

Abstract
Currents through voltage-gated Ca2+ channels (ICa) may be regulated by cytoplasmic Ca2+ levels ([Ca2+]c), producing Ca2+-dependent inactivation (CDI) or facilitation (CDF). Since ICa regulates sensory neuron excitability, altered CDI or CDF could contribute to pain generation after peripheral nerve injury. We explored this by manipulating [Ca2+]c while recording ICa in rat sensory neurons. In uninjured neurons, elevating [Ca2+]c with a conditioning prepulse (−15 mV, 2 s) inactivated ICa measured during subsequent test pulses (−15 mV, 5 ms). This inactivation was Ca2+-dependent (CDI), since it was decreased with elimination of Ca2+ influx by depolarization to above the ICa reversal potential, with high intracellular Ca2+ buffering (EGTA 10 mm or BAPTA 20 mm), and with substitution of Ba2+ for extracellular Ca2+, revealing a residual voltage-dependent inactivation. At longer latencies after conditioning (>6 s), ICa recovered beyond baseline. This facilitation also proved to be Ca2+-dependent (CDF) using the protocols limiting cytoplasmic Ca2+ elevation. Ca2+/calmodulin-dependent protein kinase II (CaMKII) blockers applied by bath (KN-93, myristoyl-AIP) or expressed selectively in the sensory neurons (AIP) reduced CDF, unlike their inactive analogues. Protein kinase C inhibition (chelerythrine) had no effect. Selective blockade of N-type Ca2+ channels eliminated CDF, whereas L-type channel blockade had no effect. Following nerve injury, CDI was unaffected, but CDF was eliminated in axotomized neurons. Excitability of sensory neurons in intact ganglia from control animals was diminished after a similar conditioning pulse, but this regulation was eliminated by injury. These findings indicate that ICa in sensory neurons is subject to both CDI and CDF, and that hyperexcitability following injury-induced loss of CDF may result from diminished CaMKII activity.
Introduction
Key functions in excitable cells are regulated by cytoplasmic Ca2+ levels ([Ca2+]c), such as contraction in myocytes and excitation and synaptic transmission in neurons (Berridge et al., 2000). This critical signal is shaped by powerful feedback control of Ca2+ entry through voltage-gated Ca2+channels (VGCCs), where two phenomena with opposing effects on [Ca2+]c have been identified. Ca2+-dependent inactivation (CDI) is evident for various VGCC subtypes in myocytes (Hadley and Lederer, 1991) and neurons (Cox and Dunlap, 1994; Forsythe et al., 1998; Meuth et al., 2002), while Ca2+-dependent facilitation (CDF) has been demonstrated for L-type VGCC in myocytes (Anderson et al., 1994; Yuan and Bers, 1994) and for P/Q channels in neurons of the CNS (Borst and Sakmann, 1998; Cuttle et al., 1998; Chaudhuri et al., 2005).
The molecular mechanism underlying CDI requires binding of Ca2+ to pre-associated calmodulin (CaM), a process conserved in all VGCCs (Zühlke et al., 1999; Pitt et al., 2001; Liang et al., 2003). A similar direct action of CaM may produce CDF in L-type and P/Q-type channels (Lee et al., 1999; DeMaria et al., 2001; Liang et al., 2003). However, other studies implicate the participation of the serine/threonine protein kinase, Ca2+/calmodulin-dependent protein kinase II (CaMKII) (Yuan and Bers, 1994; Dzhura et al., 2000), leading to binding or phosphorylation of channel targets including the pore-forming α-subunits of VGCCs (Hudmon et al., 2005; Jiang et al., 2008; Blaich et al., 2010) and the auxiliary β-subunits (Grueter et al., 2006). CaMKII contribution to the production of CDF has been implicated for both L-type and P/Q-type channels (Hudmon et al., 2005; Jiang et al., 2008), but not for N-type channels, nor for any VGCC in the peripheral nervous system.
Neuronal activity is regulated by Ca2+ entering through VGCCs, which in turn activates Ca2+-sensitive K+ currents and thereby provides a critical brake on neuronal depolarization and action potential (AP) generation (Swensen and Bean, 2003; Hogan et al., 2008; Lirk et al., 2008; Gemes et al., 2009). Painful nerve injury results in diminished ICa in sensory neurons (Baccei and Kocsis, 2000; Abdulla and Smith, 2001; McCallum et al., 2006, 2011) and reduced activity-induced cytoplasmic Ca2+ accumulation (Fuchs et al., 2007a,b; Gemes et al., 2010), which lead to enhanced sensory neuron excitability (Sapunar et al., 2005). Although the mechanism of this loss in VGCC function could involve multiple levels of regulation, we pursued the hypothesis that injury disrupts Ca2+-dependent modulation (CDI and/or CDF) of VGCCs—a model consistent with our observation of diminished levels of cytoplasmic Ca2+ and activated CaMKII in injured sensory neurons (Kojundzic et al., 2010). Accordingly, this study was designed to examine CDI and CDF in the natural physiological context of sensory neurons, to identify the influences of injury and CaMKII signaling, and to identify whether this regulation plays a role in controlling neuronal excitability.
Materials and Methods
Ethical approval.
Prior approval and ongoing oversight for the studies described below were provided by the Medical College of Wisconsin Animal Care and Use Committee. The animals were cared for in a facility accredited by the Association for Assessment and Accreditation of Laboratory Animal Care International, registered with the United States Department of Agriculture, and compliant with United States Public Health Service regulations and requirements and provisions of the Animal Welfare Act.
Animal preparation.
One hundred thirty-four unoperated male Sprague Dawley rats (Taconic) weighing 125–150 g were used for these experiments. Other rats (n = 37) were subjected to spinal nerve ligation (SNL), based on the method of Kim and Chung (1992). Briefly, during anesthesia by inhalation of isoflurane (1.5–2.5% in oxygen) without neuromuscular blockade, the right lumbar paravertebral region was exposed through a midline incision, and the sixth lumbar (L6) transverse process was removed to expose the L5 and L6 spinal nerves, which were ligated with 4-0 silk suture and sectioned ∼5 mm distal to their respective dorsal root ganglia (DRGs). The wounds were closed in layers and the skin stapled. Control rats received skin incision and closure only.
Sensory testing.
Rats underwent sensory testing for a form of hyperalgesic behavior that we have previously documented to be associated with an aversive percept (Hogan et al., 2004; Wu et al., 2010). Briefly, on three different days between 10 and 21 d after surgery, right plantar skin was mechanically stimulated with a 22G spinal needle with adequate pressure to indent but not penetrate the skin. Whereas control animals respond with only a brief reflexive withdrawal, rats following SNL may display a complex hyperalgesia response that incorporates sustained licking, chewing, grooming, and sustained elevation of the paw. The frequency of hyperalgesia responses was tabulated for each rat.
Dissociation of DRG neurons.
Between 21 and 30 d postsurgery, L5 DRGs from control or SNL rats were removed following decapitation under deep isoflurane anesthesia. In some cases, L4 DRGs from control animals were also used since there are no evident differences between them in the uninjured state. Ganglia were placed in a 35 mm dish containing Ca2+/Mg2+-free, cold HBBS (Life Technologies) and cut into four to six pieces that were incubated in 0.5 mg/ml liberase TM (Roche) in DMEM/F12 with glutaMAX (Life Technologies) for 30 min at 37°C followed with 1 mg/ml trypsin (Sigma-Aldrich) and 0.36 mg/ml DNase (Sigma-Aldrich) for another 10 min. After addition of trypsin inhibitor (type II; Sigma-Aldrich), tissues were centrifuged and lightly triturated in neural basal media (1×) (Life Technologies) containing 2% (v:v) B27 supplement (50×) (Life Technologies), 0.5 mm glutamine (Sigma-Aldrich), 0.05 mg/ml gentamicin (Life Technologies), and 10 ng/ml nerve growth factor 7S (Alomone Labs Ltd.). Cells were then plated onto poly-l-lysine (70–150 kDa; Sigma-Aldrich)-coated coverslips and cultured at 37°C in 5% CO2. All cells were studied 3–8 h after dissociation. Small- to medium-sized (30 ± 3 μm) neurons were used for this study.
Whole-cell patch-clamp recording.
Electrodes with a resistance of 2–4MΩ were pulled from borosilicate glass (Garner Glass) using a micropipette puller (P-97; Sutter Instrument) and fire polished. Recording was performed in the whole-cell configuration with an Axopatch 200B amplifier (Molecular Devices). After whole-cell configuration was established, electrical compensation for the cell membrane capacitance and series resistance were initiated. Neurons with >10 MΩ access resistance after breakthrough were discarded. Experiments were performed 5–15 min after breakthrough, and at room temperature (∼25°C). Signals were filtered at 2 kHz through a 4-pole Bessel filter, and digitized at 10 kHz with a Digidata 1320 A/D interface (Molecular Devices).
Seals were achieved in modified Tyrode's containing (in mm): 140 NaCl, 4 KCl, 2 CaCl2, 2 MgCl2, 10 d-glucose, and 10 HEPES, pH of 7.4, with an osmolarity of 300 mOsm. Voltage-induced currents flowing through Ca2+ channels were recorded using an extracellular solution containing (in mm): 2 CaCl2 or 2 BaCl2, 4-aminophyridine 1, 10 HEPES, 140 tetraethylammonium chloride (TEACl), pH of 7.4, with an osmolarity of 300 mOsm. The internal pipette solution contained (in mm): 120 CsCl, 10 TEACl, 4 Mg-ATP, 0.3 Na-GTP, 0.5 EGTA, 0.2 CaCl2, 1 MgCl2, and 10 HEPES, pH of 7.2, with an osmolarity of 300 mOsm. The free concentration for Ca2+ was calculated to be 90 nm using Maxchelator (http://maxchelator.stanford.edu). Measured voltages are corrected for the junction potential (−5 mV). Data from whole-cell ICa recordings were evaluated with Axograph X 1.3.5 (AxoGraph Scientific), with which peak inward currents and charge transfer were measured. To correct for cell size, inward currents are expressed as a function of cell capacitance (pA/pF).
Intracellular recording.
