

Abstract
The kinase MEKK2 (MAP3K2) has recently been implicated in tumor growth and metastasis. Thus, selective inhibition of MEKK2 may be a novel strategy for cancer therapy. In order to identify inhibitors of MEKK2 kinase activity, we have developed a novel activity assay for MEKK2 based on the discovery that recombinant purified MEKK2 has intrinsic ATPase activity. This MEKK2 ATPase assay was validated for enzyme identity and enzymatic purity by multiple methods including mass spectrometry analysis, testing different sources of MEKK2 and comparing ATPase assay IC50 data for multiple inhibitors to literature values and to IC50 data generated using MEKK2 binding and transphosphorylation assays. Taken together, these data indicated that genuine MEKK2 activity was being measured in this assay and no other ATPases contributed to the signal. A miniaturized version of the assay was validated for high throughput screening and compound libraries were screened. The screening hits generated comparable potencies in the MEKK2 intrinsic ATPase, binding and transphosphorylation assays. We identified a novel MEKK2 inhibitor and confirmed that crizotinib and bosutinib are potent in vitro inhibitors of MEKK2 activity with IC50 values of <100 nM. Thus, this assay has utility for the discovery of small molecule inhibitors of MEKK2 activity.
Keywords: MEKK2, MAP3K2, ATPase, crizotinib, bosutinib
INTRODUCTION
Mitogen-activated protein kinases (MAPK) are critical cell signaling components that transduce extra-cellular stimuli to the nucleus ultimately leading to changes in gene expression1–2. ERK5 is a MAPK that is activated by a number of growth factors, oxidative stress, and hyperosmolarity3. Disruption of the ERK5 gene in mice resulted in embryonic lethality due to defects in cardiac development and angiogenesis4. ERK5 protects many cell types from stress-induced apoptosis3, 5–6. MEK5 is the only MAP2K for the ERK5 pathway7. MEK5 gene knock out in mice was lethal and resulted in abnormal cardiac development, similar in phenotype to the ERK5 knockout mouse7. The only defined MAP3Ks for the ERK5 pathway are MEKK2 (MAP3K2) and MEKK3 (MAP3K3)8–9. These kinases phosphorylate MEK5 and activate its activity. Disruption of the MEKK2 gene in mice resulted in normal development and fertility10–11. In contrast, the disruption of MEKK3 resulted in embryonic lethality due to cardiac development defects that mimicked the ERK5 knockout phenotype12. Thus, the different phenotypes of these transgenic animals demonstrate that these two MAP3Ks integrate different stimuli. MEKK2 activates both the ERK5 and JNK pathways10, 13. MEKK2 has been observed to autophosphorylate in response to stimuli and this autophosphorylation is required for activation of MEKK214.
The role of the ERK5 pathway in cancer has only relatively recently been explored. Expression of ERK5 in a panel of 84 human early stage breast cancer tissue samples was examined and overexpression of ERK5 was found in 20% of the samples15. Overexpression of ERK5 correlated with a decrease in disease-free survival time. In a similar study done with prostate cancer, ERK5 expression was significantly increased in high-grade prostate cancer compared to benign prostatic hyperplasia16–17. Analysis of ERK5 expression in samples taken before and after hormone relapse showed a correlation with ERK5 activation and hormone-insensitive disease17. ERK5 overexpression in PC3 cells resulted in more efficient tumor formation in mice. In another study, a dominant negative form of ERK5 restricted the proliferation of myeloma cells and sensitized the cells to apoptosis-inducing drugs18. In a recent study, miR-143 was shown to be a tumor suppressor in prostate cancer using a mouse model19. One of the mechanisms of miR-143 appears to be suppression of ERK5 protein expression. Activation of the ERK5 pathway, but not the ERK1 pathway, was correlated with lymph node metastasis in oral squamous cell carcinoma20. A potent and selective ERK5 inhibitor has been reported that inhibited tumor growth in an in vivo mouse cancer model21. MEK5 has also been implicated in tumor development. MEK5 expression was determined in 127 cases of prostate cancer and 20 cases of benign prostatic hypertrophy22. The data indicated that elevated expression of MEK5 correlated with bone metastasis and poor prognosis. In one study linking MEKK2 to cancer, 11 prostate cancer tissue samples were compared to uninvolved prostate tissue using imaging mass spectrometry (MS)16. One peptide was found to discriminate cancer from uninvolved tissue. This peptide was a fragment of MEKK2. In confirmation of the MS data, MEKK2 was expressed at 4.4-fold higher level in prostate cancer tissue versus benign tissue using western blotting. Even higher levels of MEKK2 expression were observed in LNCaP, Du145 and PC3 prostate cell lines.
We have recently examined the role of MEKK2 in tumor growth and metastasis using an in vivo mouse xenograft model for breast cancer23. We found that shRNA-mediated knockdown of MEKK2 inhibited activation of ERK5 in response to EGF in the breast cancer cell line MDA-MB-231. Knockdown of MEKK2 expression strongly inhibited both tumor growth and metastasis. Increased apoptosis was observed with the loss of MEKK2 expression in xenografts versus size-matched control tumors, even though growth of MEKK2 knockdown cells in culture was unaffected. MEKK2 shRNA knockdown in the BT474 cell line also resulted in inhibited tumor growth in xenografts. Thus, MEKK2 is critical for EGFR- and Her2/Neu tyrosine kinase-dependent ERK5 activation, tumor growth of both MDA-MB-231 and BT474 cells, and metastasis of MDA-MB-231 cells. Because MEKK2 is required for EGFR activation of ERK5, we assessed whether knockdown of ERK5 in MDA-MB-231 cells would show similar tumor growth and metastasis phenotypes as with MEKK2 knockdown. ERK5 knockdown resulted in a decrease in metastasis without a significant effect on tumor growth. Thus, MEKK2 regulation of ERK5 is only one arm of MEKK2 signaling controlling tumor growth and metastasis. These results further support MEKK2 as a novel target for small molecule inhibitor development.
Kinase intrinsic ATPase activity is the hydrolysis of ATP to ADP and phosphate in the absence of a phosphate-accepting substrate. Intrinsic ATPase activity has been observed for a number of kinases using in vitro biochemical assays and purified kinase. This ATPase activity has been described for several MAPKs, including p38α, p38γ, ERK2, JNK3 and MEK124–25. A format comparison study compared screening results using an antibody-based disassociation-enhanced lanthanide fluoroimmunoassay (DELFIA), an ATP-consumption intrinsic ATPase assay, and a fluorescence polarization binding assay for ITK26. The authors concluded that the intrinsic ATPase assay identified the most comprehensive set of inhibitors from screening. In another study using PI3Kα, IC50 data for a panel of inhibitors generated using an intrinsic ATPase assay was similar to data generated by a lipid phosphorylation assay27.
We have discovered that MEKK2 has intrinsic ATPase activity and developed a high throughput MEKK2 activity assay based on this property. Multiple methods were employed to demonstrate that genuine MEKK2 activity was being measured and no other ATPase activities contributed to assay signal. In addition, a miniaturized version of the assay was validated for HTS and compound libraries were screened. Using this assay, a novel MEKK2 inhibitor was identified and crizotinib and bosutinib were confirmed to inhibit MEKK2 activity with IC50 values of <100 nM.
