

Abstract
We report a novel gene for a parkinsonian disorder. X-linked parkinsonism with spasticity (XPDS) presents either as typical adult onset Parkinson's disease or earlier onset spasticity followed by parkinsonism. We previously mapped the XPDS gene to a 28 Mb region on Xp11.2–X13.3. Exome sequencing of one affected individual identified five rare variants in this region, of which none was missense, nonsense or frame shift. Using patient-derived cells, we tested the effect of these variants on expression/splicing of the relevant genes. A synonymous variant in ATP6AP2, c.345C>T (p.S115S), markedly increased exon 4 skipping, resulting in the overexpression of a minor splice isoform that produces a protein with internal deletion of 32 amino acids in up to 50% of the total pool, with concomitant reduction of isoforms containing exon 4. ATP6AP2 is an essential accessory component of the vacuolar ATPase required for lysosomal degradative functions and autophagy, a pathway frequently affected in Parkinson's disease. Reduction of the full-size ATP6AP2 transcript in XPDS cells and decreased level of ATP6AP2 protein in XPDS brain may compromise V-ATPase function, as seen with siRNA knockdown in HEK293 cells, and may ultimately be responsible for the pathology. Another synonymous mutation in the same exon, c.321C>T (p.D107D), has a similar molecular defect of exon inclusion and causes X-linked mental retardation Hedera type (MRXSH). Mutations in XPDS and MRXSH alter binding sites for different splicing factors, which may explain the marked differences in age of onset and manifestations.
INTRODUCTION
Recently, we described a family (Fig. 1) affected with a distinct X-linked parkinsonian syndrome, XPDS (1). XPDS is a slowly progressive disease with considerable phenotypic variability with respect to age of onset (range: 14–58 years) and presenting symptoms. Spasticity was the initial symptom in three affected individuals who later developed a parkinsonian resting tremor, masked facies and bradykinesia. Two individuals presented with typical parkinsonian features and did not manifest spasticity, even at old age. One affected individual developed seizures and one had moderate memory loss in his 80s, but none had developmental delay. We mapped the XPDS locus to a 28 Mb region on chromosome Xp11.2–Xq13.3 that contains ∼200 protein coding genes (1). Complete or partial sequencing of 18 selected candidate genes failed to reveal a pathological change.
Figure 1.

Family with X-linked parkinsonism and spasticity (XPDS). The individual tested via exome sequencing is designated with an arrow.
Analysis of genetic variation in families via exome resequencing has proved a powerful approach for identifying genes that underlie Mendelian disorders. It has been successful even in challenging circumstances, including rare disorders with a limited number of subjects, presence of locus heterogeneity and/or reduced penetrance (2,3). These studies have relied on an exceedingly rare frequency of the causative variant in the population. Here we report functional analysis of candidate variants detected by exome resequencing that identified the genetic cause of XPDS.
RESULTS
Identification and functional analysis of candidate variants
Exome sequencing revealed 100 variants (91 SNPs, 9 indels) in the identity-by-descent (IBD) region on the X chromosome. Assuming that XPDS is an extremely rare disorder, we filtered out variants annotated in dbSNP or the 1000 Genomes Database as of June 2011, or were present in 1200 Caucasian exomes (NHLBL Exome Variant Server; UW EVS). This reduced the number of candidate variants to five, of which one was synonymous, two resided in a 3′-UTR and two were intronic indels within 30 nt from an exon–intron boundary (Table 1). Sanger sequencing was performed to validate the exome data and confirm co-transmission with disease.
Table 1.
Unique variants found in the XPDS exome within the X-chomosome linkage interval
| Variant (hg19 coordinates) | Description | Gene symbol | Gene name |
|---|---|---|---|
| 40 456 545 C>T | Coding, synonymous | ATP6AP2 | ATPase, H+ transporting, lysosomal accessory protein 2 |
| 55 250 449 A>G | 3′-UTR, non-coding | PAGE5 | P antigen family, member 5 (prostate associated) |
| 46 952 353 G>A | 3′-UTR, non-coding | RGN | regucalcin (senescence marker protein-30) |
| 40 588 605 G>GA | Intronic indel | MED14, EXLM1 | mediator complex subunit 14, transcription activator |
| 52 891 619GT>G | Intronic indel | XAGE3 | X antigen family, member 3 |
In parallel, given the pattern of decreasing age at onset and increasing severity of symptoms with successive generations in the XPDS family, we tested possible repeat expansions in the coding sequence within the linkage region. Repeat instability within non-coding regulatory sequences tends to produce very variable phenotypes, whereas repeat expansions within coding regions more typically produce a core phenotype with increased severity (4) as is manifested in this family. No expansions were detected in the re-aligned exome reads from the IBD interval. The reference genome contained ∼3700 potentially unstable repeats (total length ≥24 nt) within genic regions of the IBD interval, of which 10 resided in coding exons and/or exon–intron boundaries. All 10 sites were verified by Sanger sequencing in two affected subjects (IV-1 and IV-5) and two control subjects, but no repeat expansions were found (data not shown).
Given the absence of mutations that directly affect protein sequence, we next evaluated the effect of the five variants on gene expression and RNA splicing using patient-derived lymphoblastoid cell lines (LCL). For an X-linked disorder, even modest changes in gene expression/RNA splicing may be sufficient to cause disease in males who lack a normal allele. By qRT–PCR, we tested the two variants in 3′ UTRs for changes in the gene-expression level. We also examined an intronic variant near the exon–intron boundary and a synonymous exonic variant for evidence of altered splicing, such as changes in the balance of splice isoforms or appearance of new products.
Using LCLs from two patients and two unaffected Caucasian males, we did not find consistent effects of variants in RGN and XAGE3 on mRNA expression level or the variant in MED14/EXLM1 on splicing. PAGE5 was not expressed in LCL. In contrast, the silent exonic c.345C>T (p.S115S) mutation dramatically altered splicing of the ATP6AP2 gene (MIM 300556) (Fig. 2).
Figure 2.