Intact ganglia with dorsal roots attached were harvested by laminectomy during isoflurane anesthesia and placed in artificial CSF containing (in mm): 128 NaCl, 3.5 KCl, 1.2 MgCl2, 2.3 CaCl2, 1.2 NaH2PO4, 24.0 NaHCO3, and 11.0 glucose bubbled by 5% CO2 and 95% O2 to maintain a pH of 7.35. Intracellular recordings were performed with microelectrodes fashioned from borosilicate glass (1mm OD, 0.5 mm ID, with Omega fiber) (FHC Bowdoinham) using a P-97 programmable micropipette puller (Sutter Instrument). Pipettes were filled with 2M K+ acetate, which was buffered with 10 mm HEPES, with a resulting resistance of 60–90 MΩ. Neurons were impaled using differential interference contrast imaging and infrared illumination. Membrane potential was recorded using an active bridge amplifier (Axoclamp 2B; Molecular Devices). Voltage recordings were filtered at 10 kHz and then digitized at 40 kHz (Digidata 1322A; Molecular Devices) and analyzed off-line (AxoGraph X 1.3.5; AxoGraph Scientific). Recordings were not started until resting membrane potential had stabilized (typically within 2 min), at which time neurons were excluded if the resting membrane potential was less polarized than −45 mV. Somatic APs were generated by direct membrane depolarization with current injection through the recording electrode, for which voltage error was minimized using a discontinuous current-clamp mode with a switching rate of 2 kHz, while monitoring for complete settling of electrode potential between sampling. Excitability was assayed by counting the number of APs generated by injection of depolarizing current through the recording electrode (2 nA, 50 ms). This stimulus produced repetitive firing in approximately half of the neurons. A conditioning prepulse was produced during single-electrode voltage-clamp (0 mV, 2 s) from a holding potential that was identical to the cell's resting membrane potential, after which the recording mode was automatically switched to discontinuous current-clamp mode for determining firing pattern during subsequent test pulses (2 nA, 50 ms). Data were included only from neurons that showed depolarization to 30 mV or less.
Cytoplasmic Ca2+ microfluorometry.
Fura-2 (pentapotassium salt from Invitrogen) was dissolved in H2O to a concentration of 10 mm, and then diluted to 200 μm in internal pipette solution for whole-cell patch-clamp recordings. Experiments were started 5–10 min after the whole-cell configuration was established, during which loading was achieved by passive diffusion. The fluorophore was excited alternately with 340and 380 nm wavelength illumination (150W Xenon, Lambda DG-4; Sutter Instrument), while images were acquired at 510 nm using a cooled 12-bit digital camera (Coolsnap fx; Photometrics). Images were obtained using an inverted microscope (Diaphot 200; Nikon Instruments) and a 40× fluor oil-immersion objective. The ratio of fluorescence emission during 340 nm excitation divided by the intensity during 380 nm excitation (R340/380) was calculated as an indicator of [Ca2+]c. This ratio was calculated on a pixel-by-pixel basis following background subtraction (MetaFluor; Molecular Devices).
Agents.
Stock solutions were prepared by dissolving agents in either distilled water or dimethylsulfoxide (DMSO), at concentrations at least 1000 times the final concentration, and kept frozen in aliquots. The stock solutions were diluted in extracellular solution just before use. Solutions were delivered by gravity or through a pressure regulated microperfusion system (ALA-BP8 system; ALA Scientific) for at least 3 min before recording. Peptides were delivered in an external Ca2+ solution to which cytochrome c (0.1 mg/ml; Sigma-Aldrich) had been added to prevent nonspecific binding in the delivery system. This concentration has been shown to have no effect on ICa in cerebellar neurons (Randall and Tsien, 1995) and in sensory neurons (data not shown). KN-93, KN-92, chelerythrine, and dantrolene were purchased from EMD4 Biosciences. Nisoldipine and ω-conotoxin GVIA were purchased from Sigma-Aldrich. Myristoylated autocamtide-2-related inhibitory peptide (mAIP) was obtained from Alexis Bioscience. Effective concentrations of ω-conotoxin GVIA, nisoldipine, and dantrolene were based on prior findings in identical sensory neuron preparations (Gemes et al., 2009; McCallum et al., 2011).
Recombinant adeno-associated virus vectors.
To construct the recombinant adeno-associated virus (rAAV) vector encoding a chimeric EGFP-AIP, a DNA fragment containing the EGFP-AIP sequence from pEGFPc1-AIP (kindly provided by Dr. Steven H. Green, Department of Biological Sciences, University of Iowa) (Bok et al., 2007; Zha et al., 2009) was cloned into the NdeI/HpaI sites of pAAV-DS-CMV, a double-strand (ds) AAV plasmid (Viromics), to generate pdsAAV-CMV-EGFP-AIP. EGFP released from pEGFP-c1 was cloned into pAAV-DS-CMV to generate pdsAAV-CMV-EGFP. The AAV vectors of AAV8-EGFP-AIP and AAV8-EGFP were produced in our laboratory by the triple transfection of either pdsAAV-CMV-EGFP-AIP or pdsAAV8-CMV-GFP (control) with pRC8 and pHelpler (Viromics) into 293T cells, followed by two cycles of cesium chloride gradient purification and concentrated as previously described (Grimm et al., 2003). Encapsidated DNA was quantified by a PicoGreen assay (Invitrogen) following denaturation of the AAV particles, and the physical titer was calculated and expressed as genome copy number per milliliter (GC/ml). The titers of AAV8-EGFP-AIP and AAV8-CMV-EGFP were 3.04 × 1012 GC/ml and 9.84 × 1012 GC/ml, respectively. All vector injections were 2 μl for each DRG, using an established technique (Fischer et al., 2011).
Statistical analysis.
For comparisons between groups, matched recordings were used in which neurons from the same dissociations were mixed among the different groups. Unpaired t tests were performed between groups using Excel (Microsoft). One-way or two-way ANOVA was performed where appropriate and post hoc paired comparisons (Tukey's HSD) were performed using Prism (GraphPad Software). Comparisons of ratios of APs generated before and after conditioning in intact DRGs were compared with Mann–Whitney U test with Bonferroni correction for multiple comparisons. Data are presented as average ± SEM.
Results
Identification of CDI and CDF in sensory neurons
To explore whether ICa in sensory neurons is regulated by neuronal activity, we first used a simple repetitive pulse protocol that has been used to reveal inhibition and facilitation in various other cell types (DeMaria et al., 2001; Grueter et al., 2006; Blaich et al., 2010). Only a monotonic decrement in ICa was observed during stimulation at rates from 10–100 Hz (n = 6; Fig. 1A). While this pattern indicates a dominant effect of CDI and voltage-dependent inactivation (VDI) during tonic firing, it does not necessarily demonstrate the absence of CDF, which may be masked by concurrent CDI (Zühlke et al., 2000). Also, phasic activity may be a more relevant model of normal function in sensory neurons, which are quiescent apart from trains induced by receptive field stimulation (Ma et al., 2003). We therefore examined an alternative approach in which CDF is evaluated during recovery from a prior conditioning prepulse (Lee et al., 2000; Hudmon et al., 2005). A protocol was designed (hereafter referred to as the standard protocol; Fig. 1B,C) in which the holding potential was set at −95 mV to minimize preferential closed-state inactivation of Ca2+ channels (Patil et al., 1998). The duration of the test pulse was set at 5 ms to approximate the duration of an AP under patch recording conditions, and the voltage was stepped to −10 mV, which is between the voltages producing peak current in Ba2+ and Ca2+ (Hogan et al., 2000; McCallum et al., 2006). Immediately (10 ms) following a conditioning prepulse (2 s depolarization to −10 mV), ICa showed an initial phase of net ICa inactivation (16 ± 1% of baseline, n = 42) that was followed by recovery. A delayed phase ensued during which ICa exceeded baseline (123 ± 4% at 22 ± 1 s). In additional matched groups of neurons, shorter conditioning prepulses also produced net facilitation (ICa peaks at 115 ± 6% of baseline for 250 ms of conditioning, n = 4; 118 ± 4%, for 500 ms, n = 5; 131 ± 5%, for 2 s depolarization, n = 5). Like ICa from myocytes and CNS neurons, these findings demonstrate activity-dependent modulation of ICa in sensory neurons.
Figure 1.

Ca2+ current changes during pulse trains and in response to a prior conditioning prepulse. A, Currents elicited by trains of 60 AP waveform voltage commands at 10, 30, 60, and 100 Hz show progressive inactivation and no net facilitation. Note different time scales for each trace. B, After a sustained conditioning prepulse (2 s depolarization to −10 mV), test depolarizations (5 ms duration, −10 mV; at 10 ms, 100 ms, 300 ms, and 1 s, and every 2.5–98.5 s following the conditioning prepulse, not all are shown) produce currents (C) demonstrating initial net ICa inactivation of peak current (arrows) followed by net facilitation compared with the baseline ICa (dashed line).
To isolate Ca2+-dependent influences (CDI and CDF), we used a reference protocol in which Ca2+ entry during the conditioning prepulse was prevented by depolarization to +55 mV, which is above the ICa reversal potential in these neurons (49.4 ± 1.4 mV, n = 13). This protocol restricts Ca2+ influx (Fig. 2) without affecting resting [Ca2+]c, and retains the influences of VDI and the repeated test pulses. In contrast to the standard protocol, depolarization to +55 mV was followed by initial ICa inhibition (VDI) but no facilitation phase (Fig. 3A). This clearly demonstrates the dependence of facilitation upon Ca2+ influx, and provides a reference for selective quantification of CDI and CDF in neurons recovering from a conditioning Ca2+ influx. Specifically (Fig. 3B), the difference at the 10 ms time point (termed %CDI) represents the maximal extent of net CDI, whereas the difference when the recovery curve maximally exceeds the +55 mV control curve (termed %CDF) represents the greatest extent of net CDF.
Figure 2.

Cytoplasmic Ca2+ transients evoked by test protocols. A, The standard protocol (prepulse depolarization to −10 mV for 2 s, pipette EGTA 0.5 mm, left trace) shows elevation of Fura-2 ratio of emission during excitation at 340 and 380 nm (R340/380), which reflects cytoplasmic Ca2+ concentration. Recovery occurred despite the continuing train of test depolarizations (5 ms, −10 mV) every 2.5 s for 98.5 s. Increasing pipette EGTA (10 mm, middle trace) reduced the resting [Ca2+]c and the transient peak. Prepulse depolarization to +55 mV with standard pipette EGTA (0.5 mm, right trace) selectively reduced transient peak, with a residual transient possibly due to the tail current during repolarization. Recordings were made 5–8 min after whole-cell configurations were established. B, Summary data for baseline and peak [Ca2+]c (arrows, A) represented by fluorescence ratio. Mean ± SEM. Numbers in the bars indicate n for neurons of each group. Brackets indicate significant differences. ***p < 0.001. Baseline versus peak is also significantly different for each group.
Figure 3.

