

Abstract
Acyl homoserine lactones (AHLs) are the most common class of quorum sensing signal molecules (autoinducers) that have been reported to be essential for virulence of many relevant pathogenic bacteria such as Pseudomonas aeruginosa. New approach for controlling infections of such bacteria is through quorum quenching. In this study, the acyl homoserine lactone inhibitory activity of the crude enzyme from a Bacillus weihenstephanensis-isolate P65 was characterized. The crude enzyme was found to have relatively high thermal stability and was stable in pH range 6 to 9. The crude enzyme extract was found to have lactonase activity of 36.3 U/mg total protein. Maximum enzyme activity was achieved within a range of 28–50°C and pH 6–9. None of the metals used enhanced the activity neither did EDTA inhibit it. However, a concentration of 10 mM Fe+2 reduced the activity to 73.8%. Catalytic activity and kinetic constants were determined using hexanoyl homoserine lactone as a substrate. Studying enzyme substrate specificity using synthetic standard signals displayed broad spectrum of activity. The enzyme was found to be constitutive. Isolation and complete nucleotide sequence of the respective lactonase gene were done and submitted to the Genbank database under accession code KC823046.
1. Introduction
Bacteria communicate with each other using a mechanism known as quorum sensing, a mechanism that is dependent on their population density and which was first reported in the bioluminescent bacterium Vibrio fischeri [1].Quorum-sensing bacteria can release, detect, and respond to small signal molecules—the autoinducers (AIs)—that accumulate in the environment as the population grows [2, 3]. At some threshold concentration, the autoinducer effects a change in the gene expression of the population by reentering the cell causing a signaling cascade that ultimately leads to transcriptional regulation of target genes [4]. Quorum-sensing systems have been found in pathogenic bacteria of plants, animals, and humans [5]. Acyl homoserine lactones (AHLs) are the most common autoinducers used by Gram-negative bacteria. The majority of natural AHLs reported to date share conserved structural characteristics, a homoserine lactone ring unsubstituted at the β- and γ-positions, which is N-acylated at the α-position with an acyl group derived from fatty acid biosynthesis (a fatty acyl group) [6]. AHLs vary in their acyl chain lengths (4 to 14 carbons) and/or contain an oxo or a hydroxyl substituent in the acyl chain [2, 3]. Many of the infection-related phenotypes associated with quorum sensing are controlled by AHL/LuxR/LuxI systems: LuxR is the AHL receptor, and LuxI is the AHL synthase. The quorum-sensing systems can induce antibiotic production [7]; pigmentation [8]; biofilm formation [5]; and the expression of virulence factors, for example, proteases, lytic enzymes, exotoxins, rhamnolipids, and extracellular polysaccharides [8, 9].
For example, the opportunistic pathogen Pseudomonas aeruginosa possesses two well identified N-acyl homoserine lactone quorum-sensing systems that regulate large, overlapping sets of genes [10]. These 2 distinct lux type quorum-sensing systems are termed las and rhl, which are named after their influence on elastase and rhamnolipid production, respectively. The las system regulates the rhl system as part of a cascade of virulence regulators. Thus, LasR represents a central checkpoint with the highest degree of interconnection in the network [10, 11]. This hierarchical circuitry controls multiple virulence traits [12]. Pseudomonas aeruginosa is also known for its high antibiotic resistance which is attributable to a concerted action of multidrug efflux pumps with chromosomally encoded antibiotic resistance genes and the low permeability of the bacterial cellular envelopes [13]. From here, the targeting of quorum-sensing system represents a novel and promising means of infection control. The idea lies in the development of a drug that attenuates bacterial virulence rather than antibiotic mediated bacteria killing or growth inhibition such that the organism fails to establish successful infection. Compounds with such abilities are termed antipathogenic drugs [14].
An obvious strategy to achieve this is to screen for enzymes capable of degradation of AHL signal molecules [15]. A search for enzymes degrading the AIs of QS systems is promising for designing agents to effectively suppress bacterial infections [16]. Many different bacteria belonging to various genera have been reported to express activity degrading AHLs.
One of the first described and the best characterized enzymes was the AHL-lactonase AiiA24B1, the product of the aiiA gene from Bacillus sp. 24B1 [17]. Since then, homologues of AiiA lactonase have been discovered in many bacteria belonging to the Bacillus genus. All of them show high nucleotide sequence similarity, greater than 90% [18–20]. Lactonases hydrolyze the lactone ring opening rendering the signal molecule inactive, Figure 1.
Figure 1.
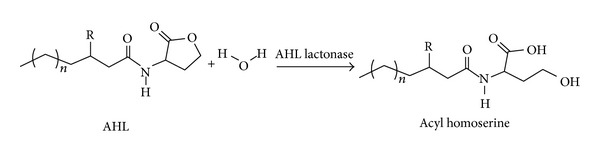
AHL lactonases hydrolyze the lactone ring in the homoserine moiety of AHLs, without affecting the rest of the signal molecule structure [21].
In this study, crude lactonase enzyme was collected from a previously identified Bacillus weihenstephanensis isolate P65 (Genbank accession code = KC899665) that was isolated from soil. This bacterium was previously screened for its quorum quenching activity against different synthetic AHLs and also against naturally produced AHLs in the extracts of 7 clinically isolated P. aeruginosa isolates. The aim of the present was to carry further studies on the enzyme of interest, visualize its stability, characterize its catalytic activity, and substrate specificity, and determine the full enzyme sequence and putative tertiary structure in order to use this enzyme as an antipathogenic drug.
2. Materials and Methods
2.1. Chemicals
All chemicals were of high quality from available grades purchased from El-Nasr Chemicals (Adwic), Egypt. Acyl homoserine lactone standards (butanoyl (C4), hexanoyl (C6), heptanoyl (C7), and octanoyl (C8) homoserine lactone) were purchased from Sigma-Aldrich, Germany. Reagents for DNA extraction and PCR were a product of Fermentas, USA.
2.2. Bacterial Strains
2.2.1. Chromobacterium violaceum CV026
CV026 is mutant strain of Chromobacterium violaceum that acts as an acyl homoserine lactone (AHL) dependent biosensor, producing the characteristic purple pigment violacein in response to the presence of the AHL [23]. It was subcultured in a medium containing 20 μg/mL kanamycin for purification as it is kanamycin resistant [24]. It could also be grown without kanamycin. CV026 was subcultured in Luria Bertani (LB) broth for maintenance and stored in slant medium or in lyophilized form for long term preservation.