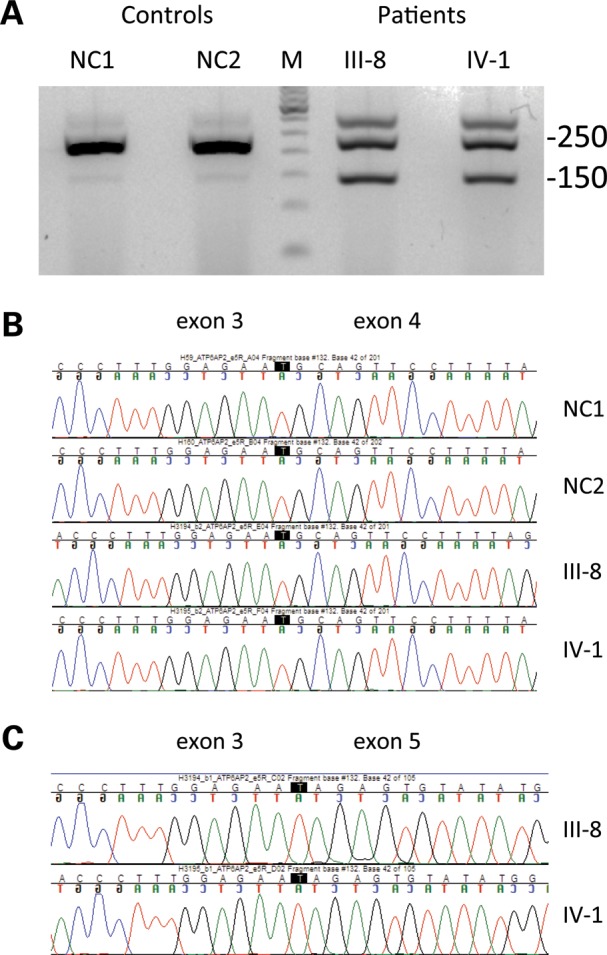
The silent c.345C>T (p.S115S) mutation results in overexpression of ATP6AP2 splice isoforms that lack exon 4. (A) RT–PCR with primers positioned in exons 3 and 5 amplifies a major 250 bp and a minor 150 bp product in controls; the minor product is highly overexpressed in patients. (B and C) Sequence analysis of splicing products from 250 bp (B) and 150 bp bands (C) confirming exon 4 skipping in the 150 bp band. III-8, IV-1: affected patients from the XPDS pedigree depicted in Fig. 1. NC1, NC2: normal controls. M: molecular weight marker.
In addition to a major splice product of the expected size that migrates at 250 bp, a faint minor band at 150 bp is seen in both controls. This 150 bp band becomes a major species in both XPDS subjects. Direct sequencing of the eluted and purified RT–PCR fragments determined that the 250 bp band contains normally spliced exons 3, 4 and 5, whereas the 150 bp band lacks exon 4. The skipping of exon 4 results in an in-frame transcript (Δe4) encoding a protein with internal deletion of 32 residues. The upper band seen in both patients consists of a heterogeneous mixture of transcripts and is likely an RT–PCR artifact.
ATP6AP2 is an essential gene with ubiquitous expression. It encodes a single-pass transmembrane domain protein that is involved in a range of processes such as intracellular pH homeostasis (5), renin–angiotensin system (6) and WNT signaling (7). Surprisingly, another mutation in this gene causes the MRXSH syndrome (OMIM #300423), a congenital mental retardation with epilepsy (8). This silent mutation, c.321C>T (p.D107D), also positioned in exon 4, significantly impairs splicing efficiency resulting in the overexpression of the Δe4 transcript.
Variants in ATP6AP2 exon 4 and their predicted effect on splicing
The nucleotide sequence of exon 4 is nearly invariant in the human population. Besides mutations found in the MRXSH and XPDS families, there is only one rare synonymous c.357G>A (p.E119E) variant (0.02% frequency) listed in the EVS. No phenotypic information was available for this sample. We found no exon 4 mutations in 1160 patients with Parkinson's disease (PD). However, only 35 male patients had a family history consistent with an X-linked disorder (e.g. two or more affected males, no male-to-male transmission) and none had a history of spasticity.
Human Splicing Finder predictions suggest that the two disease-related mutations, c.321C>T (p.D107D) and c.345C>T (p.S115S), affect different sets of splicing factors (Table 2). c.321C>T (p.D107D) disrupts enhancer sites for SRp40 and 9G8, whereas c.345C>T (p.S115S) creates a new silencer site. Interestingly, c.357G>A (p.E119E) could also affect splicing of exon 4, although through different mechanisms. c.357G>A (p.E119E) is predicted to disrupt both a potential enhancer for splicing factor SRp55 and a silencer for hnRNP A1.
Table 2.
Predicted effect of variants in exon 4 of ATP6AP2 on splicing
| Phenotype | Variant | Exon Enhancer Site | Exon silencer site |
|---|---|---|---|
| XPDS | c.345C>T | −10.01% for SRp40 | new site |
| MRXSH | c.321C>T | site broken for SRp40 site broken for 9G8 |
+22% for hnRNP A1 |
| Unknown | c.357G>A | site broken for SRp55 | site broken for hnRNP A1 |
Overproduction of minor Δe4 isoform in XPDS cells compromises the level of normal full size transcript
According to the AceView database (http://www.ncbi.nlm.nih.gov/IEB/Research/Acembly/), the human ATP6AP2 gene is alternatively spliced in multiple tissues including brain (Fig. 3A), and human ATP6AP2 mRNA splice isoforms are much more complex than those of mice (Fig. 3B). There are two major ATP6AP2 isoforms containing normally spliced exon 4 (a and d supported by 214 and 117 tissue-averaged cDNA clones, respectively, Fig. 3A), as well as several minor forms, including Δe4 (isoform c, 12 supporting clones). We used qRT–PCR quantification to measure the effect of the c.345C>T (p.S115S) variant on the relative proportion of ATP6AP2 splice isoforms, as well as on the overall transcript level (Fig. 4). In normal individuals, blood-derived cells contained <1% of Δe4 transcript (average 0.4%, range 0.07–0.8%) while brain tissues produced it at a 10-fold higher level (average 4%, range 1.5–8.4%, Fig. 4A). Strikingly, the Δe4 level is increased over 90-fold in XPDS patients, becoming a major isoform in blood cells (average 44%; patients' averages of 35% in LCL and 50% in uncultured white cells). The observed increase in Δe4 production caused by c.345C>T (p.S115S) is comparable to that of the c.321C>T (p.D107D) mutation found in MRXSH (50% in LCL) (8).
Figure 3.

Distribution of ATP6AP2 splice isoforms in humans (A) and mice (B). The shaded box highlights the minor isoform c that corresponds to the Δe4 isoform increased in XPDS patients. (NM), isoform corresponding to RefSeq gene: NM_005765 (human) and NM_027439 (mouse).
Figure 4.

qRT–PCR quantification of ATP6AP2 splice isoforms in XPDS patients and controls. (A) Percentage of the Δe4 ATP6AP2 isoform in blood cells and brain tissues. (B) Relative expression of ATP6AP2 isoforms containing exon 4. (C) Relative expression of total ATP6AP2 transcript as measured with primers for constant exons 8 and 9. (D) Relative expression of Δe4 isoform. Open circles, controls; filled circles, patients. PB, uncultured peripheral blood cells.
We were interested in whether Δe4 overexpression increases total production of ATP6AP2 mRNA or competes with the production of the isoforms that contain exon 4, resulting in their relative depletion. To account for total ATP6AP2 mRNA production, we performed an additional quantification using exons 8 and 9 present in splice isoforms a through f (Fig. 3A). The expression of e8–e9, e3–e4 and Δe4 was compared with that of external reference genes (GUSB and TBP, Fig. 4B–D). We observed no differences in the amount of total ATP6AP2 transcript between patients and controls (Fig. 4C). In XPDS patients, production of exon 4-containing isoforms was decreased, indicating that they indeed compete with Δe4 for pre-mRNA (Fig. 4B and D).