MATERIALS AND METHODS
Materials
All common reagents such as HEPES, Triton X-100, MgCl2, ethylene glycol tetraacetic acid (EGTA), sodium orthovanadate, ß-glycerophosphate, dithiothreitol (DTT) and dimethyl sulfoxide (DMSO) were reagent-grade quality and obtained from Thermo Fisher Scientific (Waltham, MA) or Sigma-Aldrich (St. Louis, MO). The ADP-Glo™ kinase assay kit (cat# V9102), containing ADP-Glo™, detection reagent and ultra pure ATP was obtained from Promega (Madison, WI). Solid white 96-well half-area plates (cat# 3693) for the manual activity assay and 384-well low volume white plates (cat# 3673) for high throughput activity assay and binding assay were from Corning Incorporated (Corning, NY). Falcon polypropylene plates (cat# 1190) used for serial dilutions of compounds for manual activity and binding assays were obtained from Becton Dickinson (Franklin Lake, NJ). Kinase inhibitors were obtained from Fisher Chemical (Waltham, MA), LC Laboratories (Woburn, MA), Selleck Chemicals (Houston, TX), AK Scientific (Union City, CA), Biovision (Milpitas, CA), Toronto Research Chemicals (Toronto Canada) and Symansis (Shanghai, China). The validation set of compounds for test screening was obtained from Prestwick Chemical (Washington DC). The kinase-focused library of compounds was obtained from The Center for Integrative Chemical Biology and Drug Discovery at the University of North Carolina at Chapel Hill (Chapel Hill, NC). Recombinant purified MEKK2 enzyme was obtained from SignalChem (Richmond, BC, Canada) (vendor 1), Invitrogen (Grand Island, NY) (vendor 2) and Carna Bioscience (Kobe, Japan) (vendor 3). We used the MEKK2 enzyme from SignalChem (vendor 1) to generate the data shown in this report unless otherwise indicated. LanthaScreen Eu-anti-GST antibody (cat# PV5594) and Kinase Tracer 236 (cat# PV5592) used in the binding assays were obtained from Invitrogen. Protease digestion and matrix-assisted laser desorption/ionization-time of flight/time of flight (MALDI-TOF/TOF) analysis of the MEKK2 solution was performed by the Proteomics Core Facility, UNC-Chapel Hill.
MEKK2 Intrinsic ATPase Activity Assay
All compound stock solutions were made in 100% DMSO. Serial dilutions of compounds for IC50 determinations were initially performed in 100% DMSO in 96-well polypropylene plates, followed by appropriate dilutions in assay buffer (50 mM HEPES, pH 7.4, 10 mM MgCl2, 0.5 mM EGTA, 0.5 mM sodium orthovanadate, 0.5 mM ß-glycerophosphate, 2.5 mM DTT and 0.01% Triton X-100), producing a constant 2.5% DMSO in all wells. Subsequently, 10 μl of the diluted compound (or just 2.5% DMSO in assay buffer for controls) were added to the wells of a 96-well plate followed by 10 μl of MEKK2 enzyme diluted in assay buffer. After a 10 minute incubation, the reactions were initiated by addition of 5 μl of ATP (150 μM ultrapure ATP from Promega kit) diluted in assay buffer and the assay plates were centrifuged at 1000 RPM for 1 minute in a Beckman Coulter Allegra X-12R centrifuge. Final concentrations in the assembled standard assay were as follows: 1x assay buffer, 1% DMSO, 30 μM ATP and 10 nM MEKK2 enzyme. The enzyme reactions were allowed to proceed protected from light for 75 minutes at 23ºC and terminated by the addition of 25 μl of solution 1 (ADP-Glo™ Reagent, Promega). At this point assay plates were centrifuged again as described above and incubated at 23°C for an hour. A 50 μL volume of solution 2 (Detection Reagent) of the Promega kit was added to each well and the plates incubated for an hour at 23ºC, protected from light. The plates were read using an endpoint luminescence protocol (measurement interval time: 1.0 s, optic module: LUM plus, gain: 3600, focal height: 13.7 mm, positional delay: 0.1 s) in a BMG PHERAstar (BMG Labtech, Cary, NC) plate reader. Luminescence data, expressed in relative luminescence units (RLU), were normalized to DMSO (100% activity) and “no enzyme” (0% activity) controls as maximum and minimum responses, respectively.
For all MEKK2 assay formats, compound concentration response curves were generated using data points that represent the average of three determinations per concentration, except for the transphosphorylation assay where duplicate determinations were performed. All IC50 values provided in this report are averages of at least three independent determinations. The IC50 values and Hill slopes were calculated from concentration response data using GraphPad Prism software (GraphPad Software Inc., La Jolla, CA) employing either a four-parameter or a three-parameter (fixed bottom) curve fit.
High Throughput MEKK2 Activity Assay
The activity assay described above was readily adapted to an automated 384-well assay version where handling of compound dilutions, delivery of reagents and mixing was performed on a Biomek® NX (Beckman Coulter Inc., Fullerton, CA) and a Nanoscreen NSX 1536 (Charleston, SC). The automated assay was carried out exactly as the manual assay including incubation times and centrifugation with the following modifications. 2.5 μL of enzyme solution was added to 384-well white solid low volume assay plates. 50nL compounds in 100% DMSO (or 100% DMSO as controls) were added to each well using a V&P Scientific pin tool (San Diego, CA). After 10 minutes 2.5 μL of ultrapure ATP was added to each well, bringing the final assay volume to 5 μL. The reaction was terminated by addition of 5 μL of solution 1 (ADP-GLO) and following 60 minutes incubation, 10 μL of solution 2 (Detection Reagent) was added. The plates were sealed with Mylar® Plate Seals (Thermo, Milford, MA) during incubations. The kinase inhibitor library was screened at 5 μM concentration and compounds with ≥50% inhibition were considered hits. Z’-factor calculations were performed as published28.
MEKK2 TR-FRET Binding Assay
Compound dilution methods and assay buffer was the same for the MEKK2 time-resolved fluorescence resonance energy transfer (TR-FRET) binding assay as in the ATPase assay. The assay was performed according to the antibody and tracer manufacturer’s recommendations (Invitrogen). Briefly, 5 μl of diluted compounds (or 3% DMSO for controls) were added to wells of a solid white low volume 384-well plate followed by 5 μl of a mixture of MEKK2 enzyme and LanthaScreen® Eu-anti-glutathione S-transferase (GST) antibody diluted in assay buffer. Immediately after addition of the enzyme-antibody mixture, the reaction mixtures were completed by the addition of 5 μl of kinase tracer 236 diluted in assay buffer. The assay plates were then centrifuged at 1000 rpm for 1 minute as before and incubated for 60 min at 23ºC. Final concentrations in the assembled assay were as follows: 1% DMSO, 2 nM LanthaScreen® Eu-anti-GST antibody, 25 nM kinase tracer and 5 nM MEKK2 enzyme. Time resolved fluorescence was measured using a BMG PHERAstar with the following parameters: number of flashes per well: 300, optic module: HTRF, excitation: 337 nm, emission A: 665 nm, emission B: 620 nm, integration start (μs):100, integration time (μs): 200, focal length: 10.3 mm, ratio multiplier: 1000, positional delay: 0.1. TR-FRET signal was calculated by determined the ratio of relative fluorescence units (RFU) obtained at 665 nm and 620 nm (665 nm/620 nm) and subsequently normalizing the value to DMSO (100% activity) and 30 μM dasatinib (0% activity) controls as maximum and minimum responses, respectively.