2.2.2. Bacillus weihenstephanensis
It was isolated on R2A agar from a soil sample in Cairo, Egypt, and maintained on nutrient agar slants. It was previously screened for its quorum quenching activity and the whole culture activity was characterized. It was identified using 16S ribosomal RNA and was submitted to Genbank under accession code KC899665.
2.3. Collection of Crude Enzyme by Sonication
The Bacillus isolate was grown for 16–18 hours in 5 mL LB broth at 28°C with shaking at 160 rpm; then the cells were collected by centrifugation and washed twice with cell washing buffer (Tris-HCl pH 7.5, 25.0 mM). Then, the cells were resuspended in cracking buffer (Tris-HCl pH 7.5, 25.0 mM and Dithiothreitol (DTT) 1.0 mM) and disrupted by 7 minutes of intermittent sonication carried out in ice bath. After sonication, centrifugation was done at 15000 rpm for 15 minutes to remove the unlysed cells and cell debris [25] and the crude enzyme extract was collected. It should be noted that sonication was also done in saline to allow for pH adjustments to test the stability of the enzyme at different pH values.
2.4. Measuring the Total Protein Concentration in the Crude Extract
Protein concentration was measured by the method of Lowry et al. [26] using bovine serum albumin as a standard. A standard curve of absorbance at 660 nm as a function of protein concentration of the standard solutions was plotted and used to determine protein concentrations of the sample. The protein concentration was adjusted to 3.5 mg/mL before carrying out further studies.
2.5. Physical Parameters That Affect the Enzyme Stability
2.5.1. Measuring the Thermal Stability
In this assay, 1 mL of the crude enzyme extract (containing about 127 enzyme units) was incubated at 50, 80, and 90°C for 30 and 60 minutes. Then, 90 μL of each aliquot was pipetted into the wells of semisolid LB agar to measure the activity using well diffusion method described by Ravn and coworkers [24]. The semisolid seeded agar was prepared as follows: a preculture of CV026 was grown overnight in 5 mL LB broth at 28°C with shaking at 160 rpm and then transferred to 100 mL molten semisolid agar (1.2% w/v) maintained at 46°C. 1 mL of 10 μM synthetic hexanoyl homoserine lactone (HHL) was also transferred to the semisolid agar. After that, 10 mL of the semisolid agar was poured over the surface of prewarmed LB agar plates. When the overlaid agar had solidified, wells were punched with a sterile Cork borer (diameter 10 mm). The plates were then incubated at 28°C for 24 hours. The growth of CV026 showed purple color in the whole plate except the zones around the wells and the residual activity was measured by comparing the inhibition zone diameters of the test to that of the control. The experiment was done in three replicates and the mean deviation and standard deviation were calculated.
2.5.2. Measuring the Stability at Different pH
The assay was carried out according to Cao and coworkers [27] with modifications as follows: 1 mL aliquots of the crude enzyme extract (containing about 127 enzyme units) were transferred to Wasserman tubes and the pH of each was changed to the desired pH using either McIlvaine buffer (prepared by mixing 0.4 mL 0.2 M Na2HPO4 and 19.6 mL 0.1 M citric acid) or Glycine-NaOH buffer (0.1 M Glycine-NaOH adjusted to pH 12). The tubes were incubated at room temperature for 30 minutes and then neutralized to pH 7 with the counter buffer. Afterwards, the volumes of all the aliquots were adjusted equally using saline. Then, 90 μL of each aliquot was pipetted into wells formed in LB agar seeded with cv026 and supplemented with 100 nM HHL for the development of inhibition zones. Control was done using 90 μL of the crude extract that was incubated at room temperature for the same time and had its volume adjusted with saline to be equal to that of test. The experiment was done in three replicates and the mean deviation and standard deviation were calculated. The stability of the enzyme was studied over a pH range of 4 to 10.
2.6. The Effect of Some Divalent Metals and EDTA on Activity
Concentrations of 1 mM and 10 mM of Fe++, Mg++, Ca++, and EDTA were added to the crude extract and incubated for 30 minutes. For Zn++ and Cu++, a concentration of 10 mM was found to have an inhibitory activity on cv026 growth so they were used in a concentration of 1 and 2 mM. A sample without an additive served as a control. Then, 90 μL of the control and the tested aliquots were pipetted into the wells of semisolid LB agar as previously described and the inhibition zone diameters were compared to that of the control. The experiment was done in three replicates and the mean deviation and standard deviation were calculated.
2.7. The Effect of Adding Signal Molecule to the Growth Medium
The isolate was grown in 5 mL LB containing 1 mM HSL then sonication was done, and the well diffusion assay was carried out as previously described to measure the zone diameters and compare them to the control grown without a signal in the growth medium.
2.8. Measuring the Catalytic Activity
This was done according to Cao and coworkers [27] with modifications as follows: aliquots of crude enzyme extract (0.5 mL) were incubated with 10 μM HHL at 28°C with shaking at 160 rpm. Then, the reaction was terminated by adding 2% SDS. This was done at 1.5, 3, 5, and 7 hours. A control was done using plain buffer containing 10 μM HSL incubated under the same conditions for the same time and also treated with SDS. After termination of the reaction, the residual amount of HHL was measured by well diffusion assay as follows: 10 mL LB agar were overlaid by 10 mL semisolid agar seeded with cv026 and then left to solidify. Wells were punctured into the agar and 60 μL of the tested samples were transferred into them. The plates were then incubated at 28°C for 24 hours for color development. The homoserine lactones (HSL) in the wells formed zones of purple color around the wells, the diameter of which was proportional to the concentration of HSL. The measured zones were then compared to standard curve showing the diameters of the purple zones formed as a function of the different concentrations of HHL (with 2% SDS) pipetted into the wells. One unit of AHL lactonase activity was defined as the amount of enzyme that hydrolyzed 1 nM HHL per minute.
2.8.1. Effect of Temperature on Catalytic Activity
This was done as described in the determination of the catalytic activity of the crude enzyme but with the incubation temperature (of the crude extract with HSL) changed once to 5, 20, 40, 50, and 70°C. A control was prepared and incubated at 28°C; then the initial rate of reaction was determined after 1.5 h for both the control and the test.
