

1. DISEASE CHARACTERISTICS
1.1 Name of the disease (synonyms)
Rothmund–Thomson syndrome
Poikiloderma atrophicans with cataract
1.2 OMIM# of the disease
#268400
1.3 Name of the analyzed genes or DNA/chromosome segments
RECQL4 (RECQ-like, type 4), RECQ4
1.4 OMIM# of the gene(s)
*603780
1.5 Mutational spectrum
Biallelic mutations in the RECQL4 gene are associated with Rothmund–Thomson (RTS) and two additional recessive disorders: RAPADILINO (RAdial hypoplasia, PAtellae hypoplasia and cleft or arched PAlate, DIarrhea and DIslocated joints, LIttle size and LImb malformation, slender NOse and NOrmal intelligence) and Baller–Gerold syndrome (BGS). More than 60 disease-causing mutations have been reported, of which at least 40 have been detected in RTS patients.1, 2 The types of observed mutations are as follows, in order of decreasing prevalence: nonsense or frameshift mutations; splicing alterations, including substitutions at canonical splice junctions or at splice-site consensus sequences and subtle intronic deletions that reduce intron size below the threshold (<80 bp) required for correct splicing;3, 4 and missense mutations. There are a few recurrent mutations, among which the most common, exon 9 c.1573delT (p.Cys525AlafsX33), has been detected in patients with all three RECQL4-associated diseases. This truncating mutation accounts for approximately one-third of RTS mutations and has only been found in compound heterozygous patients from multiple ethnic backgrounds. A deletion of the entire gene has never been identified.
1.6 Analytical methods
Bidirectional sequencing of all exonic and intronic sequences of the RECQL4 gene.
MLPA (multiplex ligation-dependent probe identification) should be applied if sequencing fails to identify both mutant alleles.
Conventional cytogenetics may unveil a high rate of spontaneous and induced chromosomal breakage, as well as mosaic trisomies and isochromosomes.5, 6, 7
1.7 Analytical validation
Bidirectional sequencing results are confirmed by sequencing using different sets of primers. Pathogenicity of novel missense alterations must be verified by testing a set of at least 100 control chromosomes of the same ethnic origin and by in silico prediction methods. RT-PCR and cDNA sequencing is performed to confirm splicing mutations and to rule out effects on splicing by missense mutations. Testing of parents for carrier status should be performed in all cases.
1.8 Estimated frequency of the disease (incidence at birth (‘birth prevalence') or population prevalence)
If known to be variable between ethnic groups, please report:
The population prevalence of RTS syndrome is unknown. RTS is a very rare disorder, with fewer than 400 cases described in the literature.
However, RTS is likely to have been under-diagnosed because of the lack of awareness of this disorder and the lack of signs unique to the syndrome. Moreover, incidences relate directly to clinicians' and clinical geneticists' knowledge about the syndrome.
1.9 Diagnostic setting

Comment: The main diagnostic sign of RTS is poikiloderma (telangiectasic lesions, reticulated areas of depigmentation, hyperpigmentation, and punctate atrophy), which appears within the first 2 years of life as a chronic lesion evolving from a previous acute erythematous rash, first affecting the face and then extending to the limbs. Another hallmark is growth delay, present in 2/3 of patients, which is noted in the prenatal setting (ie, intrauterine growth restriction) and persists harmoniously after birth along at least—2 SD (when compared with the normal population). Given the high number of genodermatoses that present with poikiloderma, the pattern of presentation should be carefully considered, as should the concomitance of other common RTS signs: radial-ray defects; growth delay; sparse hair. Although Poikiloderma with Neutropenia (PN) syndrome, in many previous cases misdiagnosed as RTS, can now be diagnosed with a specific genetic test, clinically diagnosed RTS patients still comprise subgroups of unknown molecular etiology, including those characterized by poikiloderma and cataract, often without bone defects. Ambiguities resulting from partial clinical overlap with Fanconi Anemia (FA), Werner syndrome (WS), and Dyskeratosis Congenita (DC) can be resolved by the RECQL4 test; however, this may leave open the differential diagnosis with other RECQL4-related diseases, particularly RAPADILINO syndrome.8, 9 This test should be offered to all juvenile osteosarcoma cases with poikiloderma-like lesions.
