

Abstract
Existence of a discrete new X-linked intellectual disability (XLID) syndrome due to KIAA2022 deficiency was questioned by disruption of KIAA2022 by an X-chromosome pericentric inversion in a XLID family we reported in 2004. Three additional families with likely pathogenic KIAA2022 mutations were discovered within the frame of systematic parallel sequencing of familial cases of XLID or in the context of routine array-CGH evaluation of sporadic intellectual deficiency (ID) cases. The c.186delC and c.3597dupA KIAA2022 truncating mutations were identified by X-chromosome exome sequencing, while array CGH discovered a 70 kb microduplication encompassing KIAA2022 exon 1 in the third family. This duplication decreased KIAA2022 mRNA level in patients' lymphocytes by 60%. Detailed clinical examination of all patients, including the two initially reported, indicated moderate-to-severe ID with autistic features, strabismus in all patients, with no specific dysmorphic features other than a round face in infancy and no structural brain abnormalities on magnetic resonance imaging (MRI). Interestingly, the patient with decreased KIAA2022 expression had only mild ID with severe language delay and repetitive behaviors falling in the range of an autism spectrum disorder (ASD). Since little is known about KIAA2022 function, we conducted morphometric studies in cultured rat hippocampal neurons. We found that siRNA-mediated KIAA2022 knockdown resulted in marked impairment in neurite outgrowth including both the dendrites and the axons, suggesting a major role for KIAA2022 in neuron development and brain function.
INTRODUCTION
X-linked intellectual disability (XLID) accounts for >10% of intellectually disabled male patients and explains why the sex ratio is shifted toward males in the intellectually disabled population (1). Syndromic forms have been identified for more than four decades (2), but as many of their features are nonspecific, such as seizures and hypotonia, unraveling these nonspecific entities has remained a challenge. Boundaries between specific and nonspecific forms remain scarce and evolving. Until now, more than 100 genes of XLID have been characterized. At least three main research strategies account for the rapid pace in the discovery of new XLID genes: characterization of chromosomal breakpoints of apparently balanced rearrangements involving the X-chromosome (3), classical linkage studies or locus identification through copy number variation (CNV) screening (4) and massive parallel high-throughput X-chromosome or whole exome sequencing (5). The current sequential history of the four families described in the present report reflects this course well: Family 1 represents the pioneer cytogenetic approach, allowing identification of KIAA2022 as a potential causative gene for XLID through characterization of the breakpoints of an apparently balanced X-chromosome pericentric inversion (6), while Family 3 came to attention through array-comparative genomic hybridization (CGH) screening and Families 2 and 4 through more recent X-chromosome exome sequencing. KIAA2022 has been previously proposed to be involved in XLID since an apparently balanced 46, XY invX (p22.3q13.2) pericentric inversion disrupting P2RY8 and KIAA2022 co-segregated according to an X-linked recessive mode of inheritance in a family where two males suffered severe ID and autism (6). In our previous study, based on the expression pattern of KIAA2022 in human adult and fetal brain, and assignment of P2RY8 to the pseudoautosomal region, we have suggested that KIAA2022 is a novel XLID gene whose truncation causes the severe intellectual disability observed in males harboring the X-chromosome inversion, but we could not rule out the involvement of P2RY8. Here, we describe three new families with likely pathogenic mutations of KIAA2022 (Fig. 1). We have further defined the clinical and genetic phenotypes and investigated the role of KIAA2022 in neuron development using cultured rat hippocampal neurons.
Figure 1.

Pedigrees of the four families are correlated with their molecular findings. (A) Family 1: cytogenetics breakpoints (taken from ref 6) of X-chromosome pericentric inversion. (B) Family 2: electropherogram of KIAA2022 frameshift c.3597dupA mutation. (C) Family 3: quantitative PCR performed on genomic DNA or cDNA for KIAA2022 and dosage of KIAA2022 using cDNA prepared with mRNA extracted from lymphoblasts of patient III,:3. For both the panels, each reaction was performed twice in triplicate. The expression level of the control samples was normalized using Gapdh or Adora2B. Histograms indicate KIAA2022 mRNA decreased production. (D) Family 4: Electropherogram of KIAA2022 indicating c.138 delC frameshift mutation. Amino-acid change is indicated in bold.
RESULTS
Clinical history of the patients
Family 1
Patient 1 (Fig. 1 and Fig. 2, IV: 6) was born at 38 weeks of gestation with normal parameters (2720 g; 48.5 cm; occipito-frontal circumference [OFC] 35 cm) to unrelated Caucasian parents. Hypotonia, hand stereotypies, repetitive behavior, absence of language and poor eye contact were noted during the first year of life. He acquired sitting position without support at 18 months and walked without assistance at 3 years. He had continuous stereotypic movements, self-biting, trunk rocking motions and hyperactivity. No language developed. Due to poor social interaction and difficult relationships to peers, he was admitted to a special institution for the severely retarded. Severe growth retardation (H at −5 SD at 13 years) and postnatal microcephaly (OFC −2.8 SD at 13 years) were recorded. He had mild facial dysmorphia with round face, squint, short nose and prognathism (Fig. 2, IV: 6). A brain magnetic resonance imaging (MRI) performed at 2 years indicates a small brain without any structural abnormality, except a mild enlargement of sulci in the frontal lobes.