Deficit of ATP6AP2 protein in XPDS brain
To study the expression and distribution of ATP6AP2 protein in XPDS brain, we took advantage of available brain sections from the previously characterized patient II-2 (1). Sections from this brain and two age-matched control brains were stained with polyclonal antibodies raised against the extracellular domain of the ATP6AP2 protein (Fig. 5). In the normal brain, the cytoplasm and plasma membrane of the majority of neurons were stained positively. In the XPDS case, we observed a similar distribution of immunostaining, but with marked decrease in staining intensity in both the frontal cortex and the striatum; the difference was less pronounced in the hippocampus.
Figure 5.

Reduced ATP6AP2 immunostaining in the XPDS brain. Representative immunostaining of brain sections of the XPDS case (A and B) and controls (C and D). In both the frontal cortex (A and C) and striatum (B and D), the XPDS case demonstrated normal distribution but marked reduction in the intensity of ATP6AP2 immunostaining in neurons.
On the role of ATP6AP2 in Wnt/β-catenin signaling
ATP6AP2 has been shown to modify Wnt/β-catenin signaling and may act as an adaptor between Wnt receptors, Frizzled and Lrp5/6, and the V-ATPase complex (7,9). Because deregulated Wnt signaling is a frequent finding in neurological disorders (10), we tested whether modulation of ATP6AP2 expression and/or overexpression of the Δe4 isoform could influence Wnt/β-catenin signaling. We first depleted ATP6AP2 protein in HEK293T cells harboring a Wnt/β-catenin-activated luciferase reporter (BAR) (11). Of six siRNAs shown by western blot to reduce ATP6AP2 protein, three reduced the ability of Wnt3A conditioned media to activate Wnt signaling, one further enhanced Wnt3A-dependent signaling, and two had no effect (Supplementary Material, Fig. S1).
Overexpression of ATP6AP2 is thought to repress Wnt/β-catenin signaling (9); whereas, expression of ATP6AP2 lacking its C-terminus (ΔC) has been shown to synergize with Wnt3A to activate Wnt/β-catenin signaling (7). Thus, we tested whether overexpression of the Δe4 isoform of ATP6AP2 could also influence Wnt/β-catenin activation. Unlike positive control proteins, β-catenin and constitutively active LRP6 (12), we did not observe changes in BAR activity when full length ATP6AP2, ATP6AP2 ΔC or ATP6AP2 Δe4 were overexpressed (Supplementary Material, Fig. S2). Collectively, our findings suggest that ATP6AP2 does not significantly influence Wnt/β-catenin signaling.
ATP6AP2 deficiency affects V-ATPase function with resultant impaired autophagy and lysosomal clearance
ATP6AP2 is an essential accessory unit of the V-ATPase multiprotein complex responsible for a number of processes in the eukaryotic cell including endosome acidification, endocytosis and vesicular trafficking. The engineered ablation of ATP6AP2 in cardiomyocytes leads to disassembly of the V-ATPase complex, loss of its function, impaired autophagy and eventually cell death, (13) mimicking the effect of V-ATPase inhibitors, such as bafilomycin A1 (BafA1). We were interested whether partial depletion of this essential protein by siRNA could produce a V-ATPase-deficiency related phenotype. High concentration of BafA1 (>100 nM) inhibits V-ATPase completely, induces vacuolar deacidification that impairs lysosomal protein degradation (14) and promotes apoptosis (15). We therefore investigated whether ATP6AP2 knockdown by siRNA would synergize with BafA1 at concentrations below its reported ability to inhibit vesicular acidification. All three siRNAs to ATP6AP2 shown to efficiently deplete the protein (Supplementary Material, Fig. S1) decreased cell survival at low doses of BafA1 (Fig. 6A). Next, we examined the effect of siRNA knockdown on autophagy using the expression of LC3 and p62 as the read-outs. Conversion of LC3-I (cytosolic form) to LC3-II (membrane-bound lipidated form) is a measure of autophagosomal formation. We evaluated the LC3-II/LC3-I ratio under conditions that either induce or block autophagy (Fig. 6B). In cells where autophagy was induced by starvation or blocked at the fusion step by BafA1 the ratio was increased at a comparable level; the combined treatment (starvation followed by BafA1) augmented the LC3-II/LC3-I ratio in an additive manner. We did not observe significant differences in autophagic flux between control and ATP6AP2 knockdown cells.
Figure 6.
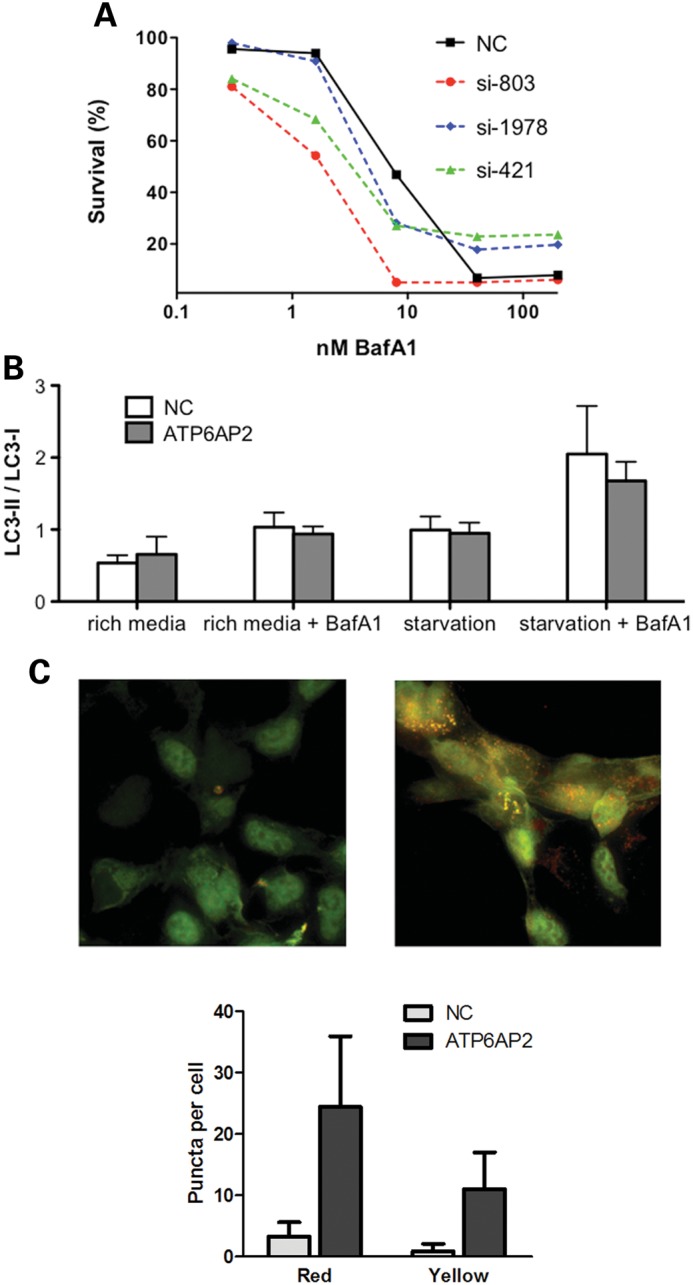
Knockdown of ATP6AP2 affects V-ATPase function and sensitizes cells to low concentrations of BafA1 (A). Dose–response of HEK293T cells to BafA1 after ATP6AP2 knockdown by siRNA. Forty-eight hours after transfection with three different siRNAs targeting ATP6AP2 (si-803, si-421, si-1978) or negative control siRNA (NC), HEK293T cells were exposed to BafA1 at the concentrations indicated. Cell-viability data are normalized to controls treated with 0.5% DMSO. (B) Autophagy flux measured in three independent experiments. HEK293T cells were transfected with negative control siRNA (NC) or with a pool of ATP6AP2 siRNA (ATP6AP2) and assayed for autophagy for a total of 6 h. BafA1 was added during the last 2 h of incubation. (C). ATP6AP2 deficiency induces accumulation of LC3-positive puncta. Shown is a representative ATP6AP2 siRNA knockdown experiment in HEK293 cells stably expressing the ptfLC3 reporter. Left panel, untransfected cells (NC); right panel, cells 96 h after transfection with a pool of ATP6AP2 siRNA (ATP6AP2); bottom, statistical analysis of puncta counts was performed on untreated (N = 31) and ATP6AP2 siRNA treated cells (N = 21) using a two-tailed t-test (P < 0.0005) for both autolysosomes (red puncta) and autophagosomes (yellow puncta).