Effects of a conditioning prepulse on ICa. A, Patterns of ICa recovery are affected by protocol conditions. The standard protocol (Ca2+ bath, pipette EGTA 0.5 mm, 2 s conditioning prepulse to −10 mV; n = 8) produced rapid initial inactivation of ICa and subsequent sustained facilitation of ICa above baseline. Depolarization to +55 mV during prepulse conditioning (n = 7) results in less initial inactivation and no facilitation. Without the conditioning prepulse (Rundown, n = 10), ICa induced by sequential test pulses developed minor rundown to 95 ± 2% of baseline, similar to the decrease observed at the end of the protocol using a +55 mV conditioning prepulse. B, Defined parameters (%CDI, %CDF) are demonstrated for a sample recovery curve by comparing to the averaged recovery curve for the +55 mV protocol (n = 12 neurons). C, Summary data for CDI and CDF. Protocols are specified according to their charge carrier (Ca2+ or Ba2+), pipette solution buffer (EGTA or BAPTA), and buffer concentration. Dantrolene (Dan, 10 μm) was compared with its vehicle (DMSO). Mean ± SEM. Numbers in the bars indicate n for neurons of each group. Brackets indicate significant differences from the control condition (Ca2+ charge carrier, 0.5 mm EGTA): *p < 0.05, **p < 0.01, ***p < 0.001. D, Compared with the standard 0.5 mm EGTA pipette buffering (gray line, same data shown for −10 mV in A), replacing bath Ca2+ with Ba2+ as the charge carrier for ICa (n = 6), increasing pipette EGTA (10 mm, n = 7) or replacing it with BAPTA (20 mm, n = 7) reduces initial inactivation and eliminates subsequent facilitation.
Characterization of CDI and CDF in sensory neurons
ICa amplitude during a test pulse immediately after the conditioning prepulse was reduced to 16 ± 1% of baseline (i.e., 84% total inactivation). The measured %CDI of 33 ± 2% (n = 8; Fig. 3C) indicates that most inactivation following depolarization under standard conditions is attributable to VDI. After recovery from inactivation, the large ensuing %CDF (34 ± 4%; Fig. 3C) identifies CDF as a powerful facilitatory influence in sensory neurons. We further examined the central question of Ca2+ dependence by replacing extracellular Ca2+ with Ba2+ as the charge carrier. This decreased CDI and prevented the development of CDF after neuronal activation (Fig. 3C,D). Delayed recovery of ICa was observed in Ba2+ bath solution, probably due to the accumulation of massive cytoplasmic Ba2+ loads secondary to the inability of the plasma membrane Ca2+-ATPase to extrude Ba2+ (Mironov and Juri, 1990). Eliminating Ca2+ accumulation by buffering the Ca2+ influx with high pipette concentrations of either EGTA (10 mm), or the more rapid Ca2+ chelator BAPTA (20 mm) (Anderson et al., 1998; Lee et al., 2009), limited the global rise in [Ca2+]c during the conditioning depolarization (Fig. 2) and decreased %CDI and %CDF (Fig. 3C,D), suggesting that a global increase in [Ca2+]c rather than a local source drives activity-related inhibition and facilitation of VGCCs in sensory neurons.
The 5 ms duration of the test pulses did not allow ICa to reach its peak for every neuron (e.g., Fig. 1C). Thus, it is possible that facilitation of ICa recorded in this way represents either increased channel conductance or accelerated channel activation, as has been noted in heterologously expressed P/Q-type channels (Liang et al., 2003). To resolve which fundamental mechanisms contribute to CDF in sensory neurons, we used a modified protocol in which the standard conditioning prepulse was followed 20 s later by a test pulse that was sustained for 50 ms to also allow evaluation of peak ICa (Fig. 4A). The rate of ICa activation, determined by measuring the time to achieve peak current, was increased after conditioning (7.2 ± 0.8 ms vs 8.1 ± 0.7 ms at baseline, n = 12, p < 0.01; Fig. 4B). This observation confirms more rapid channel opening after conditioning, which may be particularly relevant to neuron function due to the brief duration of APs. Since peak ICa in this experiment was also increased (119 ± 20 pA/pF vs 100 ± 18 pA/pF at baseline, n = 12, p < 0.001), altered channel-gating kinetics and peak conductance operate in combination to produce CDF in sensory neurons. The persistence of CDF in this protocol in the absence of repetitive test pulses (%CDF is 21 ± 6%, n = 12, when ICa is measured 5 ms after test pulse initiation, normalized against an identical protocol, but with a conditioning prepulse to +55 mV, n = 14) indicates that latent CDF persists in a quiescent neuron after intense activity.
Figure 4.

Evaluation of ICa activation kinetics after conditioning with a single test pulse. A, Baseline and test voltage commands were prolonged to 50 ms to allow current to reach a maximum, and only a single test pulse was used to identify whether CDF requires multiple pulses as used in the basic protocol. B, Current trace (typical of n = 12) shows that ICa 5 ms after initiating the test pulse is greater than the ICa at 5 ms in the baseline pulse (arrows), duplicating findings using the standard protocol. Additionally, the time to peak ICa (vertical lines) is shorter after the conditioning prepulse, and the peak ICa is greater after the conditioning prepulse compared with the baseline peak ICa (dashed line), indicating that increases in both the rate of ICa activation and the maximum ICa contribute to CDF as measured by 5 ms test pulses.
CDF outlasts elevated [Ca2+]c
Transient increase of [Ca2+]c may act through acute signaling mechanisms directly coupled to [Ca2+]c, or may have more sustained consequences via post-translational modifications of proteins that persist beyond the return of [Ca2+]c to baseline. To determine the temporal relationship between the clearance of cytoplasmic Ca2+ and Ca2+-dependent changes in ICa, we fluorometrically measured R340/380 as an indicator [Ca2+]c while simultaneously recording ICa recovery in the standard protocol (Fig. 5). Although this approach only detects global intracellular changes, the sensitivity of CDI and CDF to a slow Ca2+ chelator in the pipette (10 mm EGTA) suggests that global [Ca2+]c may be more relevant for these processes than subplasmalemmal nanodomains. The transient increase in [Ca2+]c triggered by conditioning depolarization resolved with a time constant of 2.2 ± 0.3 s (n = 6), resembling the pace of ICa recovery from initial inactivation (3.4 ± 1.5 s). Peak facilitation in these neurons (118 ± 4% of baseline) occurred at 17.3 ± 4.6 s after conditioning, by which time the depolarization-induced Ca2+ transient had fully resolved. Thus, CDI abates at approximately the same pace as the resolution of the Ca2+ transient, whereas CDF has either a delayed onset or a slower resolution than [Ca2+]c, and suggests that this form of ICa plasticity outlasts the Ca2+ transient.
Figure 5.

Time courses for ICa and cytoplasmic Ca2+ concentration ([Ca2+]c). ICa was measured during the standard protocol (Fig. 1B,C), and normalized to baseline. Fura-2 ratio of emission during excitation at 340 and 380 nm (R340/380) was recorded simultaneously as an indicator of [Ca2+]c, which was normalized such that resting baseline is 0 and peak is 100. Averaged curves (n = 6) demonstrate that net inhibition of ICa is synchronous with R340/380 elevation, but net facilitation of ICa outlasts return of R340/380 to baseline.
CDF requires N-type channel function
To determine the source of Ca2+ responsible for CDI and CDF in sensory neurons, we first considered intracellular Ca2+ stores, since Ca2+ influx in sensory neurons triggers the process of Ca2+-induced Ca2+ release (CICR) by which Ca2+ is discharged from intracellular stores through the ryanodine receptor channels (Gemes et al., 2009). This may add to the global increase in [Ca2+]c and activate CaMKII (Shakiryanova et al., 2011). To disrupt CICR, sensory neurons were pretreated with the ryanodine receptor blocker dantrolene (10 μm), using conditions we have previously shown to limit Ca2+ release from intracellular stores in dissociated sensory neurons (Gemes et al., 2009). The lack of effect of dantrolene on %CDI or %CDF (Fig. 3C) suggests that Ca2+ released via CICR does not contribute to CDI and CDF.
Sensory neurons possess multiple subtypes of VGCCs, including L-, N-, P/Q-, and R- and T-type channels (Scroggs and Fox, 1992). To identify the specific VGCC subtypes involved in CDI and CDF, we used selective pharmacological blockers to isolate effects of L-type and N-type currents, since these convey the largest portions of total voltage-gated ICa in sensory neurons (Evans et al., 1996; Abdulla and Smith, 2001; McCallum et al., 2011). Blockade of N-type current by bath application of ω-conotoxin GVIA (200 nm) reduced peak ICa by ∼50%, as expected (Fig. 6A). The effect on total Ca2+ influx during the conditioning prepulse (current integrated across time; Fig. 6B) was less, due to rapid current inactivation during the prepulse (Fig. 1C). Although CDI was not affected (Fig. 6C), the block of N-type current eliminated CDF (Fig. 6D). To determine whether reduced total ICa might itself contribute to diminished CDF regardless of the path of entry, we titrated the nonselective VGCC blocker cadmium (3–6 μm) to produce a comparable overall reduction of ICa (Fig. 6A) and found that CDF was not diminished (Fig. 6D). Since cadmium will have partially reduced N-type ICa, this finding indicates that the dependence of CDF upon N-type current is not a simple proportionate relationship. L-type blockade using 1 μm nisoldipine (Kd ∼ 2 nm) (Enyeart et al., 1986; Morel et al., 1998) also lowered total ICa to a similar extent (Fig. 6A). Although this reduced CDI by ∼25% (Fig. 6C), it had no effect on CDF (Fig. 6D). Together, these observations indicate that CDF is not sensitive to total Ca2+ influx, but rather suggest a specific role for N-type Ca2+ channels as an effector site for generating sensory neuron CDF.
Figure 6.