2. Test characteristics
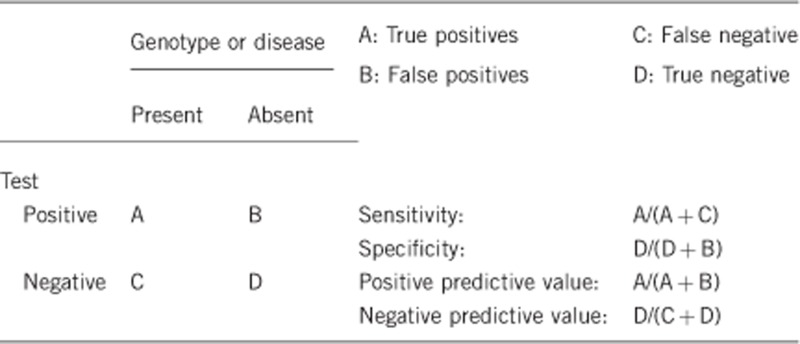
2.1 Analytical sensitivity (proportion of positive tests if the genotype is present)
Depends on the method(s) used. The analytical sensitivity could be >95%, but only if the DNA test is not restricted to exon sequencing. This applies to all three RECQL4-associated diseases.
2.2 Analytical specificity (proportion of negative tests if the genotype is not present)
>95%
2.3 Clinical sensitivity (proportion of positive tests if the disease is present)
The clinical sensitivity can be dependent on variable factors, such as age or family history. In such cases, a general statement should be given, even if quantification can only be made on a case by case basis.
The age of onset and range of clinical features is variable, but the main clinical signs should be present by the age of 2 years. By sequence analysis, including complete sequencing of all exons and introns, a disease-causing mutation is identified in ∼66% of individuals diagnosed with RTS.10 Incomplete clinical sensitivity could be explained by: (i) locus heterogeneity; (ii) mutations within the promoter of the gene; or (iii) mutations not identifiable by direct sequencing, such as deletions of entire exons or of the entire gene. Indeed, in a few patients (5 out of 64 listed in Siitonen et al.1), only one RECQL4 mutation is detectable.
2.4 Clinical specificity (proportion of negative tests if the disease is not present)
The clinical specificity can be dependent on variable factors, such as age or family history. In such cases, a general statement should be given, even if quantification can only be made on a case by case basis.
Close to 100%.
2.5 Positive clinical predictive value (life time risk to develop the disease if the test is positive)
Hundred percent for pathogenic mutations. On the basis of the literature and the reported pedigrees, all cases manifest the disease at early infancy: the penetrance is complete by at the age of 2 years, with variable expressivity. For patients who test positive for mutations, genetic counseling and surveillance should be provided for increased risk of osteosarcoma at an early age, and epithelial carcinoma of the skin in adulthood.11, 12 Prevalences of osteosarcoma and skin cancer in RTS are 30% and 5%, respectively. However, it must be considered that a few mutations detected in RTS patients are shared by RAPADILINO and BGS, which represent allelic disorders with different medical complications and cancer susceptibility.1
2.6 Negative clinical predictive value (probability not to develop the disease if the test is negative)
Assume an increased risk based on family history for a non-affected person. Allelic and locus heterogeneity may need to be considered.
Index case in that family had been tested:
If the index case in that family has been tested and found positive, a negative test in a familial non-affected person would exclude an increased risk of disease (negative clinical predictive value close to 100%).
Index case in that family had not been tested:
Under this condition, it would be inappropriate to test potentially at-risk family members before 2 years of age, by which time 90% of the cases manifest the disease. A preliminary step would be to test both parents of the index case to assess their carrier status.