Figure 2.

Facial appearance of Patients 1–7: a round face is observed in young age. All patients have strabismus. Note grimacing of Patients III 1 and IV 6.
His maternal uncle, Patient 2 (Fig. 2 III: 1), was born at 38 weeks with normal parameters (2720 g; 48.5 cm; OFC 34 cm). His development was delayed, with an ability to sit unassisted at 15 months and to walk alone at 3 years. Stereotypic movements started at 6 m, consisting of body rocking, hand stereotypies, eye staring and aggressiveness (biting). He required early admittance in a special institution for the severely retarded. He has a severe-to-profound intellectual deficiency with major anxiety, repetitive behaviors, uninterrupted stereotypic movements, stuttering and moderate spasticity of lower limbs. He has no sphincter control. He has had generalized epilepsy until age 2.5 years, with a total of a dozen crises. He learned lately (12 years) to eat without assistance. He had several episodes of fever of unknown origin. Due to growth failure (151 cm, −4 SD at 18 years), investigation of hypothalamic–pituitary–thyroid and gonadotropic axes was undertaken and proved completely normal. OFC is at the 25th centile (54 cm). There is no language ability. A brain MRI indicates enlarged Wirchow-Robin spaces. Upon clinical examination, he has a peculiar facial appearance with a short philtrum, a short nose with a thick tip, marked esotropia and a tented upper lip (Fig. 2, III: 1). The rocking truncal motion is permanent and biting frequent.
Family 2
Patient 3 (Fig. 1 and 2, V: 2) was born at term to unrelated parents with normal parameters (3900 g; 51 cm; OFC 35 cm). There is a family history of intellectual deficiency (ID) of unknown cause in a maternal grand-uncle (Fig. 1, II: 4). From the first month onwards, he developed generalized seizures of the infantile spasms type, once per month on average. EEG was indicative of hypsarrhythmia. Gastroesophageal reflux, poor eye contact and a retarded growth of head circumference (−2 SD) were observed. He sat at 12 months and walked without assistance at 34 months, losing this ability after 6 months. His vocabulary is limited to a few words. He has facial asymmetry, bitemporal narrowing, strabismus, a depressed nasal bridge and a downturned mouth (Fig. 2, V 1). Drooling is permanent. Communication is markedly impaired with stereotypic movements of hands, outbursts of laughter and screaming, repetitive rocking motions of the trunk and bruxism. These signs suggested diagnosis of an autism spectrum disorder (ASD). Postnatal growth retardation was observed (−2 SD at 7 m, −3.5 SD at 6 years). Contrasting with axial hypotonia, mild lower limb spasticity and muscle wasting of legs were noticed. The Griffiths scale of mental development (7) indicates a general quotient (GQ) of 22.8 at 6 years (−6 SD). An Agilent 180K array CGH indicated a normal male complement [arr(1-22)x2, (XY)x1] and karyotype was 46, XY(G banding, 400 bands). His younger brother, Patient 4 (Fig. 1, V: 2), was born at term with normal parameters (3080 g, 48.5 cm, OFC 33.5 cm). His records indicate poor eye contact, hypotonia, inability to sit without support at 24 months and inability to walk without assistance before 4 years. Failure to thrive (W −4.5 SD and H −6.5 SD at 3 years) and marked deceleration of head circumference growth from 4 months onwards (OFC −6 SD at 3 years) are observed. His behavioral phenotype includes drooling and repetitive movements with hand stereotypies in the context of general hypotonia, also suggesting an ASD. From 4 years onwards, hypsarrhythmia, responsive to antiepileptic drugs, developed. There is no language proficiency. Behavioral disturbances are similar to those of his brother. Due to gastroesophageal reflux, he required a gastrostomy. Facial characteristics include a round face, bitemporal narrowing and strabismus (Fig. 2, V: 2). Mild spasticity of lower limbs is also observed. His Griffith's score of mental development (7) is 29 (−5.5 SD).
Family 3
Patient 5 (Fig. 2, III: 3) was born at term with normal parameters (3200 g, 51 cm, OFC 33 cm). His mother has a history of Crohn's disease, relapsing during the second trimester of pregnancy. From 10 months onwards, a right squint was noted. He walked without assistance at 17 months. His first words were only recorded after 18 months. At 8 years, language was limited to disyllabic repeats. Eye contact is poor and there are repetitive hand movements. Behavioral disturbances were noticed from 6 years on. Attendance at a special school was required. He had frequent bouts of excitation and stiffening, suggesting a pervasive disorder. He has mild intellectual deficiency (IQ 50–70). He is unable to eat without assistance. His facial appearance is normal (Fig. 2), as was his brain MRI at 9 years. His karyotype is 46, XY (G banding, 400 bands).