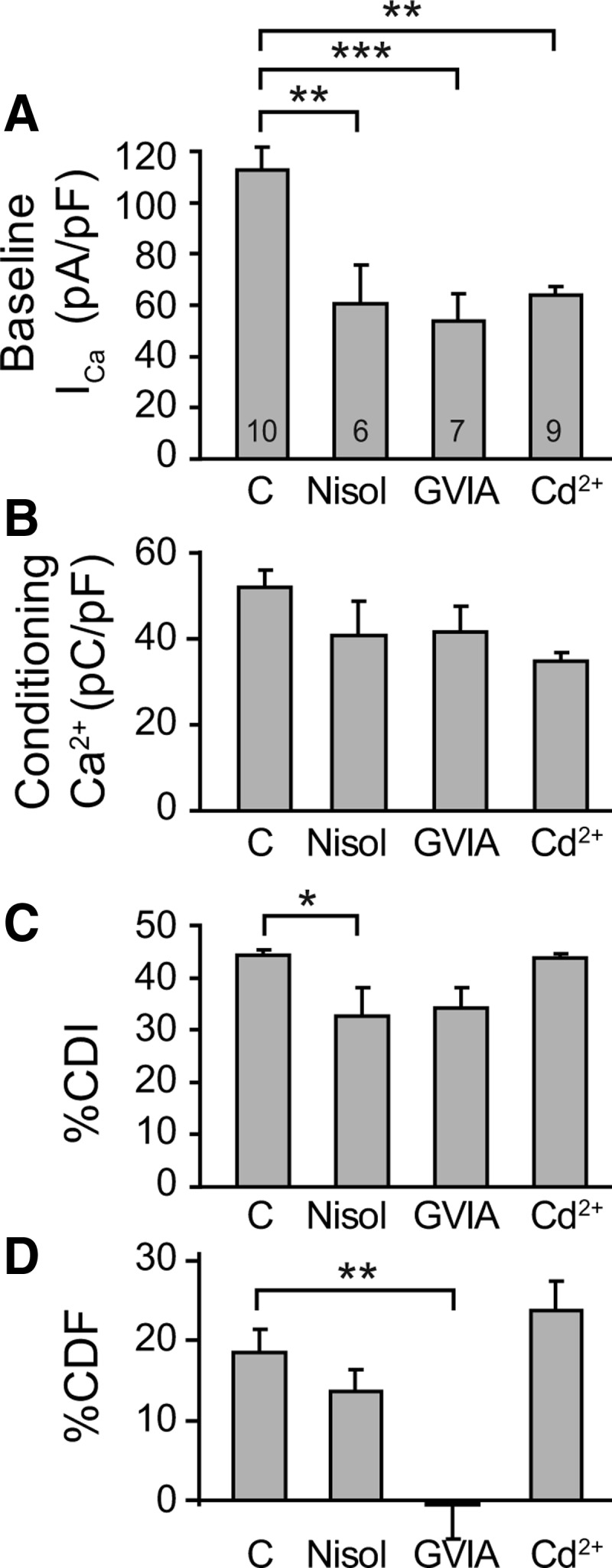
Effect of blockade of Ca2+ channel subtypes before, during, and after a conditioning prepulse. A–D, Using the standard protocol, summary data are presented for the effect of VGCC blockers on (A) baseline ICa before the conditioning prepulse, (B) Ca2+ influx during the conditioning prepulse (i.e., ICa integrated over the 2 s depolarization), (C) CDI, measured as %CDI (Fig. 3C), and (D) CDF, measured as %CDF (Fig. 3C). Responses to the standard protocol were examined in matched groups of neurons treated with the L-type channel blocker nisoldipine (Nisol, 1 μm), with the N-type channel blocker ω-conotoxin GVIA (GVIA, 200 nm), the nonspecific Ca2+ channel blocker Cd2+ (3–6 μm), compared with untreated controls (C). Number of neurons for each group is shown in the bars. Brackets indicate significant differences. *p < 0.05, **p < 0.01, ***p < 0.001.
CDF is regulated by CaMKII
CDF has been shown to involve protein phosphorylation in other systems (see above), so we explored the potential regulation of N-type CDF in sensory neurons by two Ca2+-sensitive kinases, CaMKII and protein kinase C (PKC) (Fig. 7A–G). The membrane-permeable CaMKII inhibitor KN-93 (2 μm) eliminated CDF, whereas the inactive analog KN-92 (2 μm) had no effect (Fig. 7A,H). KN-93 at this concentration additionally resulted in rundown of current in the absence of a conditioning prepulse (Fig. 7A), consistent with blockade of a potentiating effect of CaMKII on VGCCs. Alternatively, off-target direct effects of both KN-93 and KN-92 upon a variety of channels including VGCCs have been reported (Ledoux et al., 1999; Gao et al., 2006). These effects may also account for amplified inactivation during the prepulse, contributing to decreased Ca2+ influx (Fig. 7F) and apparent increased CDI (Fig. 7G), while elevated baseline ICa during KN-92 (Fig. 7E) is unexplained. Accordingly, we used a reduced KN-93 concentration (0.2 μm) to limit these off-target actions and found that this concentration of KN-93 eliminated facilitation (Fig. 7B,H) in the absence of other effects. In addition, CaMKII inhibition using a membrane-permeable peptide inhibitor of this kinase, mAIP (5 μm), also decreased facilitation, whereas myristoyl itself was inactive (Fig. 7C,H).
Figure 7.

Effect of kinase inhibition on ICa before, during, and after a conditioning prepulse. A, Using the standard protocol, the time course for recovery following a conditioning prepulse shows elimination of CDF by the CaMKII blocker KN-93 (2 μm, n = 7), but not by the inactive analog KN-92 (2 μm, n = 5), compared with matched control neurons (n = 7). In the absence of a conditioning prepulse, accelerated rundown is evident in neurons exposed to KN-93 (2 μm), suggesting a dependence of ICa upon CaMKII action. This rundown accounts for incomplete recovery after a conditioning prepulse in neurons exposed to KN-93. B, A lower concentration of KN-93 (0.2 μm, n = 8) also suppresses CDF compared with matched controls (n = 9). The reference curve (+55 mV, red) is also shown. C, The CaMKII inhibitory peptide mAIP (5 μm, n = 7) produces partial blockade of CDF compared with matched controls treated with myristoyl alone (Myr, 5 μm, n = 7). D, Neurons harvested 4 weeks after in vivo transfection by AAV vectors show the effect of AIP linked to AIP (AAV-AIP, n = 5 neurons from 4 injected DRGs in 2 rats) or GFP expression alone (AAV-GFP, n = 5 neurons from 4 injected DRGs in 2 rats). Chronic CaMKII blockade by expressed AIP eliminated CDF. The reference curve (+55 mV, red) is derived from transduced neurons. E–H, Summary data are presented for the effect of blockers on (E) baseline ICa before the conditioning prepulse, (F) Ca2+ influx during the conditioning prepulse (i.e., ICa integrated over the 2 s depolarization), (G) CDI, measured as %CDI (Fig. 3B), and (H) CDF, measured as %CDF (Fig. 3B). The PKC inhibitor chelerythrine (Chel, 10 μm) and myristoyl (Myr, 5 μm) had no effects. Separate untreated control groups (C) are shown with their matched treated groups. Negative calculated values of %CDF for neurons treated with KN-93 represent failure of ICa to recover to matched recordings using prepulse depolarization to +55 mV (data not shown). Number of neurons for each group is shown in the bars. Brackets indicate significant differences. *p < 0.05, **p < 0.01, ***p < 0.001.
To test the effect of sustained blockade of CaMKII, we expressed AIP linked to GFP (GFP-AIP), using a construct that has been proved effective in inhibiting CaMKII (Bok et al., 2007). An AAV vector expressing either GFP-AIP or GFP alone was injected into L4 and L5 DRGs, from which neurons were dissociated 4 weeks later. Transduced neurons were identified for recording by GFP fluorescence, and had diameters (30.5 ± 1.9 μm, n = 5 for GFP-AIP; 29.3 ± 1.3 μm n = 5 for GFP; no difference between groups) comparable to other recorded neurons in this study. GFP-AIP eliminated CDF, while transduced neurons expressing only GFP produced typical CDF (Fig. 7D,H). Thus, for VGCCs in the sensory neuron system, pharmacological treatment of dissociated neurons as well as selective genetic manipulation in the intact animal reveal a dependence of CDF upon CaMKII activation.
Low-dose KN-93, mAIP, and GFP-AIP expression did not affect CDI (Fig. 7G), indicating that CaMKII selectively affects CDF but not CDI. PKC can enhance sensory neuron N-type and L-type ICa (Hall et al., 1995), but inhibition with chelerythrine (10 μm) did not affect baseline ICa, CDI, or CDF (Fig. 7E–H).
Injury eliminates CDF
We have previously found that peripheral nerve injury decreases CaMKII activity in sensory neurons (Kawano et al., 2009; Kojundzic et al., 2010). We therefore hypothesized that decreased CaMKII activity associated with neuronal injury should disrupt CDF generation. Nerve injury by SNL results in axotomy of the L5 DRG neurons, while the neighboring L4 neurons are exposed to inflammation initiated by the degenerating distal L5 axon segments. Since the relative contributions of these two populations to pain behavior have not been established (Gold, 2000), we examined Ca2+-dependent regulation of ICa in neurons from both ganglia separately. Recordings were performed 21–30 d after injury since this allows nonspecific effects of surgery to subside and reflects the chronic phase of neuropathy. At that time, behavioral testing of the rats injured by SNL demonstrated hyperalgesia (39 ± 3% hyperalgesia-type response to pin touch, n = 37) in contrast to animals receiving only skin incision (0 ± 0%, n = 101; p < 0.001). Comparison of neurons from SNL and skin incision animals revealed distinct effects of injury on ICa and its regulation by neuronal activity (Fig. 8A,B). Consistent with our previous findings (McCallum et al., 2006), baseline ICa (Fig. 8C) as well as Ca2+ influx during the conditioning prepulse (Fig. 8D) were decreased in L5 neurons after SNL, compared with matched neurons from control rats. The extent of CDI was unaffected by SNL in both the L4 and L5 neurons (Fig. 8E). However, there was an almost complete loss of CDF after axotomy of the directly injured L5 neurons, and a less complete loss of CDF in the L4 neurons indirectly affected by SNL (Fig. 8F). We considered the possibility that the injury-induced reduction of Ca2+ influx during the prepulse was responsible for the disappearance of CDF, and tested this by increasing influx in SNL L5 neurons. Because of ICa inactivation, prolongation of the prepulse to 4 s minimally increased total influx (7.5 ± 2.5%, n = 5). Raising bath Ca2+ to 4 mm, however, successfully elevated influx (Fig. 8D), but this did not rescue CDF in the axotomized SNL L5 neurons (Fig. 8E). Additionally, in control neurons, there was no influence of Ca2+ influx upon %CDF (linear regression R2 = 0.02, p = 0.35). These findings show not only that CDI and CDF have distinct mechanistic pathways but also that injury disrupts the natural regulation of ICa.
Figure 8.