MEKK2 Transphosphorylation Activity Assay
The MEKK2 transphosphorylation activity assay and IC50 determinations were performed as described for the ATPase assay except the reactions were initiated by a mixture of ATP and kinase-inactive mutant MKK6 (MAP2K6, Carna Biosciences). Final concentrations in the enzyme reactions were 2 nM MEKK2, 200 nM MKK6 and 3 μM ATP. The reaction time was 25 min and the reactions were stopped by the addition of 225 μl of stop buffer (25 mM HEPES, pH 7.4, 10 mM EDTA). These reactions were then transferred to PVDF membranes using a slot blot apparatus (Hoefer PR 648) following the manufacturer’s recommendations. The membrane was allowed to dry and then probed with an anti-phosphoMKK6 antibody (Phospho-MKK3 (Ser189)/MKK6 (Ser207) (22A8) rabbit monoclonal antibody, Cell Signaling, Danvers, MA) using a standard western blotting protocol29.
RESULTS
Characterization of MEKK2 intrinsic ATPase activity
We sought to develop a high throughput activity assay for MEKK2 in order to identify small molecule inhibitors of this kinase. Using a sensitive chemiluminescent ADP detection assay (ADP-Glo kit, Promega), we observed ATPase activity using purified recombinant MEKK2 enzyme in the absence of any protein substrate. This ATPase activity increased in a linear fashion directly proportional to the concentration of MEKK2 in the reaction up to 10 nM (Fig. 1A). ATPase activity was readily detected even at 1 nM MEKK2. Reaction time course studies with 10 nM MEKK2 demonstrated linear assay signal increase from 0 – 90 min (Fig. 1B). Thus, we chose final reaction conditions of 10 nM MEKK2 and a reaction time of 75 min. The Km for ATP was determined by performing reactions with variable concentrations of ATP with and without MEKK2 (Fig. 1C). Reactions without MEKK2 were taken as background and subtracted from the signal obtained with enzyme to obtain specific RLU. The average Km value was 34 μM and thus we chose to use 30 μM ATP for the final assay. We also used pure ADP to determine a standard curve (data not shown). Under final standard assay conditions, we calculated that approximately 3 μM ADP was generated, translating into a 10% substrate conversion (ATP to ADP). The activity observed in this assay is unlikely to be MEKK2 autophosphorylation activity based on the ratio of the amount of product formed to the MEKK2 enzyme concentration. Since there is only 10 nM MEKK2 and 3,000 nM ADP generated during the course of the reaction, each molecule of MEKK2 would have to be autophosphorylated 300 times – roughly twice the sum of all the serine, threonine and tyrosine residues in the GST-MEKK2 construct. Since this assay would be used to test compounds dissolved in DMSO, the DMSO tolerance of the assay was assessed (Fig. 1D). Up to 8% DMSO was tolerated in the assay without significant effect. The final DMSO concentration in the reaction under final assay conditions was set at 1% DMSO.
Fig. 1. MEKK2 intrinsic ATPase activity assay characterization.
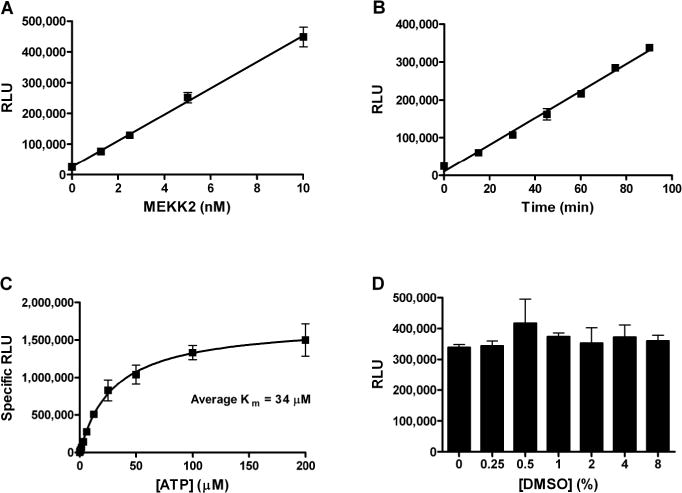
(A) Assay signal in response to MEKK2 enzyme concentration was assessed. (B) Assay signal linearity with respect to reaction times, as indicated, was determined. (C) The Km for ATP was determined using different concentrations of ATP as indicated, with and without MEKK2. Signal obtained without MEKK2 was taken as assay background for each ATP concentration tested. This value was subtracted from the respective assay signal with MEKK2 to obtain specific RLU values. (D) The assay signal at the indicated concentrations of DMSO was determined. Data points represent the average of three determinations per variable and error bars represent standard deviation. Data are representative of three independent experiments.
Validation of the identity and enzymatic purity of MEKK2 in the intrinsic ATPase activity assay
We purchased purified recombinant human MEKK2 and confirmed its identity by mass spectrometry analysis. The entire MEKK2 enzyme (vendor 1) solution was subjected to enzymatic digestion followed by MALDI TOF/TOF MS analysis of the resulting peptides. Peptides matching human MEKK2 were observed, but no other peptides were detected (data not shown).
Non-selective assay formats, such as measuring ADP generation for kinase assays, require an extra amount of enzymatic purity validation to prove that the target activity, and only that activity, is being measured. We used several experimental approaches to validate that the observed ATPase activity was due to MEKK2 and only MEKK2. In one approach, we obtained purified recombinant human MEKK2 from three different vendors since if contamination is the source of activity, then it is less likely (though possible) that the all three sources would have the same contaminating ATPase at the same concentration or activity level. We demonstrated that all three sources of MEKK2 had time dependent ATPase activity with similar specific activities, since they were all tested at 10 nM MEKK2 (Fig. 2A). Furthermore, we used sunitinib as a control inhibitor to determine its potency using all three sources of MEKK2 (Fig. 2B). The concentration response data produced average IC50 values and standard deviations (SDs) of 337 ± 157, 184 ± 80, and 315 ± 199 nM for MEKK2 from vendors 1, 2 and 3, respectively. The sunitinib potencies were all within two-fold of each other and therefore these data suggested that the same enzyme activity was being measured in all three sources of MEKK2. When inhibiting a single enzyme in any biochemical assay, a Hill slope of 1.0 is indicative of the measurement of a single enzyme – versus multiple enzymes that could be present and detected in the assay. Broad (<1) slopes may indicate that more than one enzyme is being measured in the assay. Thus, we also analyzed the Hill slope data from these IC50 curves. The average Hill slope values and SDs from these inhibitor curves were 1.06 ± 0.16, 1.01 ± 0.02 and 1.01 ± 0.05 for vendors 1, 2 and 3, respectively. The slopes for the different MEKK2 sources were very close to 1.0 indicating that a single ATPase enzyme was being measured and inhibited in all three sources of MEKK2. Sunitinib also demonstrated complete inhibition of activity at high compound concentrations (Fig. 2B) which is also expected for measurement of a single enzyme activity. It should also be noted that vendor 3 MEKK2 is different from the other two in that the vendor 3 protein construct consists of only the catalytic domain of MEKK2. These data from different sources of MEKK2 indicate that MEKK2 has intrinsic ATPase activity in vitro. Vendor 1 MEKK2 was used for all experimental data and screening shown and cited in this report except as indicated in Figure 2.