Family 4
Patient 6 (Fig. 1 and Fig. 2, III: 1) was born with normal parameters. He was able to walk unassisted at 18 months. He was slow at school and behavioral disturbances with repetitive attitudes and accesses of bulimia were noted in infancy. Due to hyperactivity and a low attention span, he required special education. At 14 years, his IQ was evaluated as 50. He is able to read and write, and has limited numeracy skills. No autistic features are noted. He has a round face (Fig. 2 III: 1). Brain MRI and EEG were normal. Patient 7 (Fig. 1 and Fig. 2, III: 2) was born at term with normal parameters, after an uneventful pregnancy. A gastroesophageal reflux was noted during the first months of life. He was able to sit without support at 9 months and to walk without assistance at 18.5 months. His language was delayed: he was unable to elaborate a three-word sentence at 10 years. He had important behavioral disturbances, consisting of aggressiveness and hyperactivity, a low attention span and repetitive behavior meeting diagnostic criteria for an ASD. His intellectual deficiency is evaluated in the severe range (IQ < 35). He required an early orientation toward special education. From the age of 7 years, he had Lennox-Gastaut type epilepsy with absences and atonic seizures responding to lamotrigine and valproate. He has no facial dysmorphia apart from a round face. At 10 years, his height was 126 cm (+1 SD), his weight 37 kg (+4 SD) and his OFC 52 cm (50th centile). Brain MRI was normal. Patient 8 (Fig. 1, II 3), maternal uncle of Patients 6 and 7, had an uneventful personal history until the age of 3 years, when hyperactivity and bulimia were noted. He had a learning disability and was integrated in specialized institutions. He presented generalized epilepsy from the age of 14 years, responsive to valproate. He has a moderate intellectual deficiency with an IQ of 46. At 40 years, height is 172 cm, weight 102 kg (+8 SD) and OFC 62 cm (+3 SD).
Molecular studies
Family 1
Both affected males are carriers of a previously described X-chromosome inversion (inv (X)(p22.3q13.2). The Xq13.2 breakpoint lies within intron 1 of KIAA2022. The Xp breakpoint is located in the pseudoautosomal region. Given the inheritance pattern of the disease phenotype, it was anticipated that disruption of KIAA2022 at Xq13.2 could be causal. Previously reported results indicated that KIAA2022 RT–PCR expression analysis using mRNA prepared from their lymphocytes revealed virtually no amplification of KIAA2022 transcripts. It was concluded that both the patients completely lack a functional KIAA2022 protein (6).
Family 2
To identify the disease-causing mutation in Family 2 (Fig. 1), we performed massive parallel sequencing of the X-chromosome–specific exons using the DNA of the index patient (Fig. 1,V: 2). After sequence analysis using in-house–developed tools and filtering against 200 Danish control individuals (8) and publicly available databases, including the 1000 genomes project database, dbSNP135 and the Exome Variant Server, 2 non-synonymous variants remained: a missense change in FATE1 (chrX:150641804-150891148(UCSC hg19) A>G, p.Thr157Ala) and the duplication of a single nucleotide in KIAA2022 (chrX:73877519-73877520(UCSC hg19) dupA, p.S1200YfsX5), the latter causing a protein frameshift and resulting in a premature stop codon. Subsequent Sanger sequencing of the KIAA2022 mutation in all available family members indicated that it co-segregated with the disease. Figure 1 shows the sequencing results for the index patient (Fig. 1, V: 2). Based on the nature of the sequence alteration and the previous report (6), it was concluded that the phenotype was the result of a KIAA2022 protein frameshift mutation.
Family 3
In this family, the proband is a carrier of an X-chromosome microduplication, dupXq13.3, identified by Agilent 180K array CGH. It spans a 70 kb segment containing KIAA2022 exon 1 (chrX: 73877519-73877520, hg19). His mother (Fig. 1, II: 2) also harbors the microduplication, which is absent in his two unaffected brothers (Fig. 1, III: 1; III: 2), and absent in his maternal grandmother (Fig. 1, I: 2), therefore likely to result from a de novo occurrence in a grand-maternal germ-cell, consistent with the fact that the four maternal uncles of the index patient are unaffected. Haplotyping with X-chromosome markers confirmed that the two unaffected brothers (Fig. 1, II:1 and II, 2) inherited their deceased maternal grandfather's X-chromosome, while the index patient inherited its maternal grandmother's one (data not shown). The duplication was subsequently confirmed using a quantitative PCR on genomic DNA prepared from peripheral lymphocytes. On cDNA, the experiments revealed a 60% decrease in the proband KIAA2022 expression (Fig. 1). Using various primer combinations, no hybrid transcript containing two copies of KIAA2022 exon 1 could be obtained from the study of the index patient's lymphoblasts. Given the nature of the microduplication, we anticipate that no fusion transcript can be produced from that locus as a consequence of impaired regulation of KIAA2022 expression. Alternatively, a diminished nonsense-mediated mRNA decay could be operating.