Effect of painful nerve injury on ICa before, during, and after a conditioning prepulse. A, In uninjured control neurons, the time course for ICa recovery was evaluated after a conditioning prepulse to either −10 mV or +55 mV (n = 12 for each). B, Matched observations made in neurons dissociated from animals subjected to SNL showed marked suppression of CDF in the axotomized neurons from the fifth lumbar level (SNL L5, n = 8) and partial suppression in the neighboring neurons (SNL L4, n = 8), compared with uninjured control neurons (dashed line, same data as in A). Prepulse depolarizations to +55 mV were recorded for SNL L5 (red, n = 8) and SNL L4 (data not shown, n = 8). C–F, Summary data are presented for the effect of injury on (C) baseline ICa before the conditioning prepulse, (D) Ca2+ influx during the conditioning prepulse (i.e., ICa integrated over the 2 s depolarization), (E) CDI, measured as ΔRCDI (Fig. 3C), and (F) CDF, measured as ΔRCDF (Fig. 3C). SNL L5 neurons show decreased baseline ICa, decreased Ca2+ influx during the conditioning prepulse, and substantial decrease of CDF compared with control neurons. Elevation of bath Ca2+ to 4 mm (SNL L5 + Ca2+) increased Ca2+ influx in SNL L5 neurons to above normal levels, but this did not rescue CDF. Number of neurons for each group is shown in the bars. Brackets indicate significant differences. *p < 0.05, ***p < 0.001.
Physiological role of CDF in sensory neurons
To place sensory neuron CDF into a natural context, we examined whether conditioning stimuli would produce CDF under conditions that duplicate a physiological setting. We first examined conditioning from a holding potential of −65 mV to replicate the resting membrane potential of sensory neurons (Sapunar et al., 2005). Under these conditions, %CDF (normalized by comparison to a reference curve using a depolarization step to +55 mV from a holding potential of −65 mV) was 19 ± 6% (Fig. 9A; n = 9). This confirms that CDF in sensory neurons evolves after neuronal activity even with a physiological resting membrane potential. We next examined the nature of the conditioning stimulus. Since depolarizations in the form of a pulse train accentuate cumulative VDI compared with sustained (square wave) depolarization (Patil et al., 1998), it is possible that CDF during recovery from neuronal activation might be seen only after nonpulsatile depolarization. To test this, we used a conditioning stimulus in the pattern of an AP train (Fig. 9B), replicating natural neuronal activity. CDF was again observed after this form of neuronal activation (facilitation to a maximum of 117% in each of two neurons). These observations indicate that CDF occurs under physiological conditions in sensory neurons.
Figure 9.

Influence of depolarization from a holding potential of −65 mV and conditioning by an impulse train. A, When a holding potential of −65 mV is used, a depolarizing conditioning prepulse (0 mV, 2 s; n = 9) is followed by facilitation, whereas a prepulse to +55 mV does not produce facilitation (n = 8). This protocol used test pulses starting at 10 ms after the prepulse and then at 2.5 s intervals. B, Facilitation also followed a conditioning stimulus composed of a train of AP waveform commands (100 APs at 100 Hz), as represented by the ICa that exceeded baseline when evoked by a test pulse 7.5 s after conditioning.
Cytoplasmic Ca2+ critically controls repetitive firing behavior in sensory neurons. Specifically, Ca2+ that enters through VGCCs opens Ca2+-sensitive K+ channels that are expressed by peripheral sensory neurons (Sarantopoulos et al., 2007), thereby inhibiting repetitive AP firing (Hogan et al., 2008; Lirk et al., 2008). We therefore directly examined whether CDI and CDF triggered by neuronal depolarization influence the subsequent generation of APs in repetitively firing small- to medium-sized sensory neurons (diameter 31 ± 1 μm; input resistance 73 ± 7 MΩ; resting membrane potential −58 ± 1 mV; no differences between the conditioned group, n = 52, and the time control group, n = 22). Recordings were performed by an intracellular technique to minimize disturbance of cytoplasmic Ca2+ signaling (Friel and Tsien, 1992) and in intact DRGs to eliminate the effects of dissociation. At various intervals following a conditioning prepulse (0 mV, 2 s, voltage-clamp), we determined the number of APs during a test pulse (2 nA, 50 ms, current-clamp), which was compared with the number of APs generated by an identical baseline pulse 15 s before conditioning (Fig. 10A). Control recordings were made in other neurons from the same ganglion, using the same protocol but without the conditioning prepulse (Fig. 10B). The number of APs produced by current injection was unchanged at 0.5, 1, and 2 s following the conditioning prepulse (Fig. 10C), indicating that repetitive firing was not affected during the initial period following the conditioning prepulse. However, AP generation decreased with longer intervals and was reduced to approximately half of baseline at 10 s following the conditioning prepulse, with subsequent recovery thereafter. This time course was comparable to that for the development of CDF following a conditioning prepulse in dissociated neurons. To confirm that regulation of excitability is attributable to the operation of CDF, we examined the Ca2+ dependence of this process using a similar protocol in dissociated sensory neurons delivered by whole-cell patch technique (Fig. 11A). Whereas excitability was suppressed by the conditioning prepulse in control neurons with standard pipette solution (0.5 mm EGTA; Fig. 11B), this effect was lost when Ca2+ influx was buffered with high pipette EGTA (10 mm; Fig. 11C). These findings suggest that under natural conditions, CDF functions as a potent throttle that suppresses sensory neuron excitability following a period of intense neuronal activity.
Figure 10.

Effect of a conditioning prepulse on excitability of nondissociated sensory neurons recorded by intracellular microelectrode. A, AP firing induced by depolarizing current test pulses was diminished 10 s after a conditioning prepulse in a control neuron (28 μm diameter) from an uninjured L5 DRG. B, In the absence of conditioning, a different control neuron (38 μm) showed no change in AP generation after a comparable interval. C, The time course for excitability (the ratio of APs generated during the test pulse normalized to baseline) is shown for test pulses applied at various time intervals after conditioning. Only a single test pulse was applied to each neuron. Control neurons (L5 DRG) show depression of excitability 10 s after conditioning compared with neurons without conditioning. D, After injury by L5 SNL, the conditioning prepulse failed to suppress excitability in axotomized L5 neurons. Note that both of the SNL data points (with and without conditioning prepulse) are at 10 s, but are displayed slightly offset for clarity. Mean ± SEM, numbers next to data points indicate n for neurons. *p < 0.05.
Figure 11.

Effect of a conditioning prepulse on excitability of dissociated sensory neurons recorded by patch technique. A, Similar to the recordings in nondissociated neurons shown in Figure 10, AP firing induced by depolarizing current test pulses was diminished 20 s after a conditioning prepulse in a control neuron (25 μm diameter) recorded with standard pipette solution (0.5 mm EGTA). Whereas conditioning decreases excitability (the ratio of APs generated during the test pulse normalized to baseline) with standard pipette buffering (B), high buffering (10 mm EGTA) eliminates the effect of conditioning on excitability (C). Note that both of the data points for 10 mm EGTA (with and without conditioning prepulse) are at 20 s, but are displayed slightly offset for clarity. Mean ± SEM, numbers next to data points indicate n for neurons. *p < 0.05.
Since our observations in dissociated neurons indicate a loss of CDF after neuronal injury, we speculated that this would compromise the control of excitability after a conditioning prepulse. At the time point (10 s) at which control neurons showed maximal suppression of repetitive firing following a conditioning prepulse, neurons subjected to SNL (diameter 32 ± 1 μm; input resistance 72 ± 8 MΩ; resting membrane potential −58 ± 2 mV; no differences between the conditioned group, n = 13, and the time control group, n = 11) showed no effect of the conditioning prepulse on AP generation (Fig. 10D). This indicates that the CDF mechanism for regulating excitability after intense neuronal activation is lost following axonal injury of sensory neurons.
Sensory neurons are exposed to repeated volleys of APs, and it is possible that the presence of CDF modulates processes underlying inactivation (VDI and CDI). We evaluated this by applying a second conditioning pulse after establishing CDF by a previous conditioning pulse in control neurons (Fig. 12). Total inactivation was independent of pre-existing CDF, and CDF was fully re-established after recovery from inactivation. Axotomized SNL L5 neurons (n = 3) similarly showed a response to a second conditioning pulse that was comparable to the first (data not shown).
Figure 12.

Influence of pre-existing CDF on the response of ICa to neuronal activation. The development of both ICa inactivation and facilitation in control neurons (n = 5) are unchanged in response to a second conditioning pulse delivered during preexisting CDF, compared with the response to the initial conditioning pulse. Mean ± SEM.
Discussion
The central role of Ca2+ in coordinating neuronal function suggests dysregulation of Ca2+ signaling as a potential cause of neuropathic pain. We have identified robust Ca2+-dependent modulation of ICa in DRG neurons that is distinct from VDI. Unlike CDI, CDF is regulated by CaMKII and is lost following neuronal injury, a process that increases neuronal excitability and may contribute to neuropathic pain.
CDI in sensory neurons
A contribution of CDI to ICa inactivation during sustained depolarization has been shown in previous investigations of N-type and P/Q-type currents in neurons (Cox and Dunlap, 1994; Chaudhuri et al., 2005) and in expressed L-, N-, R-, and P/Q-type channels (Lee et al., 1999; Liang et al., 2003), where its extent is modulated by coexpression of particular auxiliary β-subunits (Lee et al., 2000). We observed ICa inactivation that is synchronous with the maximal [Ca2+]c and is sensitive to cytoplasmic Ca2+ buffering, consistent with prior observations in a variety of expressed VGCC subtypes, for which CaM-dependent CDI is responsive to global cytoplasmic Ca2+ accumulation (Lee et al., 1999; Liang et al., 2003; Chaudhuri et al., 2007). CDI at sensory neuron central terminals may contribute to short-term synaptic depression as has been noted for P/Q-type currents in the calyx of Held (Forsythe et al., 1998; Xu and Wu, 2005), thereby enhancing the temporal contrast for sensations conveyed by high intensity afferent activity.
CDF in sensory neurons
While CDF has been observed for P/Q channels in the CNS (Borst and Sakmann, 1998; Chaudhuri et al., 2005), its presence in peripheral sensory neurons or the participation of other neuronal VGCC subtypes in CDF have not previously been reported. It is unlikely that our observations represent voltage-dependent facilitation (Kavalali and Plummer, 1994) or relief of tonic G-protein-mediated inhibition (Ikeda, 1991; Zamponi and Snutch, 1998) since depolarization to +55 mV did not produce facilitation, and sensitivity to PKC that typifies G-protein-mediated inhibition was absent (Swartz, 1993). Several lines of evidence support a direct role of Ca2+ in producing ICa facilitation. First, depolarization to a potential that admitted minimal Ca2+ eliminated facilitation. Second, replacement of extracellular Ca2+ with Ba2+ similarly reduced facilitation, which indicates a specific dependence on Ca2+ as the permeant cation. Third, we observed that CDF of N-type currents is sensitive to high cytoplasmic Ca2+ buffering, indicating that globally increased cytoplasmic Ca2+ is necessary to initiate CDF in sensory neurons. This contrasts with CDF in cerebellar P-type channels, which is dependent on Ca2+ elevation only in the nanodomain around individual channels (Chaudhuri et al., 2005), but concurs with findings from brainstem and expressed P/Q channels and myocyte L-type channels (Xiao et al., 1994; Borst and Sakmann, 1998; Lee et al., 2000).
