocked completely by silencing STAT-1 expression. Thus, eotaxin-3 expression in colonic myofibroblasts is controlled positively by Th2 cytokines (IL-4 and IL-13), and regulated negatively by Th1 cytokine (IFN-γ). It can be speculated that eosinophil recruitment via eotaxin-3 expression might be determined by a balance between Th1 and Th2 cytokine expression in the mucosa.
In conclusion, eotaxin-3 was up-regulated in active UC patients. In addition, colonic myofibroblasts have been identified as a local source for eotaxin-3 in the colonic mucosa. IL-4- and IL-13 are potent inducers of eotaxin-3, whereas IFN-γ acts as a negative regulator of eotaxin-3 expression. These findings may provide important insights into the molecular mechanisms responsible for eosinophil accumulation and activation in the inflamed mucosa of UC patients. However, a clear role for eosinophils in the pathogenesis of IBD and the precise regulatory mechanism of eotaxin-3 expression remain unclear. In future, these questions could be explored further at the experimental level using cytokine- or eosinophil-deficient mice.
Acknowledgments
This study was supported by a Grant-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology of Japan (24590940), and a grant for the Intractable Diseases from the Ministry of Health, Labor and Welfare of Japan.
Disclosures
The authors disclose no conflicts of interest.
References
- 1.Mayer L. Evolving paradigms in the pathogenesis of IBD. J Gastroenterol. 2010;45:9–16. doi: 10.1007/s00535-009-0138-3. [DOI] [PubMed] [Google Scholar]
- 2.Podolsky DK. Inflammatory bowel disease. N Engl J Med. 2002;347:417–429. doi: 10.1056/NEJMra020831. [DOI] [PubMed] [Google Scholar]
- 3.Mizoguchi A, Mizoguchi E. Inflammatory bowel disease, past, present and future: lessons from animal models. J Gastroenterol. 2008;43:1–17. doi: 10.1007/s00535-007-2111-3. [DOI] [PubMed] [Google Scholar]
- 4.Sartor RB. Mechanisms of disease: pathogenesis of Crohn's disease and ulcerative colitis. Nat Clin Pract Gastroenterol Hepatol. 2006;3:390–407. doi: 10.1038/ncpgasthep0528. [DOI] [PubMed] [Google Scholar]
- 5.Andoh A, Imaeda H, Aomatsu T, et al. Comparison of the fecal microbiota profiles between ulcerative colitis and Crohn's disease using terminal restriction fragment length polymorphism analysis. J Gastroenterol. 2011;46:479–486. doi: 10.1007/s00535-010-0368-4. [DOI] [PubMed] [Google Scholar]
- 6.Andoh A, Kuzuoka H, Tsujikawa T, et al. Multicenter analysis of fecal microbiota profiles in Japanese patients with Crohn's disease. J Gastroenterol. 2012;47:1298–1307. doi: 10.1007/s00535-012-0605-0. [DOI] [PubMed] [Google Scholar]
- 7.Franke A, McGovern DP, Barrett JC, et al. Genome-wide meta-analysis increases to 71 the number of confirmed Crohn's disease susceptibility loci. Nat Genet. 2010;42:1118–1125. doi: 10.1038/ng.717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Asano K, Matsushita T, Umeno J, et al. A genome-wide association study identifies three new susceptibility loci for ulcerative colitis in the Japanese population. Nat Genet. 2009;41:1325–1329. doi: 10.1038/ng.482. [DOI] [PubMed] [Google Scholar]
- 9.Kobori A, Yagi Y, Imaeda H, et al. Interleukin-33 expression is specifically enhanced in inflamed mucosa of ulcerative colitis. J Gastroenterol. 2010;45:999–1007. doi: 10.1007/s00535-010-0245-1. [DOI] [PubMed] [Google Scholar]
- 10.Lampinen M, Ronnblom A, Amin K, et al. Eosinophil granulocytes are activated during the remission phase of ulcerative colitis. Gut. 2005;54:1714–1720. doi: 10.1136/gut.2005.066423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Blom K, Rubin J, Halfvarson J, et al. Eosinophil associated genes in the inflammatory bowel disease 4 region: correlation to inflammatory bowel disease revealed. World J Gastroenterol. 2012;18:6409–6419. doi: 10.3748/wjg.v18.i44.6409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Coppi LC, Thomazzi SM, de Ayrizono ML, et al. Comparative study of eosinophil chemotaxis, adhesion, and degranulation in vitro in ulcerative colitis and Crohn's disease. Inflamm Bowel Dis. 2007;13:211–218. doi: 10.1002/ibd.20018. [DOI] [PubMed] [Google Scholar]
- 13.Masterson JC, McNamee EN, Jedlicka P, et al. CCR3 blockade attenuates eosinophilic ileitis and associated remodeling. Am J Pathol. 2011;179:2302–2314. doi: 10.1016/j.ajpath.2011.07.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lampinen M, Backman M, Winqvist O, et al. Different regulation of eosinophil activity in Crohn's disease compared with ulcerative colitis. J Leukoc Biol. 2008;84:1392–1399. doi: 10.1189/jlb.0807513. [DOI] [PubMed] [Google Scholar]
- 15.Berstad A, Borkje B, Riedel B, Elsayed S. Increased fecal eosinophil cationic protein in inflammatory bowel disease. Hepatogastroenterology. 1993;40:276–278. [PubMed] [Google Scholar]