Family 4
X-chromosome exome sequencing was also performed and revealed three identical variants in both affected male siblings: a single missense change in DMD and PLP2 and a frameshift mutation in KIAA2022. Among these three sequence alterations, KIAA2022 c.183 delC was the only one that was also observed in their affected uncle (Fig. 1). This mutation was confirmed by Sanger sequencing. The mutation is responsible for a protein frameshift at codon 62 (of 1516) leading to a premature stop codon 22 codons downstream, which may likely result in mRNA degradation by the nonsense-mediated mRNA decay machinery or in a total loss of protein function if the mRNA is translated (p.Arg62GlufsX22). These results, together with the clinical phenotype of the patients and the previous data, suggest that this KIAA2022 truncating mutation is causative in the brothers.
In vitro study of KIAA2022 role in neuronal development and neurite growth
We have provided evidence that intellectual deficiency is associated with a loss of function of KIAA2022 in our patients. KIAA2022 is expressed in the brain, but its neurobiological function remains unknown. To explore its involvement in neuronal development, we cultured hippocampal neurons from rat brains at embryonic day 18. After brain tissue dissociation, hippocampal neurons were transfected with siRNAs specific for rat KIAA2022 to suppress KIAA2022 expression, or scrambled siRNA (scRNA) as control. Expression of KIAA2022 siRNA, but not the scrambled scRNA, caused a dramatic reduction in the expression of KIAA2022 (Fig. 3A). As part of the normal developmental process, multiple neurites grow from the soma, one of which growing at a higher rate into the longest neurite to eventually become the axon (9,10).
Figure 3.

KIAA2022 knockdown leads to suppression of neurite growth. (A) Primary cultured hippocampal neurons were prepared from embryonic day 18 rat embryos as previously described. Neurons were transfected with KIAA2022-specific siRNA (siRNA) or a scrambled control siRNA (scRNA), together with a fluorescent protein DsRED to indicate transfection. Neurons were then maintained in Neurobasal medium in a humidified incubator containing 5% CO2. Two days after transfection, neurons were fixed, permeabilized and immunostained for KIAA2022. Whereas neurons transfected with scRNA showed normal levels of KIAA2022, a marked reduction in KIAA2022 was shown in neurons expressing siRNA. Transfected neurons were indicated by DsRED (arrows). (B) Neurons were transfected with KIAA2022-specific siRNA (siRNA) or a scrambled control siRNA (scRNA), together with enhanced green fluorescent protein (EGFP). Two days after transfection, neurons were fixed, permeabilized and immunostained for TuJ-1 (tubulin beta-III) to indicate all neuritis. Transfected neurons were indicated by EGFP (arrows). (C–F) Measurements on neurite length, including total neurites, singe neurite average, total dendrites and average length of individual dendrites. *P < 0.05, Student's t test.
Forty-eight hours after plating, neurons were fixed and immunostained with antibodies against a neuron-specific tubulin TuJ-1 to indicate neuronal structure. Measurements show that neurite growth in KIAA2022 siRNA-expressing neurons was markedly suppressed. KIAA2022 knockdown reduced the total neurite length or average neurite length to ∼60% of the control (all neurites: scRNA 177.9 ± 15.7 µm; siRNA 110.2 ± 19.1 µm; n = 18 cells for each. Average of all neurites: scRNA 55.7 ± 3.3 µm; siRNA 34.3 ± 3.6 µm; n = 18 cells for each). Total dendrite growth was measured by excluding the longest neurite, presumably the axon. We found that the total length of dendrites was also inhibited, though not statistically significant (scRNA 83.9 ± 12.7 µm, n = 18 cells; siRNA 63.2 ± 13.6 µm, n = 18 cells). When the total dendritic length was divided by dendrite branch numbers, the average length of an individual dendritic neurite was significantly inhibited (scRNA 37.4 ± 3.3 µm, n = 18 cells; siRNA 26.7 ± 2.8 µm, n = 18 cells) (Fig. 3B–F). To examine the formation and growth of axons, we immunolabeled neurons with antibodies against tau, an axon-specific marker protein. Neurons expressing siRNA still had a single tau-positive neurite, indicating that the polarization process was intact. However, siRNA knockdown of KIAA2022 dramatically suppressed axonal growth. Expression of KIAA2022 siRNA caused a 40% reduction in average axon length compared with control (scRNA 105.6 ± 6.1 µm, n = 26 cells; siRNA 63.2 ± 8.0 µm, n = 26 cells) (Fig. 4). These findings suggest a major role for KIAA2022 in neuronal development. Inhibition of KIAA2022 expression showed no obvious effects on the soma size or cell survival.
Figure 4.