Fig. 2. Intrinsic ATPase activity observed from different sources of purified MEKK2 and used to determine IC50 values for a panel of kinase inhibitors.
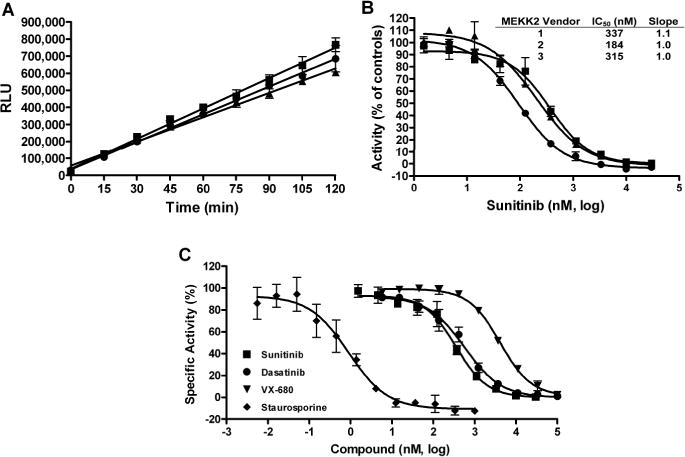
(A) Intrinsic ATPase assay reaction time courses were performed using three different sources of purified MEKK2 obtained from three different vendors: vendor 1(■), vendor 2 (●), and vendor 3 (▲). (B) Concentration response data for sunitinib was normalized to controls with and without enzyme and plotted as percent activity. These IC50 value determinations resulted in average IC50 values of 337, 184, and 315 nM using MEKK2 from vendor 1, 2, and 3, respectively, and average Hill slopes of 1.0 – 1.1 as indicated. (C) Concentration response data for a panel of kinase inhibitors was normalized to controls with and without MEKK2 and plotted as percent activity. Potency determinations for sunitinib (■), dasatinib (●), VX-680 (▼), and staurosporine (◆) resulted in average IC50 values (nM) and SDs of 337 ± 157, 753 ± 268, 3,603 ± 826, 1.0 ± 0.6, respectively. The average Hill slope values derived from the IC50 curves for these compounds ranged from 0.93 – 1.10. Data points represent the average of three determinations per concentration and error bars represent standard deviation. Data are representative of three independent experiments.
We further validated that the assay was measuring genuine MEKK2 activity and not a contaminant ATPase or kinase, by using a panel of 4 kinase inhibitors for inhibitor-based enzymatic purity studies. Inhibitor concentration response curves were generated for sunitinib, dasatinib, VX-680 and staurosporine using the intrinsic ATPase activity assay (Fig. 2C). Sunitinib, dasatinib and VX-680 generated IC50 values (nM) and SDs of 337 ± 157, 753 ± 268 and 3,603 ± 826, respectively (Table 1). These values are comparable (within 3-fold and the same rank order) to data provided by Invitrogen using their MEKK2 TR-FRET binding assay (Lanthascreen) (Table 1). Staurosporine is a very potent inhibitor of MEKK2 and behaved as a tight binding inhibitor in the ATPase assay. Therefore, the MEKK2 concentration in the reaction was reduced to 1 nM to obtain valid IC50 and Hill slope data for staurosporine. Titration of staurosporine resulted in an average IC50 and SD of 1.0 ± 0.6 nM (Table 1). This IC50 value also favorably compared to the reported Invitrogen TR-FRET binding assay IC50 of 4.2 nM for staurosporine. Using a different bead-based competition binding assay for MEKK2, Kd values for these same compounds were published as part of a large kinase profiling panel30. This published binding data gave the same rank order as we obtained and our IC50 values differed from these published Kd values by 2.4-fold to 6-fold (Table 1). Using our ATPase assay, the average Hill slopes for this panel of inhibitors ranged from 0.93 to 1.1 (see Table 1) which was indicative of a single enzyme species being measured. Furthermore, at high concentrations, all compounds were able to completely inhibit the enzyme activity (Fig. 2C). The compounds in this set were tested for assay interference by adding compound after completion of the MEKK2 enzyme reaction. None of these compounds demonstrated inhibition of the assay format (data not shown). Thus, this panel of structurally diverse and varied potency inhibitors confirmed enzymatic purity of the ATPase activity assay, i.e. a single enzyme species was generating signal. In addition, the comparable IC50 data between our MEKK2 ATPase activity assay and reported MEKK2 binding assays indicated that genuine MEKK2 activity was being measured.
Table 1.
MEKK2 Intrinsic ATPase Assay Control Inhibitor Potency Data Compared to Literature Values
| Compound | MEKK2 Intrinsic ATPase Assaya | Published MEKK2 Binding Assayb | Published MEKK2 Binding Assayc | |
|---|---|---|---|---|
|
|
||||
| IC50 ± SD (nM) | Hill Slope ± SD | IC50 (nM) | Kd (nM) | |
| Sunitinib | 337 ± 157 | 1.06 ± 0.16 | 120 | 57 |
| Dasatinib | 753 ± 268 | 1.10 ± 0.20 | 960 | 140 |
| Staurosporine | 1.0 ± 0.6d | 0.93 ± 0.13 | 4.2 | 2.4 |
| VX-680 | 3,603 ± 826 | 0.97 ± 0.13 | 2,300 | 1,100 |
IC50 value determinations were performed at least three times with average IC50 values and Hill slopes provided with standard deviations (SD).
TR-FRET Binding Assay: Invitrogen Lanthascreen™ Eu Kinase Binding Assay for MEKK2 Validation Packet (http://tools.invitrogen.com/content/sfs/manuals/MAP3K2_LanthaScreen_Binding.pdf)
Bead-based competition binding assay30.
Staurosporine IC50 determinations were performed using 1 nM (instead of 10 nM) MEKK2 enzyme in the activity assay.