Another common method of autophagy evaluation is visualization of autophagosomes as LC3-positive puncta. We used HEK293 cells stably transfected with the mRFP-GFP-LC3 reporter (16). The difference in pH sensitivity of the two fluorescent tags allows the autophagosome before fusion with the lysosome (yellow fluorescence of both RFP and GFP) to be distinguished from the autolysosome (red fluorescence of RFP). All three siRNAs to ATP6AP2 augmented the presence of punctate LC3-positive structures (Fig. 6C). Accumulation of yellow and red LC3-positive puncta indicates perturbation of autophagy and lysosomal clearance.
To examine the XPDS brain for defects in autophagy, we performed comparative immunostaining with p62/SQSTM1 antibodies, a marker of impaired autophagy in various neurodegenerative conditions (17). IHC revealed massive accumulation of p62 in the XPDS striatum (Fig. 7) but not in other regions examined (data not shown) indicating a region-specific impairment of constitutive autophagy in the XPDS brain. It is of note that ATP6AP2-deficiency was also most prominent in the XPDS striatum (Fig. 5).
Figure 7.
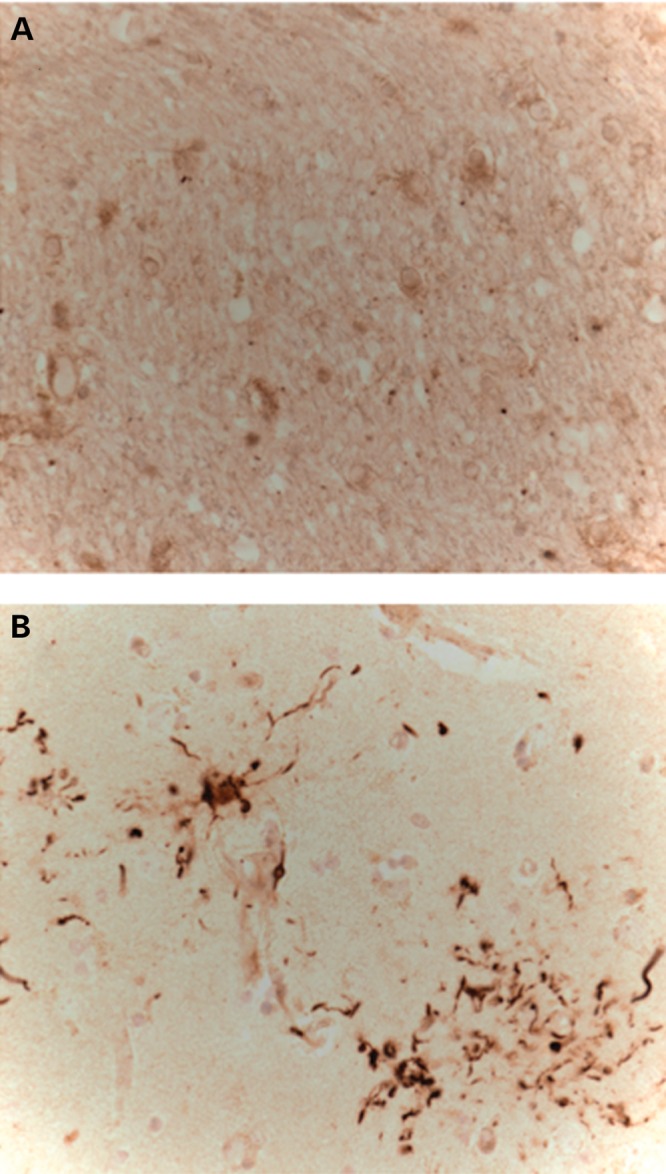
p62 immunostaining of the striatum of a control (A) and XPDS case (B). Note p62 labeling of plaque-like pathology, similar to tau immunostaining previously described in this case (1).
DISCUSSION
Herein, we show that a silent mutation in ATP6AP2, which encodes an accessory unit of an essential lysosomal enzyme, V-ATPase, is the cause of a familial parkinsonian disorder, XPDS. The availability of patient blood cells allowed detection of the underlying molecular phenotype, aberrant splicing of ATP6AP2 mRNA that results in the overexpression of its minor isoform. Another silent mutation in the same exon of ATP6AP2 that produces a similar defect in splicing causes X-linked mental retardation Hedera type (MRXSH; OMIM 300423), a distinct CNS pathology with several overlapping features with XPDS, which include spasticity and seizures (1,8,18). According to recent estimates, at least 15% of all disease-causing mutations affect RNA splicing (19,20). Many of them are silent exonic mutations with no clearly predictable effect on splicing. This highlights a necessity to procure cells or other biological material from patients for studies of genetic disorders.