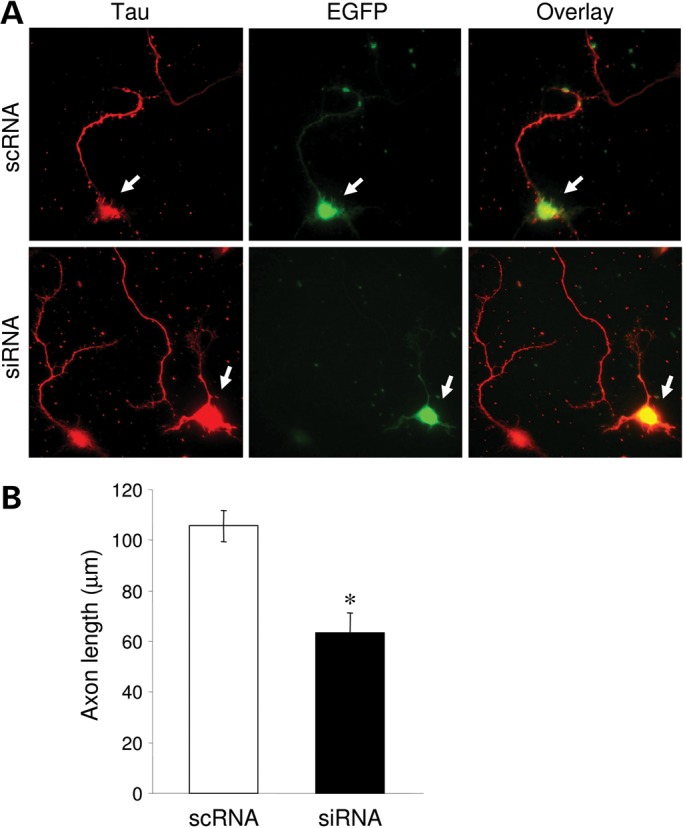
KIAA2022 knockdown inhibits axon development. (A) 48 h after transfection, neurons were immunostained for the axon-specific protein tau. (B) Axon growth was markedly reduced in siRNA-expressing neurons compared with the scRNA control. *P < 0.05, Student's t test.
DISCUSSION
By reappraisal of the family described in 2004, and by comparing clinical and molecular data with those of six new patients belonging to three additional families, we were able to provide evidence for considering KIAA2022-related XLID as a discrete entity (Table 1). This evidence comes from identification of KIAA2022 sequence alterations that are either protein truncating in Families 2 and 4 or hypomorphic in Family 3. The other batch of evidence comes from a phenotype belonging to the same spectrum among the eight affected males. Delayed motor milestones, followed by delayed, rudimentary speech or absent language, and a course eventually determining mild (one patient) to severe intellectual deficiency is the rule. Remarkably, strabismus was present in all of them. A specific facial dysmorphia does not seem to be present, although a round face is noted in most. Interestingly enough, affected males from Families 1 and 2 have apparently an endophenotype with severe autistic features comprising uninterrupted rocking motions, hand stereotypies, auto and hetero-aggressivity, in the context of post-natal growth deficiency, generalized seizures and microcephaly. A KIAA2022 complete loss of function might account for such a severe phenotype in these families with either disruption of KIAA2022 exon 1 or a c.2597dupA protein-truncating mutation. Seizures were generalized with infantile spasms, Lennox-Gastaut syndrome or grand-mal seizures without any argument on brain MRI for a cortical dysgenesis. Although apparently diverse, infantile spasms and Lennox-Gastaut syndrome belong to the same continuum of severe encephalopathies with disorganized EEG, therefore present in 3 of 5 epileptic patients in our series. The fourth patient has an EEG pattern compatible with these entities (see supplemental data). The patient from Family 3 has a reduced KIAA2022 expression, consistent with a mild delay, although associated with behavioral disturbances reminiscent of those observed in the other male patients.
Table 1.
Synopsis of clinical and molecular findings
| Ancestry |
Pt 1 | Pt 2 | Pt 3 | Pt 4 | Pt 5 | Pt 6 | Pt 7 | Pt8 |
|---|---|---|---|---|---|---|---|---|
| French | Portuguese | French | French | |||||
| KIAA2022 mutation | Intron 1 disruption by breakpoint no transcript | cf. Pt1 | c.3597insA protein frameshift p.Ser1200Tyr*fsX5 | cf. Pt3 | Exon 1 70 kb microduplication hypomorphic | c.183delC protein frameshift p.Arg62Glu*fsX22 | cf. Pt6 | cf. Pt6 |
| Devt milestones | ||||||||
| (i) Walking w/o assistance | 3 years | 3 years | 34 months (transient) | 4 years | 17 months | 18 months | 18 months | 14 months |
| (ii) Language | absent | absent | rudimentary | absent | rudimentary | rudimentary | poor | poor |
| (iii) Sphincter control | no | no | no | no | obtained | no | no | obtained |
| Degree of ID | severe to profound | severe to profound | mild | moderate to severe | ||||
| Seizures | absent | generalized | flexor spams | flexor spasms | absent | absent | Lennox-Gastaut | generalized |
| Autistic behavior | +++ | +++ | +++ | +++ | + | absent | + | absent |
| Growth parameters (Z-score) | ||||||||
| Birth W | −1 | −1 | +1 | +0.2 | −0.6 | +0.8 | ||
| H | −0.8 | −0.8 | +1.3 | −0.8 | −0.2 | ukn | +0.2 | ukn |
| OFC | +0.5 | −0.1 | +0.1 | −1 | −1.2 | −0.4 | ||
| Childhood/adulthood | ||||||||
| W | −4 | −2 | −2.3 | −3.2 | −0.5 | +1.3 | +1.2 | +2.5 |
| H | −3 | −4 | −2 | −0.1 | −2 | −2.2 | −0.8 | |
| OFC | −3 | −0.6 | −2.5 | −3.8 | mean | −1.6 | −2 | +4.2 |
| Age at the last examination | 13 years | 20 years | 6 years | 4 years | 8 years | 14 years | 10 years | 40 years |
KIAA2022 is expressed in the brain, and expression studies in mice were supportive of a role for KIAA2022 in brain development (11), but its neurobiological function remains unknown. The mRNA levels of KIAA2022 peak around the perinatal period and decline 3 days after birth (11), indicating a role for KIAA2022 in early brain development. Our in vitro study using cultured rat neurons provided some evidence regarding the neurobiological function of KIAA2022. At the beginning of neural morphogenesis, suppression of KIAA2022 expression by siRNA caused a dramatic reduction in neurite growth, including both dendrites and axons. In line with these findings, a role for KIAA2022 in neurite growth has recently been shown in PC12 cells (12). Likely, deterred growth and structural anomalies in developing neurons may