2.8.2. Effect of pH on Catalytic Activity
Aliquots of crude enzyme extract used in this assay, each of about 1 mL volume (containing about 127 enzyme units), had their pH values adjusted to 6, 7, 8, and 9 using McIlvaine and glycine-NaOH buffer. Then, they were incubated with HSL of concentration 10 μM at 28°C. After 1.5 h the reaction was stopped using SDS 2% and 60 μL was pipetted into wells of semisolid LB seeded with cv026 overlaying LB agar as previously described. The plates were then incubated at 28°C for 24 hours and the diameters of the formed zones were measured and compared to standard curve to determine the residual amount of HHL. The activity at different pH was compared to the activity at pH 7.
2.9. Determination of Kinetic Constants
Aliquots of crude enzyme extract (0.5 mL containing 63.5 enzyme units) were incubated with different concentrations of HSL: 5, 10, 15, and 20 μM at 28°C with shaking at 160 rpm. Then, the reaction was terminated by adding 2% SDS after 90 minutes and the residual amount of HHL was measured by well diffusion assay. The initial velocity of the reaction was calculated from the following equation:
| (1) |
where A is the concentration of AHL and t is the time in minutes. The data were plotted as a Lineweaver-Burk curve also called the double reciprocal curve. Afterwards, kinetic constants for the crude enzyme extract, V max, K cat, and K m, were calculated. V max represents the maximum velocity of reaction, K cat represents the turnover number and was calculated relevantly to the total protein concentration (1 mg protein is equivalent to 36.3 lactonase units), and K m represents Michaelis-Menten constant which is the concentration of the substrate at half V max.
2.10. Enzyme Substrate Specificity
The ability of the enzyme to hydrolyze different acyl homoserine lactones was assessed using standard signals, butanoyl (C4), heptanoyl (C7), and octanoyl (C8) homoserine lactones. This was done by measuring the catalytic activity of the enzyme as described above for hexanoyl homoserine lactone. The reaction was carried out for 90 min at 28°C and pH 7. Units of lactonase activity to total protein concentration was measured in terms of C4, C7, and C8 HSL and compared to that measured previously using HHL standard.
2.11. Chromosomal DNA Extraction
It was done according to Pospiech and Neumann [28], and finally, the extracted DNA was dissolved in 500 μL TE buffer containing RNase with a concentration of 100 μg/mL and placed in eppendorf tubes as aliquots of 20 μL each, stored at −20°C to be used as a template for the amplification of lactonase gene.
2.12. Amplification of Lactonase Gene
Primers were designed using data that were collected from NCBI GenBank (http://www.ncbi.nlm.nih.gov/), which were then used to design the suitable primers using ClustalW (http://www.genome.jp/tools/clustalw/) program. The primers were afterwards checked for their specificity to the required gene using Primer 3 program (http://primer3.sourceforge.net/).
The 2 primers used were as follows: PHlact-F: 5′ ATGACAGTAAAGAAGCTTTATTTCG 3′ and PHlact-R: 5′ CTATATATACTCTGGGAACACTTTAC 3′. The PCR was done according to the following thermal cycling conditions: initial denaturation was carried out at 95°C for 5 min. Then, 30 cycles of denaturation (at 95°C for 30 sec), annealing (at 50°C for 50 seconds), and extension (at 72°C for 1 min) were carried out. Final extension took place at 72°C for 5 min.
2.13. Agarose Gel Electrophoresis
Agarose gel electrophoresis was carried out as described by Sambrook and Russell using 0.8% agarose gel containing 0.1 μg/mL ethidium bromide [29].
2.14. DNA Sequencing and Accession Code of Lactonase Gene
Sequencing of PCR product was done by GATC Company through Sigma scientific company, Egypt. It was done by the use of ABI 3730xl DNA Sequencer. The obtained sequence was analyzed, and assembled using Staden package program version 3 [30]. The ORF was detected using FramePlot [31] and the full nucleotide sequence was submitted to GenBank database under the accession code KC823046.
2.15. Prediction of the Tertiary Structure of the Lactonase Enzyme
The putative tertiary structure of the lactonase enzyme was predicted using Swiss-Model software (http://swissmodel.expasy.org/) [32–34]. This was done to visualize the predicted conformation of the protein and the possible metal-binding residues which might have an effect on the enzyme activity.
3. Results
3.1. Physical Parameters That Affect the Enzyme Stability
3.1.1. Thermal Stability of the Enzyme
The crude enzyme retained >90% of its activity when incubated at 50°C for 30 and 60 minutes. When incubated at 80°C for 30 minutes, it retained about 78.5% of the activity. However, when incubated at 80°C for 60 minutes, the activity was lost completely. The same happened when the crude enzyme was incubated at 90°C for 30 minutes where the activity was completely abolished evidenced by the absence of any inhibition zones in comparison to the control. The results represented in Figure 2 show the residual activity of the enzymeafter incubation at different temperatures from which the enzyme is found to have relatively high thermal stability.
Figure 2.
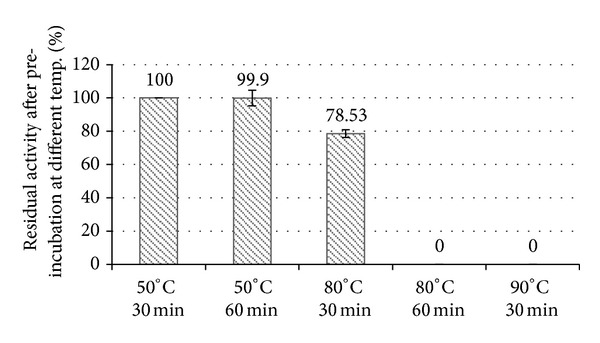
Residual activity of the enzyme after preincubation at different temperatures.
3.1.2. The Stability at Different pH
As shown from the results in Figure 3, the isolate retained >90% of the activity when preincubated at pH 6, pH 8, and pH 9 compared to the control preincubated at pH 7 while the activity was lost completely after preincubation at pH 4, pH 5, and pH 10.
Figure 3.
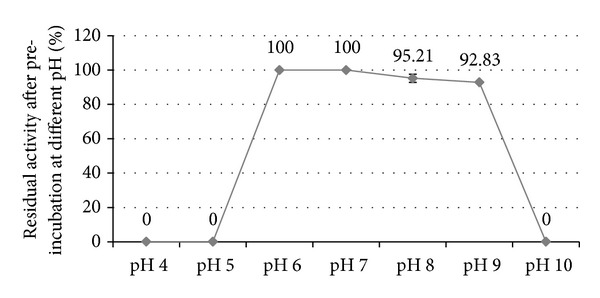
Residual activity of the enzyme after preincubation at different pH.