V-ATPase complex and its accessory protein ATP6AP2 are present within the plasma membrane and membranes of some organelles such as endosome, lysosome and secretory vesicle. V-ATPase is involved in receptor-mediated endocytosis, membrane trafficking, protein processing and degradation and nutrients uptake (21,22). V-ATPase complex is composed of an 8-subunit V1 sector that is responsible for ATP hydrolysis and a 6-subunit V0 sector that serves as a transmembrane proton channel (23,24). ATP6AP2 is specifically associated with the V0 sector ensuring its integrity. Targeted disruption of ATP6AP2 is lethal to the cell; it destabilizes V0 and prevents its assembly but has no effect on V1 (13). The resulting acute V-ATPase deficiency carries multiple consequences including impaired lysosome-mediated protein degradation and autophagy (13,25). We show that even partial depletion of ATP6AP2 by siRNA sensitizes HEK293T cells to low doses of V-ATPase pump inhibitor BafA1. The accumulation of autophagosomes and autolysosomes upon siRNA knockdown suggests a direct effect of ATP6AP2 deficiency on the autophagy process. The immunoblotting with endogenous LC3 did not detect autophagy changes; the results from the two LC3-based assays may reflect differences in assay sensitivity and/or cell-line-specific responses and warrant further investigation. Perhaps most relevant to the pathogenesis of the disorder, the most affected area of XPDS brain, the striatum, showed ATP6AP2 deficiency, pathological Tau deposits (1) and massive accumulation of p62/SQSTM1 indicating profound defect in lysosome-mediated protein degradation and autophagy.
ATP6AP2 is a ubiquitous protein as is its pathological Δe4 ATP6AP2 isoform, which is overexpressed in XPDS and MRXSH cells of non-neural origin. Yet, the effect of mutations in both disorders is CNS-confined rather than pleiotropic. This may be explained by particularly strong demand of neurons for V-ATPase related functions. Additionally, ATP6AP2 isoforms may carry out yet unidentified functions exclusive for neural cells. The complexity of alternative splicing of ATP6AP2 has greatly evolved in the human compared with the mouse (Fig. 3). In the PC-12 model of neurogenesis, overexpression of Δe4 ATP6AP2 inhibited neural differentiation (26). Besides overexpression of Δe4, a splicing mutation ultimately affects the level of the normal full size ATP6AP2 isoform. At the transcript level, the production of the normal isoform is reduced because of competition for pre-mRNA. At the protein level, the Δe4 isoform seems to have a shorter half-life (8,26) resulting in a lowered output of total ATP6AP2 protein. In normal brain, basal Δe4 expression is an order of magnitude higher than in non-neural cells so one can reasonably anticipate that the effect of the XPDS mutation on ATP6AP2 protein level in the brain will be also augmented. By IHC, we did observe a deficit of total ATP6AP2, which was most pronounced in the XPDS striatum, an area commonly involved in PD. Of note, heterozygous female carriers in both MRXSH and XPDS families do not manifest clinical symptoms even though the Δe4 transcript is overexpressed in their blood cells [(8) and our unpublished data]. The cells that express the normal ATP6AP2 allele may be sufficient to compensate for cells with the mutant allele on the active X chromosome. It is also possible that disease-free female carriers have a skewed pattern of X inactivation in neural tissues.
Why do splicing mutations that produce a similar molecular phenotype in blood cells cause such different CNS disorders? The most likely explanation is a quantitative difference in Δe4 expression level and/or in the balance of ATP6AP2 splicing isoforms in the developing brain of MRXSH and XPDS patients. Although point mutations in the canonical 5′ and 3′ splice sites often have severe splicing phenotypes, exonic point mutations that cause efficient exon skipping are rare. This suggests that exon 4, by natural design, is balanced between inclusion and exclusion that always produces transcripts with a correct reading frame. The ratio of inclusion to exclusion would then depend on the developmental and tissue specific control of splicing factors. For many pathogenic splicing mutations, change in the isoform level is the tissue-specific quantitative trait. Examples are MAPT-related tauopathies (27), in which splicing mutations in MAPT result in rather subtle changes in the balance of splice isoforms of tau protein, and the pathology is confined to the CNS. The differential effect of ATP6AP2 splicing mutations can be viewed as a result of interaction of corresponding cis-elements affected by the mutation with development stage-specific splicing factors in the brain. For instance, c.321C>T (p.D107D) disrupts a cis-element for several positive regulators and produces a permanent splicing defect in MRXSH patients. On the other hand, c.345C>T (p.S115S) creates a new silencer site. If the respective negative splicing regulator is temporarily downregulated in the developing brain, this would mitigate an early effect on neurodevelopment, but later might cause PD-like symptoms. It remains to be seen whether the affected individuals from the MRXSH family, who are currently very young, will develop PD-like features in adulthood.
MATERIALS AND METHODS
Subjects
Approval for the recruitment and genetic analysis of the XPDS family, control subjects and individuals with PD was granted by the University of Washington and VA Puget Sound Health Care System Institutional Review Boards. All tissue samples were obtained following written informed consent for autopsy and the use of the material and clinical information for research. The three living affected males and two obligate carrier females in the previously reported XPDS family were re-examined. Subjects with PD were participants in the Parkinson's Genetic Research Study (28) at the VA Puget Sound Health Care System. Unrelated participants from other studies, who provided informed consent for sharing DNA or genetic information for use in research, served as controls. These individuals were not screened for PD.
Exome sequencing
Genomic DNA isolated from the blood of the most severely affected individual, IV-5 (1) (Fig. 1), was used to prepare a shotgun sequencing library. The Nimblegen_solution_V2refseq2010.HG19 probe library was used for target enrichment, and sequencing was performed on the Illumina GAIIx platform with paired-end 76 base reads. Fastq sequence files were aligned against the human reference sequence (National Center for Biotechnology Information 37/hg19) with the Burrows-Wheeler Aligner. Duplicate paired-end reads were removed from the merged data sets. SNP and indel calling was performed with the GATK Unified Genotyper and annotated with SeattleSeq server (http://gvs.gs.washington.edu/SeattleSeqAnnotation/) according to the National Center for Biotechnology and University of California Santa Cruz (http://genome.cse.ucsc.edu/cgi-bin/hgGateway) databases. Additionally, exome reads from the linkage interval on the X chromosome were re-aligned using the Splitread algorithm (29) to identify nucleotide repeat sequences within coding sequences and near intron/exon junctions. Tandem Repeats Finder (http://tandem.bu.edu/trf/trf.html) (30) and Short Tandem Repeat DNA Internet DataBase (https://tandem.bu.edu/cgi-bin/trdb/trdb.exe) (31) were used to screen the reference genome within the IBD interval for potentially unstable repeats, defined as having length 24 nt or more.
Cell lines and brain tissues
EBV-transformed LCL from patients and controls were established and maintained according to standard protocols (32). Human embryonic kidney cells (HEK-293 and HEK-293T) were obtained from the American Type Culture Collection and cultured in DMEM supplemented by 10% FBS at 37°C and 5% CO2. Post-mortem brain tissues of neurologically normal control subjects used for RNA isolation were obtained from the Neuropathology Core Brain Bank at the University of Washington. The average age of subjects was 70 and the average post-mortem interval was 4 h. Tissue samples were flash frozen at the time of autopsy and stored at −80°C.