- 16.Wagner M, Peterson CG, Ridefelt P, Sangfelt P, Carlson M. Fecal markers of inflammation used as surrogate markers for treatment outcome in relapsing inflammatory bowel disease. World J Gastroenterol. 2008;14:5584–5589. doi: 10.3748/wjg.14.5584. discussion 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Carlson M, Raab Y, Peterson C, Hallgren R, Venge P. Increased intraluminal release of eosinophil granule proteins EPO, ECP, EPX, and cytokines in ulcerative colitis and proctitis in segmental perfusion. Am J Gastroenterol. 1999;94:1876–1883. doi: 10.1111/j.1572-0241.1999.01223.x. [DOI] [PubMed] [Google Scholar]
- 18.Stubbs VE, Power C, Patel KD. Regulation of eotaxin-3/CCL26 expression in human monocytic cells. Immunology. 2010;130:74–82. doi: 10.1111/j.1365-2567.2009.03214.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.De Lucca GV. Recent developments in CCR3 antagonists. Curr Opin Drug Discov Devel. 2006;9:516–524. [PubMed] [Google Scholar]
- 20.Hebenstreit D, Luft P, Schmiedlechner A, Duschl A, Horejs-Hoeck J. SOCS-1 and SOCS-3 inhibit IL-4 and IL-13 induced activation of Eotaxin-3/CCL26 gene expression in HEK293 cells. Mol Immunol. 2005;42:295–303. doi: 10.1016/j.molimm.2004.09.004. [DOI] [PubMed] [Google Scholar]
- 21.Hoeck J, Woisetschlager M. Activation of eotaxin-3/CCLl26 gene expression in human dermal fibroblasts is mediated by STAT6. J Immunol. 2001;167:3216–3222. doi: 10.4049/jimmunol.167.6.3216. [DOI] [PubMed] [Google Scholar]
- 22.Nakayama T, Watanabe Y, Oiso N, et al. Eotaxin-3/CC chemokine ligand 26 is a functional ligand for CX3CR1. J Immunol. 2010;185:6472–6479. doi: 10.4049/jimmunol.0904126. [DOI] [PubMed] [Google Scholar]
- 23.Mahida YR, Beltinger J, Makh S, et al. Adult human colonic subepithelial myofibroblasts express extracellular matrix proteins and cyclooxygenase-1 and -2. Am J Physiol. 1997;273:G1341–1348. doi: 10.1152/ajpgi.1997.273.6.G1341. [DOI] [PubMed] [Google Scholar]
- 24.Okuno T, Andoh A, Bamba S, et al. Interleukin-1beta and tumor necrosis factor-alpha induce chemokine and matrix metalloproteinase gene expression in human colonic subepithelial myofibroblasts. Scand J Gastroenterol. 2002;37:317–324. doi: 10.1080/003655202317284228. [DOI] [PubMed] [Google Scholar]
- 25.Dharmsathaphorn K, McRoberts JA, Mandel KG, Tisdale LD, Masui H. A human colonic tumor cell line that maintains vectorial electrolyte transport. Am J Physiol. 1984;246:G204–208. doi: 10.1152/ajpgi.1984.246.2.G204. [DOI] [PubMed] [Google Scholar]
- 26.Shimada M, Andoh A, Hata K, et al. IL-6 secretion by human pancreatic periacinar myofibroblasts in response to inflammatory mediators. J Immunol. 2002;168:861–868. doi: 10.4049/jimmunol.168.2.861. [DOI] [PubMed] [Google Scholar]
- 27.Jiang H, Harris MB, Rothman P. IL-4/IL-13 signaling beyond JAK/STAT. J Allergy Clin Immunol. 2000;105:1063–1070. doi: 10.1067/mai.2000.107604. [DOI] [PubMed] [Google Scholar]
- 28.Ohshima M, Yokoyama A, Ohnishi H, et al. Overexpression of suppressor of cytokine signalling-5 augments eosinophilic airway inflammation in mice. Clin Exp Allergy. 2007;37:735–742. doi: 10.1111/j.1365-2222.2007.02707.x. [DOI] [PubMed] [Google Scholar]
- 29.Fuss IJ. Is the Th1/Th2 paradigm of immune regulation applicable to IBD? Inflamm Bowel Dis. 2008;14(Suppl. 2):S110–112. doi: 10.1002/ibd.20683. [DOI] [PubMed] [Google Scholar]
- 30.Darnell JE, Jr, Kerr IM, Jak SGR. STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science. 1994;264:1415–1421. doi: 10.1126/science.8197455. [DOI] [PubMed] [Google Scholar]
- 31.Manousou P, Kolios G, Valatas V, et al. Increased expression of chemokine receptor CCR3 and its ligands in ulcerative colitis: the role of colonic epithelial cells in in-vitro studies. Clin Exp Immunol. 2010;162:337–347. doi: 10.1111/j.1365-2249.2010.04248.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.West GA, Matsuura T, Levine AD, Klein JS, Fiocchi C. Interleukin 4 in inflammatory bowel disease and mucosal immune reactivity. Gastroenterology. 1996;110:1683–1695. doi: 10.1053/gast.1996.v110.pm8964392. [DOI] [PubMed] [Google Scholar]
- 33.Chinen T, Kobayashi T, Ogata H, et al. Suppressor of cytokine signaling-1 regulates inflammatory bowel disease in which both IFNgamma and IL-4 are involved. Gastroenterology. 2006;130:373–388. doi: 10.1053/j.gastro.2005.10.051. [DOI] [PubMed] [Google Scholar]
- 34.Heller F, Florian P, Bojarski C, et al. Interleukin-13 is the key effector Th2 cytokine in ulcerative colitis that affects epithelial tight junctions, apoptosis, and cell restitution. Gastroenterology. 2005;129:550–564. doi: 10.1016/j.gastro.2005.05.002. [DOI] [PubMed] [Google Scholar]
- 35.Kubo M, Hanada T, Yoshimura A. Suppressors of cytokine signaling and immunity. Nat Immunol. 2003;4:1169–1176. doi: 10.1038/ni1012. [DOI] [PubMed] [Google Scholar]