3.1.3. Effect of Some Divalent Metals and EDTA on Activity
At all the concentrations of Ca++, Mg++, Zn++, and Cu++ and also EDTA used in the assay, the enzyme retained ~100% of its activity. The activity was neither stimulated nor inhibited. It also retained ~100% of the activity at a concentrated 1 mM of Fe++ while at a concentration of 10 mM, the activity was reduced to 73.8%. Results are shown in Figure 4.
Figure 4.
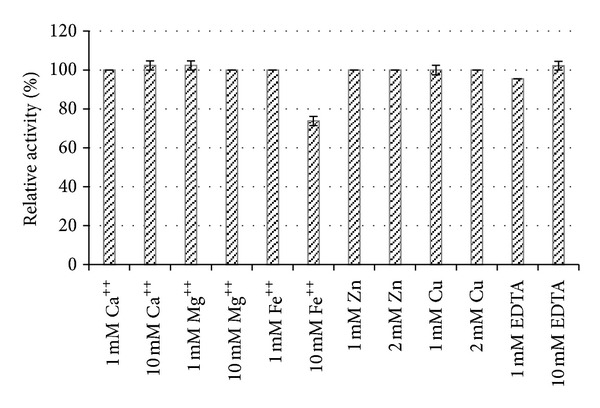
Effect of metal ions on activity.
3.1.4. The Effect of Adding Signal Molecule to the Growth Medium
Relative activity of the crude enzyme of the isolate grown in LB containing the HSL was found to be equal to 98.86% ± 1.14 of the activity of the crude enzyme of isolate grown without HSL in the medium. Activity which came almost equal indicates that the AHL degrading lactonase is a constitutive enzyme and its productivity is not enhanced by adding the substrate to the growth medium.
3.1.5. Measuring the Catalytic Activity of the Crude Enzyme
As shown in Figure 5, the initial rate of reaction (during the first 90 minutes) was 36.28 nM·min−1 mg−1 and then it dropped to 17.77 nM·min−1 mg−1. At 5 hours, no zone was formed indicating the complete degradation of HHL. According to the initial reaction velocity, the activity in terms of AHL lactonase activity to the total protein concentration was found to be equal to 36.3 U/mg total protein.
Figure 5.
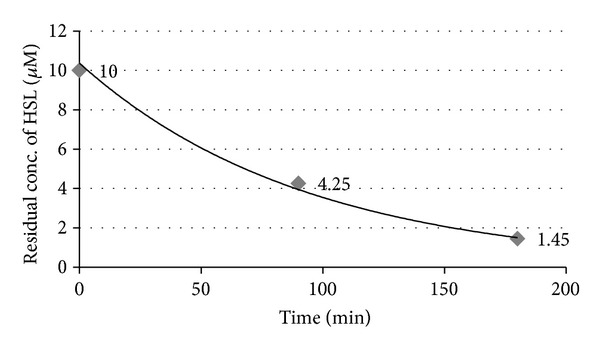
Catalytic rate of reaction using HHL as a substrate.
3.1.6. Effect of Temperature on Catalytic Activity
Results, Figure 6, show that the activity was almost not affected between 28 and 50°C. At 20°C, activity was reduced to 90.5% and to 80.7% at 5°C. However, it was greatly affected at 70°C and the activity dropped to about 41.8% of the maximum activity.
Figure 6.
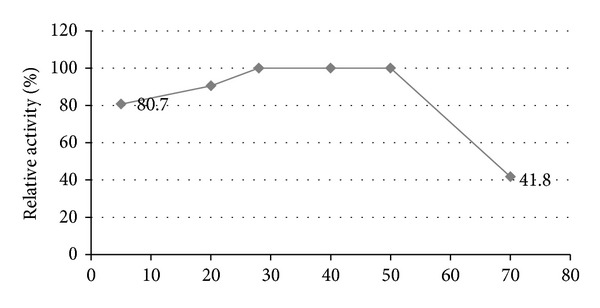
Effect of temperature on catalytic activity.
3.1.7. Effect of pH on Catalytic Activity
Result, Figure 7, showed that activity was almost unchanged over the studied pH range where the enzyme retained >90% of maximum activity between pH 6 and pH 9. The AHL Lactonase activity to the total protein concentration described as lactonase units/mg total protein remained almost unchanged within this range. The activity at pH values outside this range was not assessed as the enzyme proved to be unstable at pH 5 and pH 10 as concluded earlier.
Figure 7.
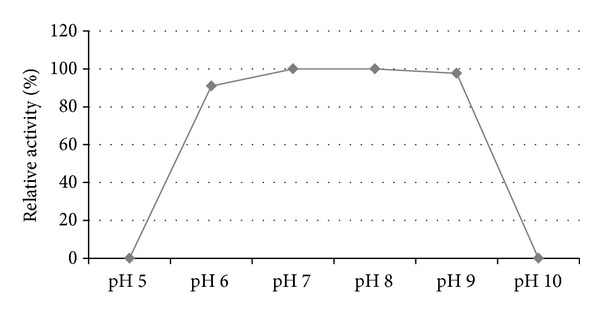
Effect of pH on catalytic activity.
3.2. Determination of Kinetic Constants
Maximum rate of reaction (V max) was found to be equal to 460.5 nM·min−1, K cat was equal to 52.4 min−1, and Michaelis-Menten constant (K m) was found to be equal to 77.13 nM.
3.3. Enzyme Substrate Specificity
By quantification of the residual AHL, we determined the relative enzyme activity of AHL-lactonase on different AHL derivatives. The data indicate that AHL lactonase has a broad substrate spectrum and digests efficiently all of the 4 tested AHL compounds used in the study, Table 1. One unit of lactonase activity was defined as the amount of enzyme that hydrolyzed one nM of the AHL tested in 1 min. Within a narrow range of difference in activity, maximum activity was observed against HHL (C6-HSL) as a substrate with slight difference when compared to activity against C4-HSL and C7-HSL while least degrading activity was observed against C8-HSL.
Table 1.