Validation of a miniaturized semi-automated high throughput MEKK2 activity assay
We converted this MEKK2 activity assay into a less expensive semi-automated high throughput assay by reducing volume size from 25 to 5 μl and using pin tools to deliver compound. We assessed assay variability by setting up whole 384-well plates of maximum and minimum signal wells by including or excluding enzyme, respectively, and using DMSO instead of compound (Suppl. Fig. S1A). ‘No enzyme’ signal mimics signal at high concentration of inhibitor in this assay (see Fig. 2C). The Z’-factors calculated from these duplicate HTS validation experiments were 0.68 and 0.77. We tested the validity of the HTS version of the assay to reproducibly and accurately determine compound potency values by performing IC50 determinations with the control inhibitors sunitinib and dasatinib (Fig. 3). This HTS version of the MEKK2 ATPase assay generated IC50 values and SDs of 963 ± 178 nM and 1,656 ± 319 nM for sunitinib and dasatinib, respectively. These IC50 values are within 3-fold of the values determined in the manual assay (see Table 1) and acceptable reproducibility with %CVs of <20%. As a final validation step, the small Prestwick collection of predominately Food and Drug Administration (FDA)-approved drugs was screened using the semi-automated HTS version of the MEKK2 ATPase assay (Suppl. Fig. S1B). The Z’-factors for this screen ranged from 0.61 to 0.78, with an average of 0.71. No actives (≥50% inhibition) were obtained.
Figure 3. Validation of the high throughput MEKK2 activity assay.
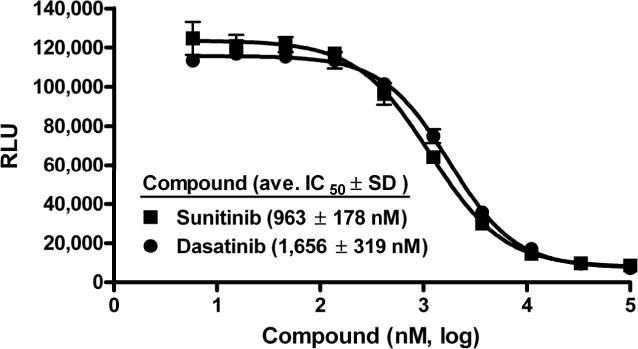
The intrinsic ATPase activity assay was performed using a low volume 384-well plate format with a total enzyme reaction volume of 5 μl. IC50 variability experiments were performed with the semi-automated high throughput screening activity assay using two control inhibitors. IC50 determinations for sunitinib (■) and dasatinib (●) resulted in average IC50 values (nM) ± SD of 963 ± 178 and 1,656 ± 319, respectively. Data are representative of three independent experiments.
Screening for MEKK2 inhibitors using the intrinsic ATPase activity assay
A kinase-focused library consisting of 4,727 compounds was screened to identify novel MEKK2 inhibitors (Suppl. Fig. S1C). The Z’-factors for this screen ranged from 0.53 to 0.86, with an average of 0.72. Only 2 actives (≥50% inhibition) were obtained. In order to identify other control inhibitors of MEKK2 kinase activity, we screened a small collection of 13 kinase inhibitors, 11 of which are FDA-approved drugs and 2 are in clinical trials. Sunitinib was employed as an internal control. These compounds were screened at 1 μM concentration in the MEKK2 intrinsic ATPase assay and 3 hits (≥50% inhibition) were obtained (Fig. 4A). The hits were pazopanib, crizotinib and bosutinib.
Figure 4. MEKK2 inhibitory activity of a panel of known kinase inhibitors and IC50 determination for screening hits.
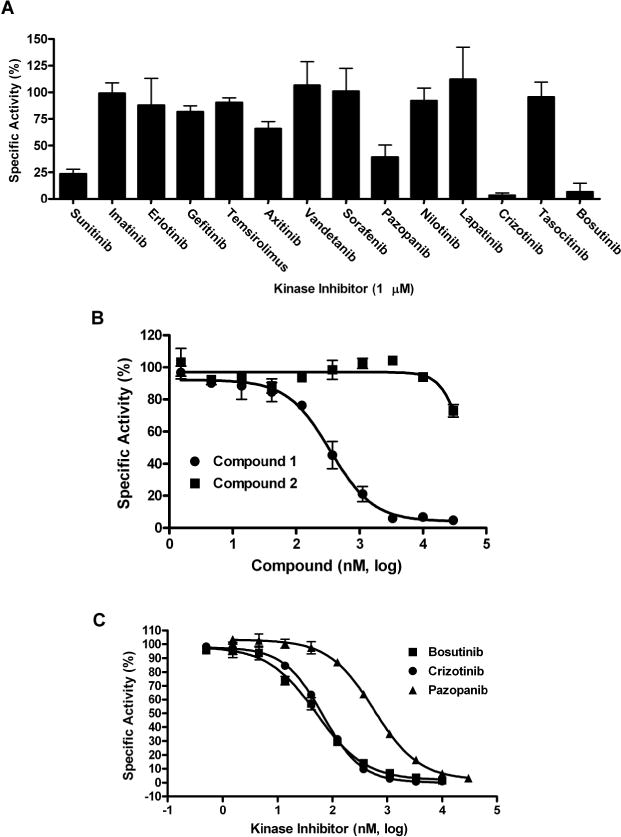
(A) A panel of marketed and clinical trial kinase inhibitors was screened at 1 μM compound concentration using the MEKK2 activity assay. The compounds were screened in duplicate wells in three independent experiments and the normalized data from all experiments was aggregated by averaging percent inhibition values obtained from all experiments and plotting them as shown. Error bars represent standard deviation of the three experiments. (B) Concentration response data for the hit compound from the large kinase-focused library screen and a structural analog was normalized to controls with and without MEKK2 and plotted as percent specific activity. One hit (compound 1, ●) confirmed activity with an average IC50 of 299 ± 71 nM and a close structural analog (compound 2, ■) resulted in an IC50 of >30 μM. IC50 data are representative of at least three independent experiments. (C) The compounds resulting in ≥50% inhibition from the known kinase inhibitor panel were tested in concentration response experiments, except for sunitinib which was used as a control. IC50 determinations for bosutinib (■), crizotinib (●), and pazopanib (▲) resulted in IC50 values (nM) and SD of 59 ± 34, 75 ± 35, 698 ± 163, respectively. IC50 data are representative of three independent experiments.
IC50 determinations of hit compounds using three different MEKK2 assay formats: intrinsic ATPase, binding, and transphosphorylation assays
The active compounds from screening were initially confirmed by IC50 determination using the intrinsic ATPase activity assay. In regard to the large kinase-focused library screen, only one of the 2 actives repeated activity in IC50 confirmation using the intrinsic ATPase assay – this hit compound was referred to as compound 1. Compound 1 generated in IC50 of 299 ± 71 nM in this assay (Fig. 4B and Table 2). A close analog of this compound (compound 2) was also tested and found to be inactive (>30 μM IC50) and thus served as a specificity control (Fig. 4B and Table 2). From the screening of 13 known kinase inhibitors, pazopanib, crizotinib and bosutinib confirmed activity with IC50 values (± SD) in the ATPase assay of 698 ± 163, 75 ± 35 and 59 ± 34 nM, respectively (Fig. 4C and Table 2).
Table 2.