lead to deficiencies in axonal wiring and synapse formation during brain maturation, causing a loss of the proper execution of and the balance in neuronal and circuit function. Indeed, aberrant neural morphogenesis and synaptogenesis are associated with a number of brain disorders, including fragile-X mental retardation, autism, CDKL5-related encephalopathy, Rett syndrome and Down syndrome (13–18). Additional expression studies, attempting to integrate different potential companion proteins, including KIAA2022, MeCP2 and FMR1, in a common developmental pathway will undoubtedly shed new light on our understanding of neurite outgrowth. The role of KIAA2022 in neuronal development, together with the clinical features of the patients observed, provides good evidence that KIAA2022 is essential for proper brain development.
MATERIALS AND METHODS
Population
Family 2 was diagnosed within the frame of a EuroMRX study of 248 families with ID segregating in a XL fashion. Family 3 was diagnosed by using array CGH in the systematic evaluation of ID patients in a regional University hospital and Family 4 was diagnosed during an experimental high coverage ES study of seven XLID families in a University hospital.
Array CGH (Family 3)
Array CGH, using the Human 180K array (Agilent Technology), was performed for Patient 5 against a male control. CNV calls were done using the ADM2 algorithm. CNV were considered when at least three markers were included and candidates CNV were confirmed by a qPCR. A qPCR was performed on genomic DNA of patients and qPCR mixtures were prepared using the Power SYBR® Green PCR Master Mix (Applied Biosystems, Foster City, USA). Two groups of primers were used to confirm the Xq13.3 duplication on Patient 5. The first group is localized in the first intron of the KIAA2022 gene. The second group of primers is localized in the same genomic region, Xq13.3, but in a position without the gene. qPCR conditions were as follows: 10 min denaturation at 95°C, then 45 cycles of 95°C for 10 s, 60°C for 30 s and 72°C for 1 min. qPCRs were performed on the LightCycler 480 (Roche). Primer sequences are available upon request.
Exome sequencing (Families 2 and 4)
Genomic DNA (3 μg) from the index patient of Family 2 (V:II in Fig. 1) was used for constructing a single-end Illumina sequencing library using the Illumina Genomic DNA Single End Sample Prep kit, according to the instructions of the manufacturer. X-chromosome exome enrichment was performed using the Agilent SureSelect Human X Chromosome kit, which contains 47 657 RNA baits for 7591 exons of the human X chromosome. Single end deep sequencing was performed on the Illumina Genome Analyzer GAIIx. The read length was 76 nt. Sequences were analyzed with in-house-developed tools. Segregation analysis in the family was confirmed by conventional Sanger sequencing using gene-specific primers (available upon request).
X-chromosome exome sequencing in Family 4 was performed using the RainDance method which results in the synthesis of 11575 amplicons corresponding to 5.9 Mb and covering 802 genes. Ninety-four percent of the target regions had five reads. Data were analyzed using MPI leup. Variants identified were confirmed by Sanger sequencing with specific primers. PCRs were performed using the ReadyMix kit (Sigma-Aldrich, St Louis, Missouri, USA) and PCR products were purified using the exonuclease Exosap (Amersham, London, UK). PCR conditions available upon request. The sequence reaction was prepared using the BigDye (Applied Biosystems) kit. Products were purified on a Sephadex column (Sigma-Aldrich, Basel, Switzerland) and analyzed on an ABI sequencer (ABI Prism 3130, PE Biosystem, Waltham, MA, USA). Data were exported for analysis using the Sequencing Analysis 3.7 software (Applied Biosystems).
cDNA preparation and quantitative PCR (Family 3)
Total RNA was extracted from lymphoblasts using the TRIzol reagent (Invitrogen) according to the manufacturer's instructions. RNA samples were treated with 0.5 unit of DNase I (Qiagen) per 1 μg RNA at 37°C for 30 min followed by an enzyme inactivation at 65°C for 5 min. Reverse transcription of 2 μg of total RNA was performed in 20 μl of Superscript reaction mix containing 12.5 ng of dN6, 40 U of RNase inhibitor (Invitrogen), 0.5 mm dNTP, 0.01 m dithiothreitol (DTT) and 200 U of Superscript II reverse transcriptase (Invitrogen). For a quantitative PCR, we used the LightCycler 480 system (Roche) and the SYBR Green I Master kit (Roche). Each reaction was performed in triplicate with three independent controls, using 2 μl of first-stand cDNA or 10 ng genomic DNA. GAPDH was used as an internal control for cDNAs and ADORA2B exon 2 for genomic DNAs. Primer sequences are available upon request.