Hydrolytic activity on different AHL standards.
| Substrate | Lactonase activity units/mg total protein |
|---|---|
| C4-HSL | 30.16 U/mg |
| C6-HSL | 36.3 U/mg |
| C7-HSL | 32.02 U/mg |
| C8-HSL | 21.2 U/mg |
3.4. Lactonase Gene Amplification
Using the extracted chromosomal DNA, an expected PCR product (the amplified gene) of 0.75 Kb was detected using agarose gel electrophoresis.
3.5. Full Lactonase Gene/Enzyme Sequence
Sequencing revealed the presence of the motif HXHXDH. The presence of a tyrosine (Y) residue at the position 194 was also observed in our sequence. Using NCBI database and ClustalW, amino acid sequence alignment of our enzyme with AHL lactonases from other species was done and is displayed in Figure 8.
Figure 8.
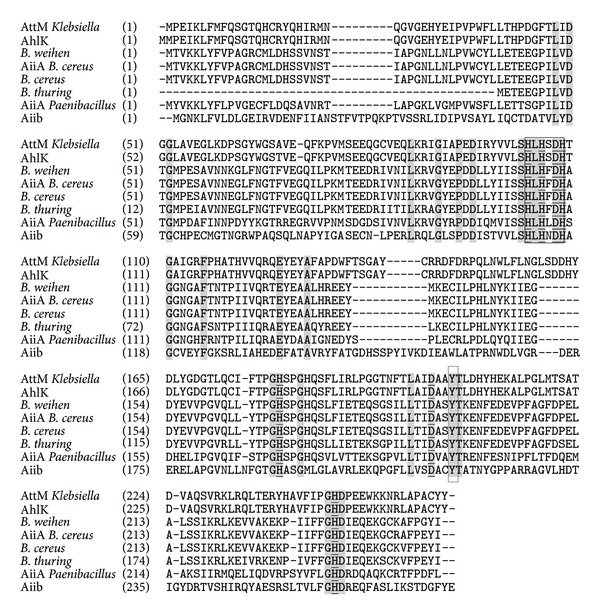
Alignment of the amino acid sequence of lactonase from B. weihenstephanensis (accession = KC823046) with other homologs using ClustalW. Lactonase was aligned with AttM lactonase of Klebsiella pneumoniae VA360 (accession code = Zp-23552218), AhlK of Klebsiella pneumonia (accession = AAO47340.1), AiiA of Bacillus cereus (accession code = AAY22194), metallo-beta-lactamase protein from Bacillus cereus (accession = ZP_03103014.1), lactonase of Bacillus thuringiensis serovar sotto (accession = ZP_04127391.1), AiiA of Paenibacillus alvei DSM 29 (accession = ZP_10863318.1), and Aiib from Agrobacterium tumefaciens (accession: 2R2D_E). HXHXDH motif and Tyr (194) were boxed, identical residues were shaded with grey, and the proposed metal ligands according to Thomas and coworkers [22] for the dinuclear zinc form of AHL lactonase were underlined.
3.6. Prediction of the Tertiary Structure of Lactonase Enzyme
The putative tertiary structure of lactonase gene, shown in Figure 9, was predicted by Swiss-Model software using N-acyl homoserine lactone hydrolase of Bacillus thuringiensis serovar kurstaki as a template (accession: 3DHA_A). The model showed a QMEAN4 score of 0.862 [35]. The model displays that our lactonase enzyme has a putative structure of a metalloenzyme that is hypothesized to contain binding sites for 2 zinc atoms where all the residues interacting with the ligand are completely conserved between model and template.
Figure 9.
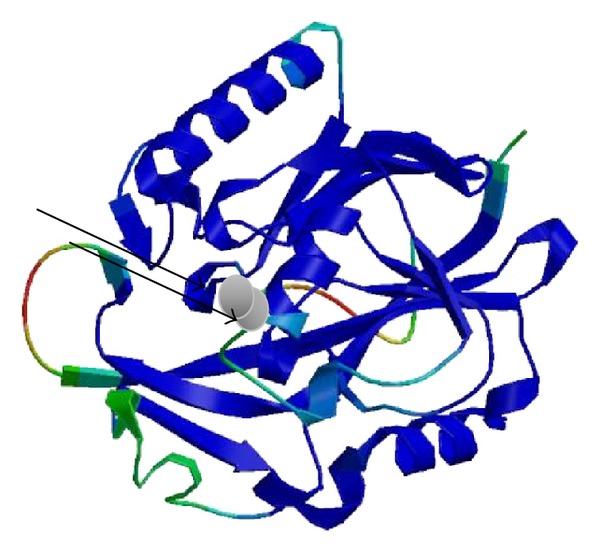
The putative tertiary structure of lactonase enzyme of B. weihenstephanensis, accession code Genbank KC823046. The two arrows show the suggested metal ligands for dinuclear zinc binding.
4. Discussion
Here, we studied the activity of crude AHL degrading enzyme from the B. weihenstephanensis which was found to be closely related to lactonases that are widespread in B. thuringiensis and Bacillus cereus strains. AHL lactonases have been used previously to demonstrate that quenching bacterial quorum sensing is a promising strategy for preventing and controlling bacterial infections. The data presented here show that the AHL lactonase encoded by B. weihenstephanensis is a good candidate for further development into an effective drug for infection control.
From results in this study, the enzyme proved to have high thermal stability retaining >78% of its activity after 30 min of preincubation at 80°C. In 2012, Cao and coworkers studied the thermal stability of a lactonase enzyme and stated that it retained about 60% of its activity after incubation for 20 min at 80°C or for 3 min at 90°C [27]. Another study on a recombinant AHL-lactonase stated that the enzyme exhibited thermal stability at 70°C, retaining more than 80% of the initial activity after preincubation at 70°C for 30 min [36]. A third study also showed that AHL-lactonase exhibited excellent thermal stability at temperatures below 37°C, and the purified enzyme, kept at 4 and 21°C for 10 days, still maintained about 99% activity. But the enzyme was less stable at higher temperatures; its activity was decreased sharply after incubation for 2 h at 45°C [37]. Another study on a lactonase enzyme isolated from the archaeon organism Sulfolobus islandicus showed that the enzyme exhibited respective half-lives of 84 ± 20 min, 8.5 ± 1.5 min, and 3.6 ± 0.4 min at 85, 90, and 95°C [38]. From the previous findings, our enzyme of interest here is believed to establish relatively excellent stability at high temperatures.