Comparison of IC50 Data Generated Using Three Different MEKK2 Assay Formats
| Intrinsic ATPase Assaya
|
Binding Assaya
|
Transphosphorylation Assaya,b
|
||||
|---|---|---|---|---|---|---|
| IC50 ± SD (nM) | Hill Slope ± SD | IC50 ± SD (nM) | Hill Slope ± SD | IC50 ± SD (nM) | Hill Slope ± SD | |
| Sunitinib | 337 ± 157 | 1.06 ± 0.16 | 143 ± 6 | 0.89 ± 0.04 | 158 ± 38 | 0.96 ± 0.15 |
| Compound 1 | 299 ± 71 | 1.32 ± 0.13 | 355 ± 134 | 1.10 ± 0.10 | 261 ± 14 | 1.36 ± 0.20 |
| Compound 2 | >30,000 | NAc | >30,000 | NA | NDd | ND |
| Pazopanib | 698 ± 163 | 1.06 ± 0.07 | 1,145 ± 211 | 1.05 ± 0.03 | ND | ND |
| Crizotinib | 75 ± 35 | 1.30 ± 0.16 | 24 ± 7 | 1.09 ± 0.01 | 37 ± 22 | 1.96 ± 0.42 |
| Bosutinib | 59 ± 34 | 1.07 ± 0.09 | 64 ± 17 | 0.93 ± 0.05 | 29 ± 10 | 1.03 ± 0.21 |
IC50 value determinations were performed at least three times with average IC50 values and Hill slopes provided with standard deviations (SD).
IC50 experiments were performed using duplicate determinations per compound concentration
Not applicable
Not determined
We employed a TR-FRET MEKK2 binding assay as an orthogonal secondary assay to further validate the intrinsic ATPase activity assay and to confirm ATPase assay screening hits. This binding assay measures the binding of compound to the ATP pocket of MEKK2. The MEKK2 binding assay was optimized as indicated by the reagent manufacturer’s instructions (Invitrogen Lanthascreen Eu Kinase Binding Assay) using the same source of MEKK2 enzyme and assay buffer. Concentration response data for sunitinib was generated using the MEKK2 binding assay (Table 2). The average IC50 value and SD in the binding assay was 143 ± 6 nM and an average Hill slope of 0.89 ± 0.04. Therefore, the IC50 values for sunitinib in the ATPase and binding assay differed by only 2.4-fold and our binding data was very close to the manufacturer’s published IC50 value (120 nM, Table 1). Compound 1 was tested in the MEKK2 binding assay resulting in an average IC50 value of 355 ± 134 nM, in close agreement with the 299 nM IC50 obtained in the ATPase assay (Table 2). The inactive control compound 2 was also inactive (>30 μM IC50) in the binding assay. Thus, compound 1 represents a novel inhibitor of MEKK2. Pazopanib, crizotinib and bosutinib were also tested in the MEKK2 binding assay resulting in IC50 values (±SD) of 1,145 ± 211, 24 ± 7 and 64 ± 17 nM, respectively (Table 2). The Hill slopes for these compounds in both binding and ATPase assays, were close to expected for single site binding, ranging from 0.93 – 1.3 (see Table 2). None of the compounds in Figure 4, Table 1 and Table 2 demonstrated any significant assay interference in either the ATPase or binding assay format (data not shown). Thus, the MEKK2 binding assay data confirmed the MEKK2 inhibitory activity of these compounds discovered with the MEKK2 intrinsic ATPase assay. Furthermore, this panel of five diverse inhibitors (Table 2) demonstrated ≤3-fold difference in IC50 values when comparing the ATPase and binding assay data.
In order to further validate the screening hits and the ATPase assay, we developed a transphosphorylation assay in which kinase-inactive mutant MKK6 was used as a substrate in MEKK2 enzyme reactions. Stopped reaction mixtures were transferred to membranes using a slot blot protocol and phosphorylation of MKK6 was measured by a phospho-MKK6 specific antibody. The average apparent Km for ATP in this assay format was determined to be 3.3 μM (data not shown) which is 10-fold lower than in the ATPase assay. Therefore, we used 3 μM ATP in the transphosphorylation assay. Average IC50 values generated using this transphosphorylation assay for sunitinib, compound 1, crizotinib and bosutinib were 158, 261, 37, 29 nM (Table 2). These values differ by ≤2.2-fold compared to the ATPase and binding assay potency values. Using a bead-based MEKK2 binding assay as part of a large kinase profiling panel, it has been reported that pazopanib, crizotinib and bosutinib have Kd values of 290, 72 and 30 nM, respectively30. Thus, our ATPase data, as well as that from two other formats, are in close agreement with this published binding data. Furthermore, our data confirm that these compounds actually inhibit MEKK2 kinase activity. This multiple format comparison data further enhanced the validation of the intrinsic ATPase assay as a useful assay for the discovery of MEKK2 inhibitors.
DISCUSSION
We sought to develop a high throughput activity assay for MEKK2. Unlike many kinases, MEKK2 and many other members of the MAPK family do not effectively use small synthetic peptides as substrates. Thus, large protein substrates are typically used to measure MEKK2 activity. However, for HTS this would require large amounts of protein substrate. In developing a biochemical activity assay for MEKK2, we discovered that purified MEKK2 had intrinsic ATPase activity. This ATPase activity was discovered using a ‘universal’ chemiluminescent ADP detection kit. Although convenient, this type of detection assay requires careful validation to ensure that the observed activity derives from the target kinase (enzyme identity) and no other kinases or ATPases (enzymatic purity). Trace amounts of contaminating ATPases or other kinases in the MEKK2 preparation could also cause the observed ATPase activity.
Multiple independent methods and data were used to verify that the observed ATPase activity derived from MEKK2. First, the identity of the commercially obtained enzyme solution was confirmed to be human MEKK2 by MS and no other proteins were identified in the solution. Second, we compared three different sources of recombinant baculovirus-expressed and purified MEKK2. All three demonstrated ATPase activity in the absence of added substrate and even the specific activity was similar amongst the MEKK2 sources. Unlike the other two vendors’ constructs which were full length proteins, one of the vendor’s MEKK2 consisted of only the kinase domain of MEKK2, yet still maintained ATPase activity. Furthermore, IC50 value determination for the control kinase inhibitor sunitinib using all three sources of MEKK2 resulted in IC50 values within two-fold of each other, suggestive of measuring the same activity. Third, inhibitor-based studies were performed using a panel of 4 inhibitors that had been tested in the MEKK2 binding assay reported by Invitrogen. IC50 values measured using the ATPase assay were comparable (within 2- to 4-fold) to Invitrogen’s MEKK2 binding assay data. In addition, our data was also consistent with published potencies using a bead-based competition binding assay used for kinome profiling30. Dasatinib was reported to have a binding Kd value that is 5.4-fold lower than the IC50 we observed in the ATPase assay (Table 1). The reason for this discrepancy may be due, in part, to the expected lower values for Kd versus IC50 values. In addition, binding assays may give different potencies compared to activity assays possibly due to different affinities for different activation states of the MEKK2 enzyme preparations used in the respective studies. This may be a compound-specific effect that is observed with some inhibitors, but not all. Fourth, format comparison studies were performed with 6 compounds for which we determined IC50 values using the luminescent ATPase and TR-FRET binding assays. This panel of five diverse inhibitors demonstrated ≤ 3-fold difference in IC50 values when comparing the ATPase and binding assay data. Thus, there was a striking correlation between inhibition of the ATPase assay and the MEKK2 binding assay, even though these assays measured two different parameters – enzyme activity and ATP pocket binding. In addition, a transphosphorylation assay was developed for MEKK2 using MKK6 as a substrate. The IC50 values for 4 of these inhibitors were tested in this activity format as well and all the values were within 2-fold of the ATPase assay values. Taken together, these data indicate that the ATPase activity measured in our assay is derived from MEKK2.