3. Clinical Utility
3.1 (Differential) diagnostics: The tested person is clinically affected
(To be answered if ‘A' was marked in 1.10)
In classic cases, the correct diagnosis is based on clinical presentation with early-onset facial poikiloderma and radial-ray defects. In borderline or atypical cases, the differential diagnosis with syndromes with overlapping features, such as PN, DC, and WS, should be considered to orient the genetic test. This problem is overlooked in cases whose clinical evaluation suggests the allelic RAPADILINO or BG disorders. Molecular testing allows for the correct diagnosis, which is necessary for accurate targeting of syndrome-specific oncosurveillance.
3.1.1 Can a diagnosis be made other than through a genetic test?

3.1.2 Describe the burden of alternative diagnostic methods to the patient
No single alternative diagnostic method can be envisaged: rather, a panel of clinical-instrumental exams, including skin inspection by a dermatologist experienced in genodermatoses, baseline skeletal radiographs, and eye examination, would allow the formulation of a correct diagnosis.
3.1.3 How is the cost effectiveness of alternative diagnostic methods to be judged?
A genetic diagnosis permits the patient to avoid continuous and inconclusive clinical evaluations accompanied by multiple instrumental examinations. The cost effectiveness of alternative diagnostic methods is lower than that of the genetic test.
3.1.4 Will disease management be influenced by the result of a genetic test?

3.2 Predictive setting: The tested person is clinically unaffected but carries an increased risk based on family history
(To be answered if ‘B' was marked in 1.10)
3.2.1 Will the result of a genetic test influence lifestyle and prevention?
If the test result is positive (please describe).
If the test result is negative (please describe).
3.2.2 Which options in view of lifestyle and prevention does an at-risk person have if no genetic test has been done (please describe)?
3.3 Genetic risk assessment in family members of a diseased person
(To be answered if ‘C' was marked in 1.10)
3.3.1 Does the result of a genetic test resolve the genetic situation in that family?
Yes, if causative mutations have been identified in RECQL4, it is possible to assess the carrier status of all unaffected family members and to offer genetic counseling to the family.
3.3.2 Can a genetic test in the index patient save genetic or other tests in family members?
Yes. However, if the result is negative or uncertain, testing of family members is not recommended.
3.3.3 Does a positive genetic test result in the index patient enable a predictive test in a family member?
Infrequently, given the early onset of the disease (before the age of 2 years).
3.4 Prenatal diagnosis
(To be answered if ‘D' was marked in 1.10)
3.4.1 Does a positive genetic test result in the index patient enable a prenatal diagnosis?
Yes, provided that both disease-causing alleles have been identified in an affected family member and their segregation from obligate carrier parents has been traced.
4. If applicable, further consequences of testing
Please assume that the result of a genetic test has no immediate medical consequences. Is there any evidence that a genetic test is nevertheless useful for the patient or his/her relatives? (Please describe)
Genetic testing has no immediate medical consequences for healthy carriers. However, carriers' awareness of their genetic status is important for family planning.
Acknowledgments
This work was supported by EuroGentest2 (Unit 2: ‘Genetic testing as part of health care'), a Coordination Action under FP7 (Grant Agreement Number 261469) and the European Society of Human Genetics.
The authors declare no conflict of interest.