Several studies reported that pH has a drastic effect on the conformational structure of AHL-lactonase. In 2004, Wang and coworkers stated that lactonase enzyme was unstable at low pH where, according to this study, the asymmetric conformational structure of AHL-lactonase remained unchanged in pH ranging from 7 to 9 and slightly changed at pH 6 but significantly changed at pH 5.5 and completely lost at pH 5 [37]. Two previous studies also found that lactonase enzyme was unstable below pH 6 [27, 36]. In the same two studies, the protein was stated to be stable between pH 6–11 and pH 6–12 (maintaining >70% of its activity), respectively [27, 36]. In our present study, the enzyme displayed high stability over a pH range of 6–9 retaining about >90% of its activity. However, it was found to be unstable at pH 10 evidenced by loss of activity after preincubation at pH 10 for 30 min.
Our study here supports the idea that lactonase activity is not enhanced by the presence of divalent metal ions nor it is inhibited by the presence of metal chelators. The activity was partially inhibited by high concentration of Fe++. Consistent with the results in our study are the findings stated by Cao and coworkers [27] who stated that none of the metal ions used in that study enhanced the activity while a concentration of 10 mM Fe++ partially inhibited the activity. In 2004, Wang tested several metal ions, including Mg++, Ca++, Mn++, Co++, Zn++, Cd++, and Ni++ which showed no effect on enzyme activity at 0.2 and 2 mM concentrations. On the other hand, AHL-lactonase was partially inhibited by Cr++, Fe++, and Pb++ and completely inhibited by Cu++ [37]. The study stated that this was possibly due to reaction with sulfhydryl groups of the enzyme. Another study also reported the inhibitory activity of Cu++ on lactonase enzyme [36]. In our study here, however, Cu++ showed no inhibitory activity on the lactonase enzyme. Another study carried out in 2010 [36] showed that activity of lactonase was enhanced upon the addition of 1 mM and 10 mM Ca++ to 106% and 117.5%, respectively, and also upon the addition of 10 mM Mg++ the activity increased to 108%. It is clear, therefore, that the effect of different metals ions on activity of lactonase is still controversial. Several studies stated that lactonase is a phosphodiesterase-like metalloprotein and clarified the importance of divalent metal ions for its activity [22, 38, 39]. Other studies, like the one carried out by Wang and coworkers, stated that although trace amounts of Zn++ were measured in the enzyme, the AHL lactonase is not a metalloenzyme [37]. In our study here, EDTA used in concentrations of 1 and 10 mM as a divalent metal ion chelator showed no effect on activity emphasizing that divalent metal ion presence does not seem to be essential for activity. According to Wang and coworkers [37], both removal of Zn++ from AHL-lactonase by the ion-chelating reagent EDTA and its addition did not affect enzyme activity and hence they stated that the HXHXDH sequence seems to serve as a zinc-binding site in some enzymes but not in others and concluded that AHL-lactonase does not belong to metallohydrolase family.
Studying the catalytic activity, the crude enzyme could effectively degrade 10 μM in 5 hours. The AHL lactonase activity of the crude extract with total protein concentration of 3.5 mg/mL was found to be equal to 36.3 U/mg total protein. In a study on a recombinant lactonase in 2012, Cao and coworkers found that an AHL lactonase activity of 2.5 U/mL was measured in the cell lysate at the end of culture period using 3-oxo-C8-HSL as a substrate as compared to 127 U/mL in our study. When purified, the activity increased to about 8.54 U/mL, more than threefold [27]. Accordingly, these findings emphasize that the enzyme in our study has high potential to be developed into an antipathogenic drug of high activity.
Catalytic activity was not affected by temperature within a range of 28–50°C displaying maximum activity in that range. Below 28°C, activity was slightly reduced. At 70°C, the enzyme activity dropped to less than 50%. According to a study carried out by Wang and coworkers, lactonase enzyme inactivation was noticed at 45°C [37].
Several previous studies displaying the effect of pH on activity were carried out. A purified recombinant lactonase (AiiAB546) had the optimum pH of 8.0 and retained more than 73% of the maximum activity at pH 6.5–8.9 [36]. Another enzyme (AiiAAI96) retained about 95% of the maximal activity between pH 6.0 and 8.5 at 30°C [27]. In another study that determined the effect of pH on AHL-lactonase activity in a range from pH 5 to pH 9 using 3-oxo-C8-HSL as a substrate, AHL-lactonase activity, enhanced with pH increasing from 6 to 8, reached the maximum at pH 8 and then declined slightly at pH 9 [37]. In our study here the enzyme activity remained almost unchanged within pH 6–9 maintaining over 90% of its maximum activity over that range.
The broad spectrum of the enzyme activity was displayed by its ability to degrade all the tested AHL synthetic standards. It is also worth mentioning here that the enzyme previously showed high activity against the naturally produced signals in the extracts of 7 clinically isolated P. aeruginosa during the screening process as mentioned earlier. Several studies support that lactonase enzyme has a nonspecific substrate activity against homoserine lactones [27, 37]. Although lactonase activity is believed somewhat to be affected by the length of the acyl chain of the substrates [37], our study here showed that highest activity was displayed against C6-HSL followed by C7-HSL then C4-HSL with very slight differences between them. Activity on C8-HSL came last but also within a narrow window of variation. In 2004, Wang and coworkers also reported that the best substrates of AHL-lactonase are C6-HSL among the reduced AHL molecules and 3-oxo-C10-HSL of the C3 substituted AHL signals [37].
Amplification and sequencing of lactonase gene were done for the sake of full data collection about the enzyme to allow for further studies. Sequence analysis revealed the presence of several highly conserved domains shared with lactonases from other species. It revealed the presence of the motif HXHXDH which is believed to play a role in metal binding in metalloproteins and is conserved among almost all metalloenzymes. Thomas and coworkers reported that comparison of AHL lactonase with other superfamily members including glyoxalase II, phosphodiesterase-ZiPD, and methyl parathion hydrolase suggests possible zinc binding residues which are totally conserved in all known AHL lactonases: His104, His106, and His169 for the metal-1 site and Asp108, His109, and His235 for the metal-2 site [22]. The presence of a tyrosine (Y) residue at the position 194 was also observed in our sequence. According to Elias and coworkers, this tyrosine residue is believed to be conserved in all lactonase sequences and is believed to play a role in the positioning of lactone ring of the substrate [40]. The study also reports that lactonase is a metalloenzymes. However, Wang and coworkers stated that although sequence alignment of AHL-lactonase with these metallohydrolases revealed that they share this consensus motif known as HXHXDH at the central region, AHL-lactonase does not belong to metallohydrolase family [37]. In our study here, the presence of HXHXDH motif, residues proposed to be Zn++ binding sites and the conserved tyrosine residue, was reported as displayed earlier. Besides, the putative tertiary structure of our enzyme also suggested the presence of 2 zinc binding metal sites. However, whether these sites are truly metal-binding residues and whether lactonase is a metalloenzyme still need to be further examined.