In our observations of sensory neurons, persistence of CDF beyond the period of elevated [Ca2+]c suggests involvement of factors other than a simple action on CaM. This sustained effect is compatible with the participation of CaMKII, which retains activity through autophosphorylation even after CaM dissociates (Saitoh and Schwartz, 1985). A role for CaMKII is further suggested by the sensitivity of CDF to Ca2+ buffering, and by the efficacy of CaMKII blockers in limiting CDF. Together, these observations indicate that CaMKII is a central component of a signaling cascade triggered by neuronal activity that ultimately activates Cav2.2 channels. While we could not discern the onset kinetics of CDF in our model due to concurrent VDI and CDI, slow activation of CaMKII may account for the delayed peak of CDF, as has been noted for the CaMKII-dependent facilitation of L-type ICa in cardiac myocytes after photorelease of Ca2+ (Anderson et al., 1994). Alternatively, CDF may activate rapidly but is obscured by CDI. Brain N-type Ca2+ channels are recognized targets of CaMKII and PKC phosphorylation (Hell et al., 1994), although our data do not reveal participation of PKC in generating CDF. Resolving the specific downstream effects of CaMKII leading to sensory neuron CDF, for instance phosphorylation of the Synprint region of α1-subunits (Yokoyama et al., 1997, 2005) or prolongation of channel open probability (Dzhura et al., 2000), awaits future studies.
The dominant path for activity-induced Ca2+ influx in small- to medium-sized sensory neurons is through L-type and N-type VGCCs (Evans et al., 1996; Abdulla and Smith, 2001; McCallum et al., 2011). In our present experiments, blockade of N-type VGCCs eliminated CDF, which suggests either that Ca2+ entering through N-type channels may be uniquely able to activate CaMKII, or that CaMKII selectively enhances N-type Ca2+ channel function in sensory neurons under these conditions. The first scenario seems unlikely since this model favors localized coupling between the N-type calcium channel and CaMKII, which is not supported by elimination of CDF with high cytoplasmic Ca2+ buffering. The alternative possibility that CaMKII produces CDF in N-type calcium channels is particularly intriguing, as this form of plasticity has never been reported in neurons.
Influence of injury
Only minimal CDF remains in neurons injured by peripheral axotomy. This cannot be attributed to exaggerated inactivation by CDI and VDI, as these were unchanged by injury, so some component of the mechanistic sequence that generates CDF must become deficient after injury. Previous findings show that axotomy reduces ICa N-type current, but this is only an incomplete effect (<50%) (Baccei and Kocsis, 2000; Abdulla and Smith, 2001; McCallum et al., 2011). An additional likely factor is impaired CaMKII signaling following nerve injury, which we have found to occur selectively in animals that develop pain behavior (Kawano et al., 2009; Kojundzic et al., 2010). Although our present experiments focused on ICa responses to phasic [Ca2+]c changes evoked by neuronal activation, the failure of GFP-AIP expression to lower baseline ICa indicates that tonically lowered CaMKII activity is not likely to account for depressed baseline ICa after injury.
Potential consequences of CDF in sensory neurons: divergent physiological effects at different cellular sites
Our data indicate that Ca2+ signaling via CaMKII enhances VGCC function in the sensory neuron soma after intense neuronal stimulation. Although confirmation that a similar phenomenon occurs in neurites and synaptic terminals awaits further study, we can consider what physiological effects may be expected. CaMKII is known to be present in presynaptic terminals in other tissues such as hippocampus (Nayak et al., 1996), and N-type Ca2+ channels are the dominant presynaptic Ca2+ influx pathway for synaptic transmission of nociceptive activity in the spinal dorsal horn (Matthews and Dickenson, 2001; Heinke et al., 2004; Rycroft et al., 2007). If these elements interact in the sensory neuron central terminals as we have observed in the soma, CDF may be a key process in pain signaling. Since neurotransmitter release is proportional to the third or fourth power of Ca2+ entry (Dodge and Rahamimoff, 1967; Sakaba and Neher, 2001), CDF would be a powerful mechanism for amplifying presynaptic function. For instance, by increasing ICa 34%, the CDF that we observed would elevate neurotransmitter release threefold. This would enhance synaptic transmission after intense sensory stimulation, resulting in sensory sensitization during the time period of CDF action. Activity-induced enhanced synaptic transmission in dorsal horn pain pathways is thought to be a critical feature contributing to some chronic pain states (Latremoliere and Woolf, 2009).
Since Ca2+ regulates numerous cellular processes, the net influence of CDF on sensory neuron excitability is hard to predict and may differ in various subgroups and in different parts of the neuron. In the specific setting of peripheral nerve trauma, our finding that CDF is lost suggests that the dominant site of CDF action may not be the synaptic terminal. In the peripheral nervous system, our data show decreased AP generation in sensory neuron somata following a conditioning stimulus that induces CDF. Thus, CDF appears to provide a negative feedback upon membrane excitability after a period of intense activity. This is explainable by the rapid activation of dominant K+ currents by Ca2+ influx (Raman and Bean, 1999), producing a net outward current and decreased membrane resistance that suppress repetitive firing (Scholz et al., 1998; Swensen and Bean, 2003; Lirk et al., 2008). Similar CDF in the peripheral termini of sensory neurons would contribute to sensory adaptation, since a stimulus-induced pulse train would be followed by a period in which CDF produces relative suppression of repetitive AP generation. CDF in the membrane of the sensory neuron T-junction would be expected to intensify the Ca2+-regulated filtering of afferent AP trains, which would also diminish the amount of sensory traffic reaching the spinal dorsal horn following intense afferent activity (Stoney, 1990; Lüscher et al., 1994, 1996). The overall resulting effect of CDF acting at peripheral sites could therefore be sensory desensitization following intense activation, for instance the temporary relief from itch provided by intense counterstimuli (Ward et al., 1996).
At the sensory neuron soma, initiation of APs occurs only under pathological conditions, including peripheral nerve injury (Devor, 2006). Our present findings indicate that CDF at this site is lost after peripheral axotomy, which could further amplify ectopic activity originating in DRG somata due to loss of the braking action that CDF applies to repetitive AP generation. This can be expected to result in longer and more frequent bursts of activity, and therefore could be an important element contributing to spontaneous pain and paresthesias following peripheral nerve injury.
Footnotes
This work was supported by National Institutes of Health Grants NS-42150 (Q.H.H.) and DA-K01 02475 (H.-E.W.).
The authors declare no competing financial interests.
References
- Abdulla FA, Smith PA. Axotomy- and autotomy-induced changes in Ca2+ and K+ channel currents of rat dorsal root ganglion neurons. J Neurophysiol. 2001;85:644–658. doi: 10.1152/jn.2001.85.2.644. [DOI] [PubMed] [Google Scholar]
- Anderson ME, Braun AP, Schulman H, Premack BA. Multifunctional Ca2+/calmodulin-dependent protein kinase mediates Ca(2+)-induced enhancement of the L-type Ca2+ current in rabbit ventricular myocytes. Circ Res. 1994;75:854–861. doi: 10.1161/01.res.75.5.854. [DOI] [PubMed] [Google Scholar]
- Anderson ME, Braun AP, Wu Y, Lu T, Schulman H, Sung RJ. KN-93, an inhibitor of multifunctional Ca++/calmodulin-dependent protein kinase, decreases early afterdepolarizations in rabbit heart. J Pharmacol Exp Ther. 1998;287:996–1006. [PubMed] [Google Scholar]
- Baccei ML, Kocsis JD. Voltage-gated calcium currents in axotomized adult rat cutaneous afferent neurons. J Neurophysiol. 2000;83:2227–2238. doi: 10.1152/jn.2000.83.4.2227. [DOI] [PubMed] [Google Scholar]
- Berridge MJ, Lipp P, Bootman MD. The versatility and universality of calcium signalling. Nat Rev Mol Cell Biol. 2000;1:11–21. doi: 10.1038/35036035. [DOI] [PubMed] [Google Scholar]
- Blaich A, Welling A, Fischer S, Wegener JW, Köstner K, Hofmann F, Moosmang S. Facilitation of murine cardiac L-type Ca(v)1.2 channel is modulated by calmodulin kinase II-dependent phosphorylation of S1512 and S1570. Proc Natl Acad Sci U S A. 2010;107:10285–10289. doi: 10.1073/pnas.0914287107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bok J, Wang Q, Huang J, Green SH. CaMKII and CaMKIV mediate distinct prosurvival signaling pathways in response to depolarization in neurons. Mol Cell Neurosci. 2007;36:13–26. doi: 10.1016/j.mcn.2007.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borst JG, Sakmann B. Facilitation of presynaptic calcium currents in the rat brainstem. J Physiol. 1998;513:149–155. doi: 10.1111/j.1469-7793.1998.149by.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhuri D, Alseikhan BA, Chang SY, Soong TW, Yue DT. Developmental activation of calmodulin-dependent facilitation of cerebellar P-type Ca2+ current. J Neurosci. 2005;25:8282–8294. doi: 10.1523/JNEUROSCI.2253-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhuri D, Issa JB, Yue DT. Elementary mechanisms producing facilitation of Cav2.1 (P/Q-type) channels. J Gen Physiol. 2007;129:385–401. doi: 10.1085/jgp.200709749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox DH, Dunlap K. Inactivation of N-type calcium current in chick sensory neurons: calcium and voltage dependence. J Gen Physiol. 1994;104:311–336. doi: 10.1085/jgp.104.2.311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuttle MF, Tsujimoto T, Forsythe ID, Takahashi T. Facilitation of the presynaptic calcium current at an auditory synapse in rat brainstem. J Physiol. 1998;512:723–729. doi: 10.1111/j.1469-7793.1998.723bd.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeMaria CD, Soong TW, Alseikhan BA, Alvania RS, Yue DT. Calmodulin bifurcates the local Ca2+ signal that modulates P/Q-type Ca2+ channels. Nature. 2001;411:484–489. doi: 10.1038/35078091. [DOI] [PubMed] [Google Scholar]