References
- Siitonen HA, Sotkasiira J, Biervliet M, et al. The mutation spectrum in RECQL4 diseases. Eur J Hum Genet. 2009;17:151–158. doi: 10.1038/ejhg.2008.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larizza L, Roversi G, Volpi L. Rothmund-Thomson syndrome. Orphanet J Rare Dis. 2010;5:2. doi: 10.1186/1750-1172-5-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang LL, Worley K, Gannavarapu A, et al. Intron-size constraint as a mutational mechanism in Rothmund-Thomson syndrome. Am J Hum Genet. 2002;71:165–167. doi: 10.1086/341234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balraj P, Concannon P, Jamal R, et al. An unusual mutation in RECQ4 gene leading to Rothmund-Thomson syndrome. Mutat Res. 2002;508:99–105. doi: 10.1016/s0027-5107(02)00189-6. [DOI] [PubMed] [Google Scholar]
- Miozzo M, Castorina P, Riva P, et al. Chromosomal instability in fibroblasts and mesenchymal tumors from 2 sibs with Rothmund-Thomson syndrome. Int J Cancer. 1998;77:504–510. doi: 10.1002/(sici)1097-0215(19980812)77:4<504::aid-ijc5>3.0.co;2-y. [DOI] [PubMed] [Google Scholar]
- Beghini A, Castorina P, Roversi G, Modiano P, Larizza L. RNA processing defects of the helicase gene RECQL4 in a compound heterozygous Rothmund-Thomson patient. Am J Med Genet A. 2003;120A:395–399. doi: 10.1002/ajmg.a.20154. [DOI] [PubMed] [Google Scholar]
- Larizza L, Magnani I, Roversi G. Rothmund-Thomson syndrome and RECQL4 defect: splitting and lumping. Cancer Lett. 2006;232:107–120. doi: 10.1016/j.canlet.2005.07.042. [DOI] [PubMed] [Google Scholar]
- Kellermayer R, Siitonen HA, Hadzsiev K, Kestilä M, Kosztolányi G. A patient with Rothmund-Thomson syndrome and all features of RAPADILINO. Arch Dermatol. 2005;141:617–620. doi: 10.1001/archderm.141.5.617. [DOI] [PubMed] [Google Scholar]
- Sznajer Y, Siitonen HA, Roversi G, et al. Atypical Rothmund-Thomson syndrome in a patient with compound heterozygous mutations in RECQL4 gene and phenotypic features in RECQL4 syndromes. Eur J Pediatr. 2008;167:175–181. doi: 10.1007/s00431-007-0447-6. [DOI] [PubMed] [Google Scholar]
- Wang LL, Gannavarapu A, Kozinetz CA, et al. Association between osteosarcoma and deleterious mutations in the RECQL4 gene in Rothmund-Thomson syndrome. J Natl Cancer Inst. 2003;95:669–674. doi: 10.1093/jnci/95.9.669. [DOI] [PubMed] [Google Scholar]
- Stinco G, Governatori G, Mattighello P, Patrone P. Multiple cutaneous neoplasms in a patient with Rothmund–Thomson syndrome: Case report and published work review. J Dermatol. 2008;35:154–161. doi: 10.1111/j.1346-8138.2008.00436.x. [DOI] [PubMed] [Google Scholar]
- Wang LL, Levy ML, Lewis RA, et al. Clinical manifestations in a cohort of 41 Rothmund-Thomson syndrome patients. Am J Med Genet. 2001;102:11–17. doi: 10.1002/1096-8628(20010722)102:1<11::aid-ajmg1413>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- Jin W, Liu H, Zhang Y, Otta SK, Plon SE, Wang LL. Sensitivity of RECQL4-deficient fibroblasts from Rothmund-Thomson syndrome patients to genotoxic agents. Hum Genet. 2008;123:643–653. doi: 10.1007/s00439-008-0518-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hicks MJ, Roth JR, Kozinetz CA, Wang LL. Clinicopathologic features of osteosarcoma in patients with Rothmund-Thomson syndrome. J Clin Oncol. 2007;25:370–375. doi: 10.1200/JCO.2006.08.4558. [DOI] [PubMed] [Google Scholar]
- De S, Kumari J, Mudgal R, et al. RECQL4 is essential for the transport of p53 to mitochondria in normal human cells in the absence of exogenous stress J Cell Science 2012. advanced online publication125(Part 10:2509–2522. [DOI] [PubMed] [Google Scholar]
- Mehollin-Ray AR, Kozinetz CA, Schlesinger AE, et al. Radiographic abnormalities in Rothmund-Thomson syndrome and genotype-phenotype correlation with RECQL4 mutation status. Am J Roentgenol. 2008;191:W62–W66. doi: 10.2214/AJR.07.3619. [DOI] [PubMed] [Google Scholar]