5. Conclusion
AHL lactonase in this study appears to be a potent enzyme, demonstrating excellent thermal stability and retaining maximum activity over the studied pH range. It also has strong catalytic activity and a broad spectrum of activity against AHL signal molecules. Other than partial reduction in activity by 10 mM Fe++, lactonase enzyme in this study proved to be resistant to the effect of heavy metals and metal-chelating reagent, EDTA. Unlike other lactonases, the enzyme activity is not inhibited by the presence of copper. The enzyme contains the conserved HXHXDH sequence resembling the zinc binding motif of several groups of metallohydrolase family and shares other sequences highly conserved among lactonases from other species. Putative tertiary structure of the enzyme also suggests the presence of binding sites for two zinc atoms. Further studies on this enzyme are recommended as it represents a new outstanding tool for quorum quenching and, consequently, virulence suppression and infection control.
Conflict of Interests
The authors declare that there is no conflict of interests.
Acknowledgment
The authors acknowledge the Department of Microbiology and Immunology, Faculty of Pharmacy, Ain Shams University, for purchasing the biosensor strain and the standard signals used in this work.
References
- 1.Nealson KH, Hastings JW. Bacterial bioluminescence: its control and ecological significance. Microbiological Reviews. 1979;43(4):496–518. doi: 10.1128/mr.43.4.496-518.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fuqua C, Parsek MR, Greenberg EP. Regulation of gene expression by cell-to-cell communication: acyl-homoserine lactone quorum sensing. Annual Review of Genetics. 2001;35:439–468. doi: 10.1146/annurev.genet.35.102401.090913. [DOI] [PubMed] [Google Scholar]
- 3.Miller MB, Bassler BL. Quorum sensing in bacteria. Annual Review of Microbiology. 2001;55:165–199. doi: 10.1146/annurev.micro.55.1.165. [DOI] [PubMed] [Google Scholar]
- 4.Fuqua WC, Winans SC, Greenberg EP. Quorum sensing in bacteria: the LuxR-LuxI family of cell density-responsive transcriptional regulators. Journal of Bacteriology. 1994;176(2):269–275. doi: 10.1128/jb.176.2.269-275.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.De Kievit TR, Iglewski BH. Bacterial quorum sensing in pathogenic relationships. Infection and Immunity. 2000;68(9):4839–4849. doi: 10.1128/iai.68.9.4839-4849.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Galloway WRJD, Hodgkinson JT, Bowden SD, Welch M, Spring DR. Quorum sensing in gram-negative bacteria: small-molecule modulation of AHL and AI-2 quorum sensing pathways. Chemical Reviews. 2011;111(1):28–67. doi: 10.1021/cr100109t. [DOI] [PubMed] [Google Scholar]
- 7.Henke JM, Bassler BL. Bacterial social engagements. Trends in Cell Biology. 2004;14(11):648–656. doi: 10.1016/j.tcb.2004.09.012. [DOI] [PubMed] [Google Scholar]
- 8.Juhas M, Eberl L, Tümmler B. Quorum sensing: the power of cooperation in the world of Pseudomonas . Environmental Microbiology. 2005;7(4):459–471. doi: 10.1111/j.1462-2920.2005.00769.x. [DOI] [PubMed] [Google Scholar]
- 9.Diggle SP, West SA, Gardner A, Griffin AS. Communication in bacteria. In: d’Ettorre P, Hughes DP, editors. Sociobiology of Communication: An Interdisciplinary Perspective. Oxford University Press; 2008. pp. 11–31. [Google Scholar]
- 10.Schuster M, Greenberg EP. A network of networks: quorum-sensing gene regulation in Pseudomonas aeruginosa . International Journal of Medical Microbiology. 2006;296(2-3):73–81. doi: 10.1016/j.ijmm.2006.01.036. [DOI] [PubMed] [Google Scholar]
- 11.Viretta AU, Fussenegger M. Modeling the quorum sensing regulatory network of human-pathogenic Pseudomonas aeruginosa . Biotechnology Progress. 2004;20(3):670–678. doi: 10.1021/bp034323l. [DOI] [PubMed] [Google Scholar]
- 12.van Delden C, Iglewski BH. Cell-to-cell signaling and Pseudomonas aeruginosa infections. Emerging Infectious Diseases. 1998;4(4):551–560. doi: 10.3201/eid0404.980405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Poole K. Efflux-mediated multiresistance in Gram-negative bacteria. Clinical Microbiology and Infection. 2004;10(1):12–26. doi: 10.1111/j.1469-0691.2004.00763.x. [DOI] [PubMed] [Google Scholar]
- 14.Hentzer M, Givskov M. Pharmacological inhibition of quorum sensing for the treatment of chronic bacterial infections. Journal of Clinical Investigation. 2003;112(9):1300–1307. doi: 10.1172/JCI20074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rasmussen TB, Givskov M. Quorum-sensing inhibitors as anti-pathogenic drugs. International Journal of Medical Microbiology. 2006;296(2-3):149–161. doi: 10.1016/j.ijmm.2006.02.005. [DOI] [PubMed] [Google Scholar]
- 16.Khmel IA, Metlitskaya AZ. Quorum sensing regulation of gene expression: a promising target for drugs against bacterial pathogenicity. Molecular Biology. 2006;40(2):169–182. [Google Scholar]