- Devor M. Response of nerves to injury in relation to neuropathic pain. In: McMahon S, Koltzenburg M, editors. Wall and Melzack's textbook of pain. Ed 5. London: Churchill Livingstone; 2006. pp. 905–927. [Google Scholar]
- Dodge FA, Jr, Rahamimoff R. Co-operative action a calcium ions in transmitter release at the neuromuscular junction. J Physiol. 1967;193:419–432. doi: 10.1113/jphysiol.1967.sp008367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dzhura I, Wu Y, Colbran RJ, Balser JR, Anderson ME. Calmodulin kinase determines calcium-dependent facilitation of L-type calcium channels. Nat Cell Biol. 2000;2:173–177. doi: 10.1038/35004052. [DOI] [PubMed] [Google Scholar]
- Enyeart JJ, Aizawa T, Hinkle PM. Interaction of dihydropyridine Ca2+ agonist Bay K 8644 with normal and transformed pituitary cells. Am J Physiol. 1986;250:C95–102. doi: 10.1152/ajpcell.1986.250.1.C95. [DOI] [PubMed] [Google Scholar]
- Evans AR, Nicol GD, Vasko MR. Differential regulation of evoked peptide release by voltage-sensitive calcium channels in rat sensory neurons. Brain Res. 1996;712:265–273. doi: 10.1016/0006-8993(95)01447-0. [DOI] [PubMed] [Google Scholar]
- Fischer G, Kostic S, Nakai H, Park F, Sapunar D, Yu H, Hogan Q. Direct injection into the dorsal root ganglion: technical, behavioral, and histological observations. J Neurosci Methods. 2011;199:43–55. doi: 10.1016/j.jneumeth.2011.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forsythe ID, Tsujimoto T, Barnes-Davies M, Cuttle MF, Takahashi T. Inactivation of presynaptic calcium current contributes to synaptic depression at a fast central synapse. Neuron. 1998;20:797–807. doi: 10.1016/s0896-6273(00)81017-x. [DOI] [PubMed] [Google Scholar]
- Friel DD, Tsien RW. A caffeine- and ryanodine-sensitive Ca2+ store in bullfrog sympathetic neurones modulates effects of Ca2+ entry on [Ca2+]i. J Physiol. 1992;450:217–246. doi: 10.1113/jphysiol.1992.sp019125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuchs A, Rigaud M, Hogan QH. Painful nerve injury shortens the intracellular Ca2+ signal in axotomized sensory neurons of rats. Anesthesiology. 2007a;107:106–116. doi: 10.1097/01.anes.0000267538.72900.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuchs A, Rigaud M, Sarantopoulos CD, Filip P, Hogan QH. Contribution of calcium channel subtypes to the intracellular calcium signal in sensory neurons: the effect of injury. Anesthesiology. 2007b;107:117–127. doi: 10.1097/01.anes.0000267511.21864.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao L, Blair LA, Marshall J. CaMKII-independent effects of KN93 and its inactive analog KN92: reversible inhibition of L-type calcium channels. Biochem Biophys Res Commun. 2006;345:1606–1610. doi: 10.1016/j.bbrc.2006.05.066. [DOI] [PubMed] [Google Scholar]
- Gemes G, Rigaud M, Weyker PD, Abram SE, Weihrauch D, Poroli M, Zoga V, Hogan QH. Depletion of calcium stores in injured sensory neurons: anatomic and functional correlates. Anesthesiology. 2009;111:393–405. doi: 10.1097/ALN.0b013e3181ae63b0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gemes G, Rigaud M, Koopmeiners AS, Poroli MJ, Zoga V, Hogan QH. Calcium signaling in intact dorsal root ganglia: new observations and the effect of injury. Anesthesiology. 2010;113:134–146. doi: 10.1097/ALN.0b013e3181e0ef3f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gold MS. Spinal nerve ligation: what to blame for the pain and why. Pain. 2000;84:117–120. doi: 10.1016/s0304-3959(99)00309-7. [DOI] [PubMed] [Google Scholar]
- Grimm D, Zhou S, Nakai H, Thomas CE, Storm TA, Fuess S, Matsushita T, Allen J, Surosky R, Lochrie M, Meuse L, McClelland A, Colosi P, Kay MA. Preclinical in vivo evaluation of pseudotyped adeno-associated virus vectors for liver gene therapy. Blood. 2003;102:2412–2419. doi: 10.1182/blood-2003-02-0495. [DOI] [PubMed] [Google Scholar]
- Grueter CE, Abiria SA, Dzhura I, Wu Y, Ham AJ, Mohler PJ, Anderson ME, Colbran RJ. L-type Ca2+ channel facilitation mediated by phosphorylation of the beta subunit by CaMKII. Mol Cell. 2006;23:641–650. doi: 10.1016/j.molcel.2006.07.006. [DOI] [PubMed] [Google Scholar]
- Hadley RW, Lederer WJ. Ca2+ and voltage inactivate Ca2+ channels in guinea-pig ventricular myocytes through independent mechanisms. J Physiol. 1991;444:257–268. doi: 10.1113/jphysiol.1991.sp018876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall KE, Browning MD, Dudek EM, Macdonald RL. Enhancement of high threshold calcium currents in rat primary afferent neurons by constitutively active protein kinase C. J Neurosci. 1995;15:6069–6076. doi: 10.1523/JNEUROSCI.15-09-06069.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinke B, Balzer E, Sandkühler J. Pre- and postsynaptic contributions of voltage-dependent Ca2+ channels to nociceptive transmission in rat spinal lamina I neurons. Eur J Neurosci. 2004;19:103–111. doi: 10.1046/j.1460-9568.2003.03083.x. [DOI] [PubMed] [Google Scholar]
- Hell JW, Appleyard SM, Yokoyama CT, Warner C, Catterall WA. Differential phosphorylation of two size forms of the N-type calcium channel alpha 1 subunit which have different COOH termini. J Biol Chem. 1994;269:7390–7396. [PubMed] [Google Scholar]
- Hogan QH, McCallum JB, Sarantopoulos C, Aason M, Mynlieff M, Kwok WM, Bosnjak ZJ. Painful neuropathy decreases membrane calcium current in mammalian primary afferent neurons. Pain. 2000;86:43–53. doi: 10.1016/s0304-3959(99)00313-9. [DOI] [PubMed] [Google Scholar]
- Hogan Q, Sapunar D, Modric-Jednacak K, McCallum JB. Detection of neuropathic pain in a rat model of peripheral nerve injury. Anesthesiology. 2004;101:476–487. doi: 10.1097/00000542-200408000-00030. [DOI] [PubMed] [Google Scholar]
- Hogan Q, Lirk P, Poroli M, Rigaud M, Fuchs A, Fillip P, Ljubkovic M, Gemes G, Sapunar D. Restoration of calcium influx corrects membrane hyperexcitability in injured rat dorsal root ganglion neurons. Anesth Analg. 2008;107:1045–1051. doi: 10.1213/ane.0b013e31817bd1f0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudmon A, Schulman H, Kim J, Maltez JM, Tsien RW, Pitt GS. CaMKII tethers to L-type Ca2+ channels, establishing a local and dedicated integrator of Ca2+ signals for facilitation. J Cell Biol. 2005;171:537–547. doi: 10.1083/jcb.200505155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda SR. Double-pulse calcium channel current facilitation in adult rat sympathetic neurones. J Physiol. 1991;439:181–214. doi: 10.1113/jphysiol.1991.sp018663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang X, Lautermilch NJ, Watari H, Westenbroek RE, Scheuer T, Catterall WA. Modulation of CaV2.1 channels by Ca2+/calmodulin-dependent protein kinase II bound to the C-terminal domain. Proc Natl Acad Sci U S A. 2008;105:341–346. doi: 10.1073/pnas.0710213105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kavalali ET, Plummer MR. Selective potentiation of a novel calcium channel in rat hippocampal neurones. J Physiol. 1994;480:475–484. doi: 10.1113/jphysiol.1994.sp020376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawano T, Zoga V, Gemes G, McCallum JB, Wu HE, Pravdic D, Liang MY, Kwok WM, Hogan Q, Sarantopoulos C. Suppressed Ca2+/CaM/CaMKII-dependent K(ATP) channel activity in primary afferent neurons mediates hyperalgesia after axotomy. Proc Natl Acad Sci U S A. 2009;106:8725–8730. doi: 10.1073/pnas.0901815106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SH, Chung JM. An experimental model for peripheral neuropathy produced by segmental spinal nerve ligation in the rat. Pain. 1992;50:355–363. doi: 10.1016/0304-3959(92)90041-9. [DOI] [PubMed] [Google Scholar]
- Kojundzic SL, Puljak L, Hogan Q, Sapunar D. Depression of Ca(2+)/calmodulin-dependent protein kinase II in dorsal root ganglion neurons after spinal nerve ligation. J Comp Neurol. 2010;518:64–74. doi: 10.1002/cne.22209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latremoliere A, Woolf CJ. Central sensitization: a generator of pain hypersensitivity by central neural plasticity. J Pain. 2009;10:895–926. doi: 10.1016/j.jpain.2009.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ledoux J, Chartier D, Leblanc N. Inhibitors of calmodulin-dependent protein kinase are nonspecific blockers of voltage-dependent K+ channels in vascular myocytes. J Pharmacol Exp Ther. 1999;290:1165–1174. [PubMed] [Google Scholar]
- Lee A, Wong ST, Gallagher D, Li B, Storm DR, Scheuer T, Catterall WA. Ca2+/calmodulin binds to and modulates P/Q-type calcium channels. Nature. 1999;399:155–159. doi: 10.1038/20194. [DOI] [PubMed] [Google Scholar]
- Lee A, Scheuer T, Catterall WA. Ca2+/calmodulin-dependent facilitation and inactivation of P/Q-type Ca2+ channels. J Neurosci. 2000;20:6830–6838. doi: 10.1523/JNEUROSCI.20-18-06830.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SJ, Escobedo-Lozoya Y, Szatmari EM, Yasuda R. Activation of CaMKII in single dendritic spines during long-term potentiation. Nature. 2009;458:299–304. doi: 10.1038/nature07842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang H, DeMaria CD, Erickson MG, Mori MX, Alseikhan BA, Yue DT. Unified mechanisms of Ca2+ regulation across the Ca2+ channel family. Neuron. 2003;39:951–960. doi: 10.1016/s0896-6273(03)00560-9. [DOI] [PubMed] [Google Scholar]