Neuronal culture, transfection and morphometry
Primary cultured hippocampal neurons were prepared from embryonic day 18 rat embryos as previously described (19,20). Briefly, brain regions were dissected and digested with papain at 37°C. Dissociated neurons were transfected with KIAA-specific siRNA or scrambled control siRNA, respectively, together with EGFP to indicate transfection. Cells were seeded onto poly-l-lysine-coated coverslips at ∼0.3 × 106 cells per 60 mm dish, each containing five coverslips. Neurons were maintained in Neurobasal medium (Gibco) supplemented with 2% B27, 1% horse serum, 1% penicillin/streptomycin and 0.4% l-glutamine until use. All cells were maintained in a humidified incubator at 37°C in an atmosphere containing 5% CO2. Transfections were performed with Lipofectamine 2000 (Invitrogen) according to manufacturer's instructions.
Immunocytochemistry for structural examination of cultured neurons
Two days after transfection, neurons were fixed for 10 min in an ice-cold solution of 4% paraformaldehyde, washed twice briefly with 1× ACSF (150 mm NaCl, 10 mm HEPES pH 7.4, 3 mm KCl, 2 mm CaCl2, 10 mm glucose) and blocked for 1 h in 10% NGS/ACSF. Coverslips were then incubated for 2 h with primary antibodies anti-TuJ-1 (tubulin beta-III) (1:500, Millipore) to label all neurites, or anti-tau (1:500, Millipore) to selectively label axons. Cells were then washed three times for 5 min each with 1× ACSF and then incubated in the dark for 1 h with Alexa Fluor-conjugated secondary antibodies (1:700). Coverslips were washed an additional three times for 5 min to remove unbound secondary antibodies before being mounted with ProlongGold Antifade (Invitrogen).
Image collection and data analysis
Immunostained coverslips were mounted onto slides using ProlongGold Antifade reagent (Invitrogen) and kept in the dark for more than 4 h before imaging. Images were collected with an inverted fluorescence microscope at a 63× oil objective (Zeiss Axiovert 200M). The exposure time for a fluorescence signal was first set automatically by the software then adjusted manually so that the signals were within the full dynamic range. The neurite length was analyzed using Image J, a free software available for download from the NIH website (see electronic resources section). The total length was the addition of all neurites in a neuron, and the dendritic length included all the neurites except the longest one. The axon length was measured from the tau-positive neurites. All values were reported as mean ± SEM. Statistical analysis was performed using the two-population Student's t test.
ONLINE RESOURCES
1000 genomes Project Consortium: http://1000genomes.org/data.
NHLBI Exome Variant Server (ESP6500), http://evs.gs.washington.edu/EVS/dbSNP135, http://www.ncbi.nlm.nih.gov/snp.
NIH Image J software: http://rsbweb.nih.gov/ij/download.html.
FUNDING
This work was supported in part by the US National Institutes of Health grant R01 MH079407 (H.-Y.M.), the Brain and Behavior Research Foundation (NARSAD Young Investigator Award) (H.-Y.M.), a grant from the German Ministry of Education and Research through the MRNET (V.M.K.), the Project GENCODYS (241995) (V.M.K.), which is funded by the European Union Framework Programme 7, and Fondation de la Recherche Médicale (S.M.).
Supplementary Material
ACKNOWLEDGEMENTS
We are grateful to the patients and their parents for valuable contribution to this study. We thank Amy Lin and Céline Populaire for assistance in manuscript preparation.
Conflict of Interest statement. None declared.