RNA isolation and cDNA synthesis
Total RNA was extracted from whole blood using Pure Link Total RNA Blood Kit (Invitrogen, Carlsbad, CA, USA), from cultured LCL or fresh-frozen human cerebella using RNeasy Mini Kit (Qiagen, Valencia, CA, USA). cDNA synthesis with poly-dT primer was performed using the SuperScript III RT–PCR kit (Invitrogen). Primers for detection of splice variants were designed using Primer3 and are available on request. Effect of variants on splicing was evaluated using Human Splicing Finder, http://www.umd.be/HSF/ (33).
Quantitative reverse transcription PCR
RNA was isolated from cultured cells at least two times per sample. Quantitative reverse transcription (qRT)–PCR was performed on a 7500 Real-Time PCR System (Applied Biosystems, Foster City, CA, USA) using ABI gene-expression assays. Experiments were carried out in triplicate with two external control genes (GUSB (Hs00939627_m1, ABI) and TBP (Hs00427620_m1, ABI) for normalization of gene expression. To quantify the expression of ATP6AP2 isoforms, the following ABI qPCR assays were used: (i) Hs00997140_g1 to amplify e3–e4 junction; (ii) Hs00997145_m1 to amplify the invariant e8–e9 junction; (iii) a custom made ATP6AP2_ Δe4 assay to amplify e3–e5 junction lacking e4.
The following primers/probes were used for ATP6AP2_ Δe4 assay: Forward primer 5′-AGGGAGTGAACAAACTGGCTCTA-3′; Probe 5′-6FAM_CCCCAGGCAGTGTC_MGB-3′; Reverse primer 5′- taccatatacactctattctccaaagggta-3′.
Immunohistochemistry of brain sections
Post-mortem brain tissue from one XPDS case (pedigree position II-2) and two neurologically normal control subjects was obtained from the Neuropathology Core Brain Bank at the University of Washington. Formaldehyde-fixed paraffin-embedded sections from the frontal lobe, striatum and hippocampus were de-paraffinized and autoclaved at 15 psi, 121°C for 20 min either in citrate buffer, pH 6.0 (ATP6AP2) or in Tris/EDTA, pH 9.0 (p62/SQSTM1) for antigen retrieval. Immunodetection was performed with goat polyclonal antibodies against ATP6AP2 (AF5716, R&D Systems, Minneapolis, MN, USA), mouse monoclonal antibodies against p62/SQSTM1 (D-3, sc-28359, Santa-Cruz, Dallas, TX, USA) and secondary anti-mouse and anti-goat antibodies (Vector Laboratories, Burlingame, CA, USA). The specificity of the antigen detection was ascertained by omitting the primary antibody.
LC3 immunodetection by western blotting
Forty-eight hours after siRNA transfection, HEK293T cells were cultured in conventional or nutrient-deprived media (Eagle's balanced salt solution) for 6 h total time; where indicated, 400 nM BafA1 was added during the last 2 h of incubation. Cells were lysed in 2% SDS sample buffer followed by sonication twice for 5 s on ice to obtain whole-cell lysate. The lysates were heated at 100°C for 5 min and cleared by centrifugation at 20 000 rpm for 5 min at 4°C. Lysates were resolved fractionated on Novex® Tris–Glycine polyacrylamide gel (Life Technology). After transfer to PDVF membrane (Life Technology, CA, USA), immunoblotting with following primary antibodies: rabbit anti-LC3 (Thermo Scientific, IL, USA), rabbit anti-SQSTM1/p62 (Cell Signaling, MA, USA), rabbit anti-GAPDH (Sigma-Aldrich, St Louis, MO, USA) and with anti-rabbit HRP-conjugated secondary antibodies (Life Technology, CA, USA) were performed. The protein bands were visualized by enhanced chemiluminescence (Thermo Scientific, IL, USA) under densitometry (BioChemi digital imaging system, UVP, CA, USA) and quantified with ImageJ software (NIH). GAPDH expression level served as a loading control. The data were averaged from triplicate experiments and analyzed by T-test.
siRNA knockdowns
Reverse transfections of HEK-293T cells in 24-well plates were performed using Lipofectamine™ RNAiMAX reagent (Invitrogen) and siRNAs at 10 nM concentration. We used three siRNAs targeting various ATP6AP2 exons: NM_005765_stealth_1978; NM_005765_stealth_421; NM_005765_stealth_803; and Negative control No. 2 siRNA. Sequences are provided in the Supplementary Material and Methods.
Cell-viability assay
The luminescent CellTiter-Glo cell viability assay (Promega, Madison, WI, USA) measuring cellular ATP level was performed in duplicate according to the manufacturer's protocol. Briefly, 5 × 103 cells were seeded on wells in white clear bottom 96-well plates (Greiner Bio-One, Germany). The next day, a series of concentrations of BafA1 (Sigma-Aldrich, St Louis, MO, USA) or DMSO was added to the wells. After 48 h of culture, CellTiter-Glo reagent was added to the wells; luciferase luminescence was measured on a FluoStar Omega plate reader (BMG LABTECH, Germany).
Fluorescence microscopy
ptfLC3, the plasmid encoding the tandem fluorescent reporter was obtained from AddGene (Addgene plasmid 21074) (16). tfLC3 expressing stable cell lines were constructed by transfecting HEK293 with GenePorter 2 (Genlantis) using the manufacturer's protocol (34). Geneticin (400 μg/ml) and Zeocin (100 mg/ml) were used to maintain selections.
Cells were seeded onto poly-d-Lysine coated (Sigma-Aldrich, St Louis, MO, USA) 12 mm round glass cover slips in 24-well plates. Cells were fixed for imaging 96 h after siRNA transfection. Microscopy was performed on a Delta Vision microscope (Applied Precision, Inc.) using a 60x oil-immersion objective, a sCMOS camera, and 2 × 2 binning. Image analysis, volume rendering and isolation of red/green co-localization into a blue color channel were performed using softWorx 6.0 Beta software. Image segmentation, color channel separation and puncta counts were conducted in Adobe Photoshop CS5.