In this type of non-selective kinase assay format, it is always possible that multiple enzymes contribute to assay signal – such as the intended target and trace impurities with high specific activity. The inhibition of two or more enzymes by an inhibitor generally results in a blended IC50 curve, due to different binding affinities, resulting in a shallow slope (< 1). The Hill slopes across all 8 diverse inhibitors tested in this study were close to the ideal (1.0) for a single enzyme species, ranging from 0.93 to 1.3. A panel of inhibitors such as this provides strong evidence for enzymatic purity since even similar kinases would be expected to have slightly different affinities for some of the inhibitors. It is also possible that potential contaminating ATPase activity is not inhibited by the inhibitors, yet contributes to assay signal. However, this is not the case since all compounds demonstrated complete inhibition of activity at high compound concentrations, consistent with only one homogenous enzyme activity detected.
A screen of a kinase-focused set of compounds with the MEKK2 ATPase assay resulted in one confirmed hit (IC50 = 299 nM), representing a hit rate of 0.02%. Further screening of a set of known FDA-approved and clinical trial kinase inhibitors generated a hit rate of 23% (3 hits out of 13 compounds). The dramatically different hit rates in these screens is likely due to the types of compounds in the respective libraries – one being un-optimized theoretical inhibitors and the other consisting of potent “optimized” kinase inhibitor scaffolds. Bosutinib and crizotinib were identified as potent (<100 nM IC50) in vitro inhibitors of MEKK2 kinase activity, confirming and extending a published report on the binding activity of these compounds for MEKK2. In addition, a novel inhibitor (compound 1) was identified through screening using the ATPase assay and its activity confirmed in the MEKK2 binding and transphosphorylation formats.
The described MEKK2 ATPase assay has several advantages as a HTS assay. This assay does not require large amounts of expensive and difficult to produce protein substrate and it can be miniaturized. Unlike the binding assay, this assay has the potential to identify non-ATP competitive inhibitors. It should be noted that the Km for ATP differed by 10-fold between the ATPase and the transphosphorylation assay (30 vs. 3 μM), highlighting the importance of experimentally determining Km for ATP when developing an ATPase kinase assay. This magnitude of shift in ATP Km for kinase intrinsic ATPase activity has been previously observed25.
We have developed and optimized a high throughput activity assay for MEKK2 based on the discovery that MEKK2 has intrinsic ATPase activity. This activity assay for MEKK2 was validated for enzyme identity and enzymatic purity by multiple methods to demonstrate that genuine MEKK2 activity was being measured and no other contaminating activities contribute to the signal. Furthermore, a miniaturized version of the assay was validated for HTS and compound libraries screened. MEKK2 inhibitors were identified that may be useful as research tools or lead to potent and selective MEKK2 inhibitors.
Supplementary Material
Acknowledgments
This work was supported by National Institutes of Health (NIH) grant U54CA156735 (JES and GLJ) and in part by a grant from the Golden LEAF Foundation (JES) and funds from the State of North Carolina (JES). We acknowledge the NCCU/BRITE high throughput screening core facility, The Center for Integrative Chemical Biology and Drug Discovery at UNC-Chapel Hill and the UNC Proteomics core facility for support of this research.
List of abbreviations
- DMSO
dimethyl sulfoxide
- RLU
relative luminescence units
- RFU
relative fluorescence units
- MAPK
mitogen-activated protein kinase
- MS
mass spectrometry
- ATP
adenosine triphosphate
- ADP
adenosine diphosphate
- GST
glutathione S-transferase
- SD
standard deviation
- TR-FRET
time-resolved fluorescence resonance energy transfer
- HTS
high throughput screening
References
- 1.Whitmarsh AJ. Regulation of gene transcription by mitogen-activated protein kinase signaling pathways. Biochim Biophys Acta. 2007;1773(8):1285–98. doi: 10.1016/j.bbamcr.2006.11.011. [DOI] [PubMed] [Google Scholar]
- 2.Kim EK, Choi EJ. Pathological roles of MAPK signaling pathways in human diseases. Biochim Biophys Acta. 2010;1802(4):396–405. doi: 10.1016/j.bbadis.2009.12.009. [DOI] [PubMed] [Google Scholar]
- 3.Drew BA, Burow ME, Beckman BS. MEK5/ERK5 pathway: the first fifteen years. Biochim Biophys Acta. 2012;1825(1):37–48. doi: 10.1016/j.bbcan.2011.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yan L, Carr J, Ashby PR, Murry-Tait V, Thompson C, Arthur JS. Knockout of ERK5 causes multiple defects in placental and embryonic development. BMC Dev Biol. 2003;3:11. doi: 10.1186/1471-213X-3-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang X, Tournier C. Regulation of cellular functions by the ERK5 signalling pathway. Cell Signal. 2006;18(6):753–60. doi: 10.1016/j.cellsig.2005.11.003. [DOI] [PubMed] [Google Scholar]
- 6.Pi X, Yan C, Berk BC. Big mitogen-activated protein kinase (BMK1)/ERK5 protects endothelial cells from apoptosis. Circ Res. 2004;94(3):362–9. doi: 10.1161/01.RES.0000112406.27800.6F. [DOI] [PubMed] [Google Scholar]
- 7.Wang X, Merritt AJ, Seyfried J, Guo C, Papadakis ES, Finegan KG, Kayahara M, Dixon J, Boot-Handford RP, Cartwright EJ, Mayer U, Tournier C. Targeted deletion of mek5 causes early embryonic death and defects in the extracellular signal-regulated kinase 5/myocyte enhancer factor 2 cell survival pathway. Mol Cell Biol. 2005;25(1):336–45. doi: 10.1128/MCB.25.1.336-345.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sun W, Kesavan K, Schaefer BC, Garrington TP, Ware M, Johnson NL, Gelfand EW, Johnson GL. MEKK2 associates with the adapter protein Lad/RIBP and regulates the MEK5-BMK1/ERK5 pathway. J Biol Chem. 2001;276(7):5093–100. doi: 10.1074/jbc.M003719200. [DOI] [PubMed] [Google Scholar]
- 9.Chao TH, Hayashi M, Tapping RI, Kato Y, Lee JD. MEKK3 directly regulates MEK5 activity as part of the big mitogen-activated protein kinase 1 (BMK1) signaling pathway. J Biol Chem. 1999;274(51):36035–8. doi: 10.1074/jbc.274.51.36035. [DOI] [PubMed] [Google Scholar]