REFERENCES
- 1.Ropers H.H., Hamel B.C. X-linked mental retardation. Nat. Rev. Genet. 2005;6:46–57. doi: 10.1038/nrg1501. [DOI] [PubMed] [Google Scholar]
- 2.Stevenson R.E., Schwartz C.E., Rogers R.C. X-linked Intellectual Disabilty Syndromes. New-York, USA: Oxford University Press; 2012. pp. 1–432. [Google Scholar]
- 3.Vandeweyer G., Kooy R.F. Balanced translocations in mental retardation. Hum. Genet. 2009;126:133–147. doi: 10.1007/s00439-009-0661-6. [DOI] [PubMed] [Google Scholar]
- 4.Koolen D.A., Pfundt R., de Leeuw N., Hehir-Kwa J.Y., Nillesen W.M., Neefs I., Scheltinga I., Sistermans E., Smeets D., Brunner H.G., et al. Genomic microarrays in mental retardation: a practical workflow for diagnostic applications. Hum. Mutat. 2009;30:283–292. doi: 10.1002/humu.20883. [DOI] [PubMed] [Google Scholar]
- 5.Hu H., Wrogemann K., Kalscheuer V., Tzschach A., Richard H., Haas S.A., Menzel C., Bienek M., Froyen G., Raynaud M., et al. Mutation screening in 86 known X-linked mental retardation genes by droplet-based multiplex PCR and massive parallel sequencing. Hugo J. 2009;3:41–49. doi: 10.1007/s11568-010-9137-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cantagrel V., Lossi A.M., Boulanger S., Depetris D., Mattei M.G., Gecz J., Schwartz C.E., Van Maldergem L., Villard L. Disruption of a new X-linked gene highly expressed in brain in a family with two mentally retarded males. J. Med. Genet. 2004;41:736–742. doi: 10.1136/jmg.2004.021626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Luiz D.M., Foxcroft C.D., Stewart R. The construct validity of the Griffiths scales of mental development. Child Care Health Dev. 2001;27:73–83. doi: 10.1046/j.1365-2214.2001.00158.x. [DOI] [PubMed] [Google Scholar]
- 8.Li Y., Vinckenbosch N., Tian G., Huerta-Sanchez E., Jiang T., Jiang H., Albrechtsen A., Andersen G., Cao H., Korneliussen T., et al. Resequencing of 200 human exomes identifies an excess of low-frequency non-synonymous coding variants. Nat. Genet. 2010;42:969–972. doi: 10.1038/ng.680. [DOI] [PubMed] [Google Scholar]
- 9.Amato S., Liu X., Zheng B., Cantley L., Rakic P., Man H.Y. AMP-activated protein kinase regulates neuronal polarization by interfering with PI 3-kinase localization. Science. 2011;332:247–251. doi: 10.1126/science.1201678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Amato S., Man H.Y. Bioenergy sensing in the brain: the role of AMP-activated protein kinase in neuronal metabolism, development and neurological diseases. Cell Cycle. 2011;10:3452–3460. doi: 10.4161/cc.10.20.17953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cantagrel V., Haddad M.R., Ciofi P., Andrieu D., Lossi A.M., Van Maldergem L., Roux J.C., Villard L. Spatiotemporal expression in mouse brain of KIAA2022, a gene disrupted in two patients with severe mental retardation. Gene Expr. Patterns. 2009;9:423–429. doi: 10.1016/j.gep.2009.06.001. [DOI] [PubMed] [Google Scholar]
- 12.Ishikawa T., Miyata S., Koyama Y., Yoshikawa K., Hattori T., Kumamoto N., Shingaki K., Katayama T., Tohyama M. Transient expression of Xpn, an XLMR protein related to neurite extension, during brain development and participation in neurite outgrowth. Neuroscience. 2012;214:181–191. doi: 10.1016/j.neuroscience.2012.04.030. [DOI] [PubMed] [Google Scholar]
- 13.Belmonte M.K., Allen G., Beckel-Mitchener A., Boulanger L.M., Carper R.A., Webb S.J. Autism and abnormal development of brain connectivity. J. Neurosci. 2004;24:9228–9231. doi: 10.1523/JNEUROSCI.3340-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Humeau Y., Gambino F., Chelly J., Vitale N. X-linked mental retardation: focus on synaptic function and plasticity. J. Neurochem. 2009;109:1–14. doi: 10.1111/j.1471-4159.2009.05881.x. [DOI] [PubMed] [Google Scholar]
- 15.Chapleau C.A., Larimore J.L., Theibert A., Pozzo-Miller L. Modulation of dendritic spine development and plasticity by BDNF and vesicular trafficking: fundamental roles in neurodevelopmental disorders associated with mental retardation and autism. J. Neurodev. Disord. 2009;1:185–196. doi: 10.1007/s11689-009-9027-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ricciardi S., Ungaro F., Hambrock M., Rademacher N., Stefanelli G., Brambilla D., Sessa A., Magagnotti C., Bachi A., Giarda E., et al. CDKL5 ensures excitatory synapse stability by reinforcing NGL-1 interaction in the postsynaptic compartment and is impaired in patient iPSC-derived neurons. Nat. Cell Biol. 2012;14:911–923. doi: 10.1038/ncb2566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shepherd G.M., Katz D.M. Synaptic microcircuit dysfunction in genetic models of neurodevelopmental disorders: focus on Mecp2 and Met. Curr. Opin. Neurobiol. 2011;21:827–833. doi: 10.1016/j.conb.2011.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Garner C.C., Wetmore D.Z. Synaptic pathology of Down syndrome. Adv. Exp. Med. Biol. 2012;970:451–468. doi: 10.1007/978-3-7091-0932-8_20. [DOI] [PubMed] [Google Scholar]
- 19.Man H.Y., Sekine-Aizawa Y., Huganir R.L. Regulation of {alpha}-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor trafficking through PKA phosphorylation of the Glu receptor 1 subunit. Proc. Natl. Acad. Sci. USA. 2007;104:3579–3584. doi: 10.1073/pnas.0611698104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hou Q., Zhang D., Jarzylo L., Huganir R.L., Man H.Y. Homeostatic regulation of AMPA receptor expression at single hippocampal synapses. Proc. Natl Acad.Sci.USA. 2008;105:775–780. doi: 10.1073/pnas.0706447105. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.