WEB RESOURCES
The URLs for data presented herein are as follows:
1000 Genomes Project, http://www.1000genomes.org/page.php
Database of Genomic Variants, http://projects.tcag.ca/variation/
dbSNP homepage, http://www.ncbi.nlm.nih.gov/SNP/
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/
SeattleSeq Annotation, http://gvs.gs.washington.edu/SeattleSeqAnnotation/
University of California Santa Cruz Human Genome Browser, http://genome.cse.ucsc.edu/cgi-bin/hgGateway
Tandem Repeats Finder, http://tandem.bu.edu/trf/trf.html
Short Tandem Repeat DNAInternet DataBase, https://tandem.bu.edu/cgi-bin/trdb/
Human Splicing Finder, http://www.umd.be/HSF
AceView database, http://www.ncbi.nlm.nih.gov/IEB/Research/Acembly/
NHLBI Exome Sequencing Project Exome Variant Server, http://evs.gs.washington.edu/EVS/
SUPPLEMENTARY MATERIAL
FUNDING
This work was supported by the Department of Veteran Affairs (VISN-20 MIRECC VA Puget Sound Health Care System and VA Merit Award for T.D.B. and W.H.R., GRECC and PADRECC VA Puget Sound Health Care System, VA Merit Award I01BX000877 for B.C.K. and VA Merit Award 1I01BX000531 for C.P.Z.), the American Recovery and Reinvestment Act funds [through RC2 HG005608 from the National Institutes of Health to D.A.N. and W.H.R.] and the National Institutes of Health (T32 GM00727 and P01 GM081619 to N.S.S., R01NS069719 to W.H.R., R01NS064131 to B.C.K. and R01 NS065070 to C.P.Z.). We would also like to recognize the following ongoing studies that produced and provided exome variant calls for comparison: the National Heart, Lung and Blood Institute [Lung GO Cohort Sequencing Project (HL 1029230) and the Women's Health Initiative Sequencing Project (HL 102924)], the Broad GO Sequencing Project (HL-102925), the Seattle GO Sequencing Project (HL 102926) and the Heart GO Sequencing Project (HL-103010).
Supplementary Material
ACKNOWLEDGEMENTS
We are grateful to the family for their participation in this study. Mark Matsushita, Peggy Robertson, John Wolff, and Dora Yearout provided expert technical support. We would like to thank Drs Cristina-Maria Cruciat and Christof Niehrs, who provided the constructs expressing ATP6AP2 and LRP6, Dr Lynn Bekris, who provided control brain cDNAs and Dr Pamela McMillan for her help with the IHC. We are appreciative of Drs Alan Weiner's and Leo Pallanck's thoughtful comments.
Conflict of Interest statement. None declared.
REFERENCES
- 1.Poorkaj P., Raskind W.H., Leverenz J.B., Matsushita M., Zabetian C.P., Samii A., Kim S., Gazi N., Nutt J.G., Wolff J., et al. A novel X-linked four-repeat tauopathy with Parkinsonism and spasticity. Mov. Disord. 2010;25:1409–1417. doi: 10.1002/mds.23085. doi:10.1002/mds.23085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ng S.B., Bigham A.W., Buckingham K.J., Hannibal M.C., McMillin M.J., Gildersleeve H.I., Beck A.E., Tabor H.K., Cooper G.M., Mefford H.C., et al. Exome sequencing identifies MLL2 mutations as a cause of Kabuki syndrome. Nat. Genet. 2010;42:790–793. doi: 10.1038/ng.646. doi:10.1038/ng.646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chen Y.Z., Matsushita M.M., Robertson P., Rieder M., Girirajan S., Antonacci F., Lipe H., Eichler E.E., Nickerson D.A., Bird T.D., et al. Autosomal dominant familial dyskinesia and facial myokymia: single exome sequencing identifies a mutation in adenylyl cyclase 5. Arch. Neurol. 2012;69:630–635. doi: 10.1001/archneurol.2012.54. doi:10.1001/archneurol.2012.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cummings C.J., Zoghbi H.Y. Trinucleotide repeats: mechanisms and pathophysiology. Annu. Rev. Genomics Hum. Genet. 2000;1:281–328. doi: 10.1146/annurev.genom.1.1.281. doi:10.1146/annurev.genom.1.1.281. [DOI] [PubMed] [Google Scholar]
- 5.Ludwig J., Kerscher S., Brandt U., Pfeiffer K., Getlawi F., Apps D.K., Schagger H. Identification and characterization of a novel 9.2-kDa membrane sector-associated protein of vacuolar proton-ATPase from chromaffin granules. J. Biol. Chem. 1998;273:10939–10947. doi: 10.1074/jbc.273.18.10939. doi:10.1074/jbc.273.18.10939. [DOI] [PubMed] [Google Scholar]
- 6.Nguyen G., Delarue F., Burckle C., Bouzhir L., Giller T., Sraer J.D. Pivotal role of the renin/prorenin receptor in angiotensin II production and cellular responses to renin. J. Clin. Invest. 2002;109:1417–1427. doi: 10.1172/JCI14276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cruciat C.M., Ohkawara B., Acebron S.P., Karaulanov E., Reinhard C., Ingelfinger D., Boutros M., Niehrs C. Requirement of prorenin receptor and vacuolar H+-ATPase-mediated acidification for Wnt signaling. Science. 2010;327:459–463. doi: 10.1126/science.1179802. doi:10.1126/science.1179802. [DOI] [PubMed] [Google Scholar]
- 8.Ramser J., Abidi F.E., Burckle C.A., Lenski C., Toriello H., Wen G., Lubs H.A., Engert S., Stevenson R.E., Meindl A., et al. A unique exonic splice enhancer mutation in a family with X-linked mental retardation and epilepsy points to a novel role of the renin receptor. Hum. Mol. Genet. 2005;14:1019–1027. doi: 10.1093/hmg/ddi094. doi:10.1093/hmg/ddi094. [DOI] [PubMed] [Google Scholar]
- 9.Bernhard S.M., Seidel K., Schmitz J., Klare S., Kirsch S., Schrezenmeier E., Zaade D., Meyborg H., Goldin-Lang P., Stawowy P., et al. The (pro)renin receptor ((P)RR) can act as a repressor of Wnt signalling. Biochem. Pharmacol. 2012;84:1643–1650. doi: 10.1016/j.bcp.2012.09.020. [DOI] [PubMed] [Google Scholar]
- 10.De Ferrari G.V., Moon R.T. The ups and downs of Wnt signaling in prevalent neurological disorders. Oncogene. 2006;25:7545–7553. doi: 10.1038/sj.onc.1210064. doi:10.1038/sj.onc.1210064. [DOI] [PubMed] [Google Scholar]
- 11.Biechele T.L., Moon R.T. Assaying beta-catenin/TCF transcription with beta-catenin/TCF transcription-based reporter constructs. Methods Mol. Biol. 2008;468:99–110. doi: 10.1007/978-1-59745-249-6_8. doi:10.1007/978-1-59745-249-6_8. [DOI] [PubMed] [Google Scholar]