- 10.Garrington TP, Ishizuka T, Papst PJ, Chayama K, Webb S, Yujiri T, Sun W, Sather S, Russell DM, Gibson SB, Keller G, Gelfand EW, Johnson GL. MEKK2 gene disruption causes loss of cytokine production in response to IgE and c-Kit ligand stimulation of ES cell-derived mast cells. EMBO J. 2000;19(20):5387–95. doi: 10.1093/emboj/19.20.5387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Guo Z, Clydesdale G, Cheng J, Kim K, Gan L, McConkey DJ, Ullrich SE, Zhuang Y, Su B. Disruption of Mekk2 in mice reveals an unexpected role for MEKK2 in modulating T-cell receptor signal transduction. Mol Cell Biol. 2002;22(16):5761–8. doi: 10.1128/MCB.22.16.5761-5768.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yang J, Boerm M, McCarty M, Bucana C, Fidler IJ, Zhuang Y, Su B. Mekk3 is essential for early embryonic cardiovascular development. Nat Genet. 2000;24(3):309–13. doi: 10.1038/73550. [DOI] [PubMed] [Google Scholar]
- 13.Kesavan K, Lobel-Rice K, Sun W, Lapadat R, Webb S, Johnson GL, Garrington TP. MEKK2 regulates the coordinate activation of ERK5 and JNK in response to FGF-2 in fibroblasts. J Cell Physiol. 2004;199(1):140–8. doi: 10.1002/jcp.10457. [DOI] [PubMed] [Google Scholar]
- 14.Cheng J, Yu L, Zhang D, Huang Q, Spencer D, Su B. Dimerization through the catalytic domain is essential for MEKK2 activation. J Biol Chem. 2005;280(14):13477–82. doi: 10.1074/jbc.M414258200. [DOI] [PubMed] [Google Scholar]
- 15.Montero JC, Ocana A, Abad M, Ortiz-Ruiz MJ, Pandiella A, Esparis-Ogando A. Expression of Erk5 in early stage breast cancer and association with disease free survival identifies this kinase as a potential therapeutic target. PLoS One. 2009;4(5):e5565. doi: 10.1371/journal.pone.0005565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cazares LH, Troyer D, Mendrinos S, Lance RA, Nyalwidhe JO, Beydoun HA, Clements MA, Drake RR, Semmes OJ. Imaging mass spectrometry of a specific fragment of mitogen-activated protein kinase/extracellular signal-regulated kinase kinase kinase 2 discriminates cancer from uninvolved prostate tissue. Clin Cancer Res. 2009;15(17):5541–51. doi: 10.1158/1078-0432.CCR-08-2892. [DOI] [PubMed] [Google Scholar]
- 17.McCracken SR, Ramsay A, Heer R, Mathers ME, Jenkins BL, Edwards J, Robson CN, Marquez R, Cohen P, Leung HY. Aberrant expression of extracellular signal-regulated kinase 5 in human prostate cancer. Oncogene. 2008;27(21):2978–88. doi: 10.1038/sj.onc.1210963. [DOI] [PubMed] [Google Scholar]
- 18.Carvajal-Vergara X, Tabera S, Montero JC, Esparis-Ogando A, Lopez-Perez R, Mateo G, Gutierrez N, Parmo-Cabanas M, Teixido J, San Miguel JF, Pandiella A. Multifunctional role of Erk5 in multiple myeloma. Blood. 2005;105(11):4492–9. doi: 10.1182/blood-2004-08-2985. [DOI] [PubMed] [Google Scholar]
- 19.Clape C, Fritz V, Henriquet C, Apparailly F, Fernandez PL, Iborra F, Avances C, Villalba M, Culine S, Fajas L. miR-143 interferes with ERK5 signaling, and abrogates prostate cancer progression in mice. PLoS One. 2009;4(10):e7542. doi: 10.1371/journal.pone.0007542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sticht C, Freier K, Knopfle K, Flechtenmacher C, Pungs S, Hofele C, Hahn M, Joos S, Lichter P. Activation of MAP kinase signaling through ERK5 but not ERK1 expression is associated with lymph node metastases in oral squamous cell carcinoma (OSCC) Neoplasia. 2008;10(5):462–70. doi: 10.1593/neo.08164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yang Q, Deng X, Lu B, Cameron M, Fearns C, Patricelli MP, Yates JR, 3rd, Gray NS, Lee JD. Pharmacological inhibition of BMK1 suppresses tumor growth through promyelocytic leukemia protein. Cancer Cell. 2010;18(3):258–67. doi: 10.1016/j.ccr.2010.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mehta PB, Jenkins BL, McCarthy L, Thilak L, Robson CN, Neal DE, Leung HY. MEK5 overexpression is associated with metastatic prostate cancer, and stimulates proliferation, MMP-9 expression and invasion. Oncogene. 2003;22(9):1381–9. doi: 10.1038/sj.onc.1206154. [DOI] [PubMed] [Google Scholar]
- 23.Cronan MR, Nakamura K, Johnson NL, Granger DA, Cuevas BD, Wang JG, Mackman N, Scott JE, Dohlman HG, Johnson GL. Defining MAP3 kinases required for MDA-MB-231 cell tumor growth and metastasis. Oncogene. 2011 doi: 10.1038/onc.2011.544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fox T, Fitzgibbon MJ, Fleming MA, Hsiao HM, Brummel CL, Su MS. Kinetic mechanism and ATP-binding site reactivity of p38gamma MAP kinase. FEBS Lett. 1999;461(3):323–8. doi: 10.1016/s0014-5793(99)01488-x. [DOI] [PubMed] [Google Scholar]
- 25.Rominger CM, Schaber MD, Yang J, Gontarek RR, Weaver KL, Broderick T, Carter L, Copeland RA, May EW. An intrinsic ATPase activity of phospho-MEK-1 uncoupled from downstream ERK phosphorylation. Arch Biochem Biophys. 2007;464(1):130–7. doi: 10.1016/j.abb.2007.04.004. [DOI] [PubMed] [Google Scholar]
- 26.Kashem MA, Nelson RM, Yingling JD, Pullen SS, Prokopowicz AS, 3rd, Jones JW, Wolak JP, Rogers GR, Morelock MM, Snow RJ, Homon CA, Jakes S. Three mechanistically distinct kinase assays compared: Measurement of intrinsic ATPase activity identified the most comprehensive set of ITK inhibitors. J Biomol Screen. 2007;12(1):70–83. doi: 10.1177/1087057106296047. [DOI] [PubMed] [Google Scholar]
- 27.Klink TA, Kleman-Leyer KM, Kopp A, Westermeyer TA, Lowery RG. Evaluating PI3 kinase isoforms using Transcreener ADP assays. J Biomol Screen. 2008;13(6):476–85. doi: 10.1177/1087057108319864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang JH, Chung TD, Oldenburg KR. A Simple Statistical Parameter for Use in Evaluation and Validation of High Throughput Screening Assays. J Biomol Screen. 1999;4(2):67–73. doi: 10.1177/108705719900400206. [DOI] [PubMed] [Google Scholar]
- 29.Ahmad S, Carter JJ, Scott JE. A homogeneous cell-based assay for measurement of endogenous paraoxonase 1 activity. Anal Biochem. 2010;400(1):1–9. doi: 10.1016/j.ab.2010.01.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Davis MI, Hunt JP, Herrgard S, Ciceri P, Wodicka LM, Pallares G, Hocker M, Treiber DK, Zarrinkar PP. Comprehensive analysis of kinase inhibitor selectivity. Nat Biotechnol. 2011;29(11):1046–51. doi: 10.1038/nbt.1990. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.