- Lirk P, Poroli M, Rigaud M, Fuchs A, Fillip P, Huang CY, Ljubkovic M, Sapunar D, Hogan Q. Modulators of calcium influx regulate membrane excitability in rat dorsal root ganglion neurons. Anesth Analg. 2008;107:673–685. doi: 10.1213/ane.0b013e31817b7a73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lüscher C, Streit J, Lipp P, Lüscher HR. Action potential propagation through embryonic dorsal root ganglion cells in culture. II. Decrease of conduction reliability during repetitive stimulation. J Neurophysiol. 1994;72:634–643. doi: 10.1152/jn.1994.72.2.634. [DOI] [PubMed] [Google Scholar]
- Lüscher C, Lipp P, Lüscher HR, Niggli E. Control of action potential propagation by intracellular Ca2+ in cultured rat dorsal root ganglion cells. J Physiol. 1996;490:319–324. doi: 10.1113/jphysiol.1996.sp021146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma C, Shu Y, Zheng Z, Chen Y, Yao H, Greenquist KW, White FA, LaMotte RH. Similar electrophysiological changes in axotomized and neighboring intact dorsal root ganglion neurons. J Neurophysiol. 2003;89:1588–1602. doi: 10.1152/jn.00855.2002. [DOI] [PubMed] [Google Scholar]
- Matthews EA, Dickenson AH. Effects of spinally delivered N- and P-type voltage-dependent calcium channel antagonists on dorsal horn neuronal responses in a rat model of neuropathy. Pain. 2001;92:235–246. doi: 10.1016/s0304-3959(01)00255-x. [DOI] [PubMed] [Google Scholar]
- McCallum JB, Kwok WM, Sapunar D, Fuchs A, Hogan QH. Painful peripheral nerve injury decreases calcium current in axotomized sensory neurons. Anesthesiology. 2006;105:160–168. doi: 10.1097/00000542-200607000-00026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCallum JB, Wu HE, Tang Q, Kwok WM, Hogan QH. Subtype-specific reduction of voltage-gated calcium current in medium-sized dorsal root ganglion neurons after painful peripheral nerve injury. Neuroscience. 2011;179:244–255. doi: 10.1016/j.neuroscience.2011.01.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meuth S, Pape HC, Budde T. Modulation of Ca2+ currents in rat thalamocortical relay neurons by activity and phosphorylation. Eur J Neurosci. 2002;15:1603–1614. doi: 10.1046/j.1460-9568.2002.01999.x. [DOI] [PubMed] [Google Scholar]
- Mironov SL, Juri MU. Sr and Ba transients in isolated snail neurones studied with fura-2. The recovery from depolarization induced load and modulation of Ca release from intracellular stores. Neurosci Lett. 1990;112:184–189. doi: 10.1016/0304-3940(90)90200-s. [DOI] [PubMed] [Google Scholar]
- Morel N, Buryi V, Feron O, Gomez JP, Christen MO, Godfraind T. The action of calcium channel blockers on recombinant L-type calcium channel alpha1-subunits. Br J Pharmacol. 1998;125:1005–1012. doi: 10.1038/sj.bjp.0702162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nayak AS, Moore CI, Browning MD. Ca2+/calmodulin-dependent protein kinase II phosphorylation of the presynaptic protein synapsin I is persistently increased during long-term potentiation. Proc Natl Acad Sci U S A. 1996;93:15451–15456. doi: 10.1073/pnas.93.26.15451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patil PG, Brody DL, Yue DT. Preferential closed-state inactivation of neuronal calcium channels. Neuron. 1998;20:1027–1038. doi: 10.1016/s0896-6273(00)80483-3. [DOI] [PubMed] [Google Scholar]
- Pitt GS, Zühlke RD, Hudmon A, Schulman H, Reuter H, Tsien RW. Molecular basis of calmodulin tethering and Ca2+-dependent inactivation of L-type Ca2+ channels. J Biol Chem. 2001;276:30794–30802. doi: 10.1074/jbc.M104959200. [DOI] [PubMed] [Google Scholar]
- Raman IM, Bean BP. Ionic currents underlying spontaneous action potentials in isolated cerebellar Purkinje neurons. J Neurosci. 1999;19:1663–1674. doi: 10.1523/JNEUROSCI.19-05-01663.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Randall A, Tsien RW. Pharmacological dissection of multiple types of Ca2+ channel currents in rat cerebellar granule neurons. J Neurosci. 1995;15:2995–3012. doi: 10.1523/JNEUROSCI.15-04-02995.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rycroft BK, Vikman KS, Christie MJ. Inflammation reduces the contribution of N-type calcium channels to primary afferent synaptic transmission onto NK1 receptor-positive lamina I neurons in the rat dorsal horn. J Physiol. 2007;580:883–894. doi: 10.1111/j.1469-7793.2000.t01-1-02117.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saitoh T, Schwartz JH. Phosphorylation-dependent subcellular translocation of a Ca2+/calmodulin-dependent protein kinase produces an autonomous enzyme in Aplysia neurons. J Cell Biol. 1985;100:835–842. doi: 10.1083/jcb.100.3.835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakaba T, Neher E. Quantitative relationship between transmitter release and calcium current at the calyx of held synapse. J Neurosci. 2001;21:462–476. doi: 10.1523/JNEUROSCI.21-02-00462.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sapunar D, Ljubkovic M, Lirk P, McCallum JB, Hogan QH. Distinct membrane effects of spinal nerve ligation on injured and adjacent dorsal root ganglion neurons in rats. Anesthesiology. 2005;103:360–376. doi: 10.1097/00000542-200508000-00020. [DOI] [PubMed] [Google Scholar]
- Sarantopoulos CD, McCallum JB, Rigaud M, Fuchs A, Kwok WM, Hogan QH. Opposing effects of spinal nerve ligation on calcium-activated potassium currents in axotomized and adjacent mammalian primary afferent neurons. Brain Res. 2007;1132:84–99. doi: 10.1016/j.brainres.2006.11.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholz A, Gruss M, Vogel W. Properties and functions of calcium-activated K+ channels in small neurones of rat dorsal root ganglion studied in a thin slice preparation. J Physiol. 1998;513:55–69. doi: 10.1111/j.1469-7793.1998.055by.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scroggs RS, Fox AP. Calcium current variation between acutely isolated adult rat dorsal root ganglion neurons of different size. J Physiol. 1992;445:639–658. doi: 10.1113/jphysiol.1992.sp018944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shakiryanova D, Morimoto T, Zhou C, Chouhan AK, Sigrist SJ, Nose A, Macleod GT, Deitcher DL, Levitan ES. Differential control of presynaptic CaMKII activation and translocation to active zones. J Neurosci. 2011;31:9093–9100. doi: 10.1523/JNEUROSCI.0550-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoney SD., Jr Limitations on impulse conduction at the branch point of afferent axons in frog dorsal root ganglion. Exp Brain Res. 1990;80:512–524. doi: 10.1007/BF00227992. [DOI] [PubMed] [Google Scholar]
- Swartz KJ. Modulation of Ca2+ channels by protein kinase C in rat central and peripheral neurons: disruption of G protein-mediated inhibition. Neuron. 1993;11:305–320. doi: 10.1016/0896-6273(93)90186-u. [DOI] [PubMed] [Google Scholar]
- Swensen AM, Bean BP. Ionic mechanisms of burst firing in dissociated Purkinje neurons. J Neurosci. 2003;23:9650–9663. doi: 10.1523/JNEUROSCI.23-29-09650.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward L, Wright E, McMahon SB. A comparison of the effects of noxious and innocuous counterstimuli on experimentally induced itch and pain. Pain. 1996;64:129–138. doi: 10.1016/0304-3959(95)00080-1. [DOI] [PubMed] [Google Scholar]
- Wu HE, Gemes G, Zoga V, Kawano T, Hogan QH. Learned avoidance from noxious mechanical simulation but not threshold semmes weinstein filament stimulation after nerve injury in rats. J Pain. 2010;11:280–286. doi: 10.1016/j.jpain.2009.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao RP, Cheng H, Lederer WJ, Suzuki T, Lakatta EG. Dual regulation of Ca2+/calmodulin-dependent kinase II activity by membrane voltage and by calcium influx. Proc Natl Acad Sci U S A. 1994;91:9659–9663. doi: 10.1073/pnas.91.20.9659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Wu LG. The decrease in the presynaptic calcium current is a major cause of short-term depression at a calyx-type synapse. Neuron. 2005;46:633–645. doi: 10.1016/j.neuron.2005.03.024. [DOI] [PubMed] [Google Scholar]
- Yokoyama CT, Sheng ZH, Catterall WA. Phosphorylation of the synaptic protein interaction site on N-type calcium channels inhibits interactions with SNARE proteins. J Neurosci. 1997;17:6929–6938. doi: 10.1523/JNEUROSCI.17-18-06929.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokoyama CT, Myers SJ, Fu J, Mockus SM, Scheuer T, Catterall WA. Mechanism of SNARE protein binding and regulation of Cav2 channels by phosphorylation of the synaptic protein interaction site. Mol Cell Neurosci. 2005;28:1–17. doi: 10.1016/j.mcn.2004.08.019. [DOI] [PubMed] [Google Scholar]
- Yuan W, Bers DM. Ca-dependent facilitation of cardiac Ca current is due to Ca-calmodulin-dependent protein kinase. Am J Physiol. 1994;267:H982–H993. doi: 10.1152/ajpheart.1994.267.3.H982. [DOI] [PubMed] [Google Scholar]
- Zamponi GW, Snutch TP. Decay of prepulse facilitation of N type calcium channels during G protein inhibition is consistent with binding of a single Gbeta subunit. Proc Natl Acad Sci U S A. 1998;95:4035–4039. doi: 10.1073/pnas.95.7.4035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zha XM, Dailey ME, Green SH. Role of Ca2+/calmodulin-dependent protein kinase II in dendritic spine remodeling during epileptiform activity in vitro. J Neurosci Res. 2009;87:1969–1979. doi: 10.1002/jnr.22033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zühlke RD, Pitt GS, Deisseroth K, Tsien RW, Reuter H. Calmodulin supports both inactivation and facilitation of L-type calcium channels. Nature. 1999;399:159–162. doi: 10.1038/20200. [DOI] [PubMed] [Google Scholar]
- Zühlke RD, Pitt GS, Tsien RW, Reuter H. Ca2+-sensitive inactivation and facilitation of L-type Ca2+ channels both depend on specific amino acid residues in a consensus calmodulin-binding motif in the(alpha)1C subunit. J Biol Chem. 2000;275:21121–21129. doi: 10.1074/jbc.M002986200. [DOI] [PubMed] [Google Scholar]