- 12.Brennan K., Gonzalez-Sancho J.M., Castelo-Soccio L.A., Howe L.R., Brown A.M. Truncated mutants of the putative Wnt receptor LRP6/Arrow can stabilize beta-catenin independently of Frizzled proteins. Oncogene. 2004;23:4873–4884. doi: 10.1038/sj.onc.1207642. doi:10.1038/sj.onc.1207642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kinouchi K., Ichihara A., Sano M., Sun-Wada G.H., Wada Y., Kurauchi-Mito A., Bokuda K., Narita T., Oshima Y., Sakoda M., et al. The (pro)renin receptor/ATP6AP2 is essential for vacuolar H+-ATPase assembly in murine cardiomyocytes. Circ. Res. 2010;107:30–34. doi: 10.1161/CIRCRESAHA.110.224667. doi:10.1161/CIRCRESAHA.110.224667. [DOI] [PubMed] [Google Scholar]
- 14.Yoshimori T., Yamamoto A., Moriyama Y., Futai M., Tashiro Y. Bafilomycin A1, a specific inhibitor of vacuolar-type H(+)-ATPase, inhibits acidification and protein degradation in lysosomes of cultured cells. J. Biol. Chem. 1991;266:17707–17712. [PubMed] [Google Scholar]
- 15.Xu J., Feng H.T., Wang C., Yip K.H., Pavlos N., Papadimitriou J.M., Wood D., Zheng M.H. Effects of Bafilomycin A1: an inhibitor of vacuolar H (+)-ATPases on endocytosis and apoptosis in RAW cells and RAW cell-derived osteoclasts. J. Cell. Biochem. 2003;88:1256–1264. doi: 10.1002/jcb.10477. doi:10.1002/jcb.10477. [DOI] [PubMed] [Google Scholar]
- 16.Kimura S., Noda T., Yoshimori T. Dissection of the autophagosome maturation process by a novel reporter protein, tandem fluorescent-tagged LC3. Autophagy. 2007;3:452–460. doi: 10.4161/auto.4451. [DOI] [PubMed] [Google Scholar]
- 17.Kuusisto E., Kauppinen T., Alafuzoff I. Use of p62/SQSTM1 antibodies for neuropathological diagnosis. Neuropathol. Appl. Neuro. 2008;34:169–180. doi: 10.1111/j.1365-2990.2007.00884.x. doi:10.1111/j.1365-2990.2007.00884.x. [DOI] [PubMed] [Google Scholar]
- 18.Hedera P., Alvarado D., Beydoun A., Fink J.K. Novel mental retardation-epilepsy syndrome linked to Xp21.1-p11.4. Ann. Neurol. 2002;51:45–50. doi: 10.1002/ana.10051. doi:10.1002/ana.10051. [DOI] [PubMed] [Google Scholar]
- 19.Ward A.J., Cooper T.A. The pathobiology of splicing. J. Pathol. 2010;220:152–163. doi: 10.1002/path.2649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lopez-Bigas N., Audit B., Ouzounis C., Parra G., Guigo R. Are splicing mutations the most frequent cause of hereditary disease? FEBS Lett. 2005;579:1900–1903. doi: 10.1016/j.febslet.2005.02.047. doi:10.1016/j.febslet.2005.02.047. [DOI] [PubMed] [Google Scholar]
- 21.Nishi T., Forgac M. The vacuolar (H+)-ATPases–nature's most versatile proton pumps. Nat. Rev. Mol. Cell. Biol. 2002;3:94–103. doi: 10.1038/nrm729. doi:10.1038/nrm729. [DOI] [PubMed] [Google Scholar]
- 22.Marshansky V., Futai M. The V-type H+-ATPase in vesicular trafficking: targeting, regulation and function. Curr. Opin. Cell. Biol. 2008;20:415–426. doi: 10.1016/j.ceb.2008.03.015. doi:10.1016/j.ceb.2008.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yokoyama K., Imamura H. Rotation, structure, and classification of prokaryotic V-ATPase. J. Bioenerg. Biomembr. 2005;37:405–410. doi: 10.1007/s10863-005-9480-1. doi:10.1007/s10863-005-9480-1. [DOI] [PubMed] [Google Scholar]
- 24.Peters C., Bayer M.J., Buhler S., Andersen J.S., Mann M., Mayer A. Trans-complex formation by proteolipid channels in the terminal phase of membrane fusion. Nature. 2001;409:581–588. doi: 10.1038/35054500. doi:10.1038/35054500. [DOI] [PubMed] [Google Scholar]
- 25.Riediger F., Quack I., Qadri F., Hartleben B., Park J.K., Potthoff S.A., Sohn D., Sihn G., Rousselle A., Fokuhl V., et al. Prorenin receptor is essential for podocyte autophagy and survival. J. Am. Soc. Nephrol. 2011;22:2193–2202. doi: 10.1681/ASN.2011020200. doi:10.1681/ASN.2011020200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Contrepas A., Walker J., Koulakoff A., Franek K.J., Qadri F., Giaume C., Corvol P., Schwartz C.E., Nguyen G. A role of the (pro)renin receptor in neuronal cell differentiation. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2009;297:R250–257. doi: 10.1152/ajpregu.90832.2008. doi:10.1152/ajpregu.90832.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dickson D.W., Kouri N., Murray M.E., Josephs K.A. Neuropathology of frontotemporal lobar degeneration-tau (FTLD-tau) J. Mol. Neurosci. 2011;45:384–389. doi: 10.1007/s12031-011-9589-0. doi:10.1007/s12031-011-9589-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zabetian C.P., Samii A., Mosley A.D., Roberts J.W., Leis B.C., Yearout D., Raskind W.H., Griffith A. A clinic-based study of the LRRK2 gene in Parkinson disease yields new mutations. Neurology. 2005;65:741–744. doi: 10.1212/01.wnl.0000172630.22804.73. doi:10.1212/01.WNL.0000172630.22804.73. [DOI] [PubMed] [Google Scholar]
- 29.Karakoc E., Alkan C., O'Roak B.J., Dennis M.Y., Vives L., Mark K., Rieder M.J., Nickerson D.A., Eichler E.E. Detection of structural variants and indels within exome data. Nat. Methods. 2012;9:176–178. doi: 10.1038/nmeth.1810. doi:10.1038/nmeth.1810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Benson G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 1999;27:573–580. doi: 10.1093/nar/27.2.573. doi:10.1093/nar/27.2.573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gelfand Y., Rodriguez A., Benson G. TRDB–the Tandem Repeats Database. Nucleic Acids Res. 2007;35:D80–87. doi: 10.1093/nar/gkl1013. doi:10.1093/nar/gkl1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Anderson M.A., Gusella J.F. Use of cyclosporin A in establishing Epstein-Barr virus-transformed human lymphoblastoid cell lines. In Vitro. 1984;20:856–858. doi: 10.1007/BF02619631. doi:10.1007/BF02619631. [DOI] [PubMed] [Google Scholar]
- 33.Desmet F.O., Hamroun D., Lalande M., Collod-Beroud G., Claustres M., Beroud C. Human Splicing Finder: an online bioinformatics tool to predict splicing signals. Nucleic Acids Res. 2009;37:e67. doi: 10.1093/nar/gkp215. doi:10.1093/nar/gkp215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Guthrie C.R., Greenup L., Leverenz J.B., Kraemer B.C. MSUT2 is a determinant of susceptibility to tau neurotoxicity. Hum. Mol. Genet. 2011;20:1989–1999. doi: 10.1093/hmg/ddr079. doi:10.1093/hmg/ddr079. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.