- 17.Dong Y-H, Xu J-L, Li X-Z, Zhang L-H. AiiA, an enzyme that inactivates the acylhomoserine lactone quorum-sensing signal and attenuates the virulence of Erwinia carotovora . Proceedings of the National Academy of Sciences of the United States of America. 2000;97(7):3526–3531. doi: 10.1073/pnas.060023897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dong Y-H, Gusti AR, Zhang Q, Xu J-L, Zhang L-H. Identification of quorum-quenching N-acyl homoserine lactonases from Bacillus species. Applied and Environmental Microbiology. 2002;68(4):1754–1759. doi: 10.1128/AEM.68.4.1754-1759.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lee SJ, Park S-Y, Lee J-J, Yum D-Y, Koo B-T, Lee J-K. Genes encoding the N-acyl homoserine lactone-degrading enzyme are widespread in many subspecies of Bacillus thuringiensis . Applied and Environmental Microbiology. 2002;68(8):3919–3924. doi: 10.1128/AEM.68.8.3919-3924.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kim MH, Choi W-C, Kang HO, et al. The molecular structure and catalytic mechanism of a quorum-quenching N-acyl-L-homoserine lactone hydrolase. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(49):17606–17611. doi: 10.1073/pnas.0504996102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Czajkowski R, Jafra S. Quenching of acyl-homoserine lactone-dependent quorum sensing by enzymatic disruption of signal molecules. Acta Biochimica Polonica. 2009;56(1):1–16. [PubMed] [Google Scholar]
- 22.Thomas PW, Stone EM, Costello AL, Tierney DL, Fast W. The quorum-quenching lactonase from Bacillus thuringiensis is a metalloprotein. Biochemistry. 2005;44(20):7559–7569. doi: 10.1021/bi050050m. [DOI] [PubMed] [Google Scholar]
- 23.McClean KH, Winson MK, Fish L, et al. Quorum sensing and chromobacterium violaceum: exploitation of violacein production and inhibition for the detection of N-acylhomoserine lactones. Microbiology. 1997;143(12):3703–3711. doi: 10.1099/00221287-143-12-3703. [DOI] [PubMed] [Google Scholar]
- 24.Ravn L, Christensen AB, Molin S, Givskov M, Gram L. Methods for detecting acylated homoserine lactones produced by Gram-negative bacteria and their application in studies of AHL-production kinetics. Journal of Microbiological Methods. 2001;44(3):239–251. doi: 10.1016/s0167-7012(01)00217-2. [DOI] [PubMed] [Google Scholar]
- 25.Boyer M, Bally R, Perrotto S, Chaintreuil C, Wisniewski-Dyé F. A quorum-quenching approach to identify quorum-sensing-regulated functions in Azospirillum lipoferum . Research in Microbiology. 2008;159(9-10):699–708. doi: 10.1016/j.resmic.2008.08.003. [DOI] [PubMed] [Google Scholar]
- 26.Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. Protein measurement with the folin phenol reagent. The Journal of Biological Chemistry. 1951;193(1):265–275. [PubMed] [Google Scholar]
- 27.Cao Y, He S, Zhou Z, et al. Orally administered thermostable N-acyl homoserine lactonase from Bacillus sp. strain AI96 attenuates Aeromonas hydrophila infection in zebrafish. Applied and Environmental Microbiology. 2012;78(6):1899–1908. doi: 10.1128/AEM.06139-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pospiech A, Neumann B. A versatile quick-prep of genomic DNA from gram-positive bacteria. Trends in Genetics. 1995;11(6):217–218. doi: 10.1016/s0168-9525(00)89052-6. [DOI] [PubMed] [Google Scholar]
- 29.Sambrook J, Russell DW. Molecular Cloning: A Laboratory Manual. 3rd edition. Cold Spring Harbor, NY, USA: Cold Spring Harbor Laboratory Press; 2001. [Google Scholar]
- 30.Staden R. The staden sequence analysis package. Applied Biochemistry and Biotechnology. 1996;5(3):233–241. doi: 10.1007/BF02900361. [DOI] [PubMed] [Google Scholar]
- 31.Ishikawa J, Hotta K. FramePlot: a new implementation of the Frame analysis for predicting protein-coding regions in bacterial DNA with a high G+C content. FEMS Microbiology Letters. 1999;174(2):251–253. doi: 10.1111/j.1574-6968.1999.tb13576.x. [DOI] [PubMed] [Google Scholar]
- 32.Arnold K, Bordoli L, Kopp J, Schwede T. The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling. Bioinformatics. 2006;22(2):195–201. doi: 10.1093/bioinformatics/bti770. [DOI] [PubMed] [Google Scholar]
- 33.Guex N, Peitsch MC. SWISS-MODEL and the Swiss-pdb viewer: an environment for comparative protein modeling. Electrophoresis. 1997;18(15):2714–2723. doi: 10.1002/elps.1150181505. [DOI] [PubMed] [Google Scholar]
- 34.Schwede T, Kopp J, Guex N, Peitsch MC. SWISS-MODEL: an automated protein homology-modeling server. Nucleic Acids Research. 2003;31(13):3381–3385. doi: 10.1093/nar/gkg520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Benkert P, Biasini M, Schwede T. Toward the estimation of the absolute quality of individual protein structure models. Bioinformatics. 2011;27(3):343–350. doi: 10.1093/bioinformatics/btq662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chen R, Zhou Z, Cao Y, Bai Y, Yao B. High yield expression of an AHL-lactonase from Bacillus sp. B546 in Pichia pastoris and its application to reduce Aeromonas hydrophila mortality in aquaculture. Microbial Cell Factories. 2010;9, article 39:1–10. doi: 10.1186/1475-2859-9-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang L-H, Weng L-X, Dong Y-H, Zhang L-H. Specificity and enzyme kinetics of the quorum-quenching N-Acyl homoserine lactone lactonase (AHL-lactonase) Journal of Biological Chemistry. 2004;279(14):13645–13651. doi: 10.1074/jbc.M311194200. [DOI] [PubMed] [Google Scholar]
- 38.Hiblot J, Gotthard G, Chabriere E, Elias M. Structural and enzymatic characterization of the lactonase SisLac from Sulfolobus islandicus . Plos One. 2012;7(10, article e47028) doi: 10.1371/journal.pone.0047028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Momb J, Thomas PW, Breece RM, Tierney DL, Fast W. The quorum-quenching metallo-γ-lactonase from Bacillus thuringiensis exhibits a leaving group thio effect. Biochemistry. 2006;45(44):13385–13393. doi: 10.1021/bi061238o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Elias M, Dupuy J, Merone L, et al. Structural basis for natural lactonase and promiscuous phosphotriesterase activities. Journal of Molecular Biology. 2008;379(5):1017–1028. doi: 10.1016/j.jmb.2008.04.022. [DOI] [PubMed] [Google Scholar]