

Abstract
An asymmetric intermolecular reaction between enals and nitroalkenes to yield δ-nitroesters has been developed, catalyzed by a novel chiral N-heterocyclic carbene. Key to this work was the development of a catalyst that favors the δ-nitroester pathway over the established Stetter pathway. The reaction proceeds in high stereoselectivity and affords the previously unreported syn diastereomer. We also report an operationally facile two-step, one-pot procedure for the synthesis of δ-lactams.
Recently, N-heterocyclic carbenes (NHC) have been shown to be powerful catalysts for a variety of useful transformations.1 NHC catalysis traditionally operates via the acyl anion equivalent (Figure 1A) and this manifold has received a great deal of attention. When an enal is employed, both the acyl anion and homoenolate pathways (Figure 1B) become accessible.2 In 2004, Bode and Glorius independently reported the first NHC generated homoenolate in their annulations between enals and aldehydes to synthesize γ-lactones.3 Since those initial reports the application of the NHC homoenolate has grown tremendously. A variety of annulations have been developed to synthesize lactones, lactams, and carbocycles.4 Despite these advances, challenges in stereoinduction and differentiation between the acyl anion and homoenolate pathways remain. Stereoinduction is challenging due to the distal relationship between the NHC and the β-carbon. Differentiation between the acyl anion and homoenolate is problematic, as the majority of electrophiles used in homoenolate pathways have also been shown competent acceptors in the acyl anion pathway.
Figure 1.

Background
Nitroalkenes represent attractive electophiles for the homoenolate; the δ-nitroesters produced in this reaction are useful synthons for δ-lactams and piperidines, common motifs in drug targets and natural products.5 In 2009, Nair reported the NHC-catalyzed homoenolate reaction between enals and nitroalkenes. With the use of an achiral imidazolium precatalyst, aromatic enals and nitrostyrene derivatives were coupled in good yield and good anti selectivity.6 Recently, Liu and coworkers rendered the reaction asymmetric by employing an aminoindanol-based triazolium precatalyst. Liu’s work showed a variety of enals, both aromatic and aliphatic, to be competent coupling partners for aromatic nitrodienes, nitroenynes, and nitrostryenes in excellent ee and good anti diastereoselectivity (Figure 1).7 Herein we report our own concurrent studies on this reaction that delivers complimentary stereoselectivity and allows coupling with aliphatic nitroalkenes.
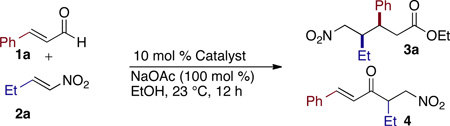
During the course of our previous investigations into the asymmetric intermolecular Stetter reaction, we observed homoenolate addition to nitroalkenes as a side product8 Intrigued by this reactivity, we began our investigation with the addition of cinnamaldehyde 1a to (E)-1-nitrobut-1-ene 2a. Our exploration of chiral catalysts began with aminoindanol-derived 5a9 (Table 1) and found it provides desired product with excellent syn diastereoselectivity (17:1), but yield and ee were unsatisfactory (25 % yield, 40 % ee). The selective formation of the syn diastereomer was unexpected in light of Nair6 and Liu’s7 previous reports which delivered the anti diastereomer. The incorporation of an aliphatic nitroalkene in this reaction is also noteworthy as only activated and aryl nitroalkenes have been previously shown. We continued our investigation by employing fluorinated pre-catalyst 5b8a and were pleased to observe product in 42 % yield, 83 % ee, and in excellent diastereoselectivity (17:1). Unfortunately this system provides no preference for the homoenolate pathway and a 1:1 mixture of the Stetter product 4 and desired nitroester 3a is formed. Efforts to improve enantioselectivity with 5b via reaction optimization proved fruitless, and yield of 3a never surpassed 50 %. With these results in hand, we chose to focus on developing conditions to promote the homoenolate pathway over the Stetter pathway.
Table1.
Chiral Catalyst Screena
 | |||||
|---|---|---|---|---|---|
| entry | Catalyst | 3a:4 | Yield 3a (%)b | dr (syn/anti)c | ee (%)d |
| 1 | 5a | 2:1 | 25 | 17:1 | −40 |
| 2 | 5b | 1:1 | 42 | 17:1 | −83 |
| 3 | 5c | 5:1 | 70 | 2:1 | −22 |
| 4 | 5d | 6:1 | 60 | 15:1 | −30 |
| 5 | 5e | >20:1 | 16 | 17:1 | 95 |
| 6 | 5f | >20:1 | 49 | 17:1 | 93 |
Reactions were conducted with 1.5 equiv of 1 and 1.0 equiv of 2.
Isolated yield after chromatography.
Diastereoselectivity determined by 1H NMR of the unpurified reaction mixture.
Enantiomeric excess was determined by HPLC analysis on a chiral stationary phase.
We hypothesized that bulky substituents in the ortho/ortho’ position of the N-aryl ring of the NHC would shift product distribution towards desired nitroester by partially blocking the acyl anion position. Precatalyst 5c was synthesized and a considerable increase in product selectivity (5:1 3a:4, 70 % yield) was observed, but enantioselectivity and diastereoselectivity suffered considerably (2:1 dr, 30 % ee). Bis-trifluoromethyl triazolium 5d showed good selectivity for the nitroester (6:1 3a:4, 60 % yield) and diastereoselectivity was excellent (15:1) but enantioselectivity was low (22 % ee).
Given the partial success with larger substituents on the aryl side, we hypothesized that an increase in sterics on the alkyl side of the azolium may deliver similar levels of selectivity favoring the homoenolate product with the potential for improved stereoselectivity. With that in mind, we examined Ye’s bis-phenyl O-TMS triazolium precatalyst 5e which provided excellent selectivity for the nitroester (>20:1 3a:4, 17 % yield) and excellent stereoselectivity (17:1 dr, 95 % ee).10 The remainder of the mass balance with catalyst 5e was enal, suggesting the catalyst was either prohibitively slow or was undergoing decomposition. We postulated that replacing the bis-phenyl moiety with an aliphatic group may lead to a more efficient catalyst. In pursuit of this belief we synthesized 5f and were pleased to observe a substantial increase in yield while product selectivity (>20:1 3a:4, 49 % yield) and stereoselectivity remained excellent (17:1 dr, 93 % ee). A brief screen of bases and base equivalents led us to our optimized conditions of 50 mol % sodium acetate (see SI).
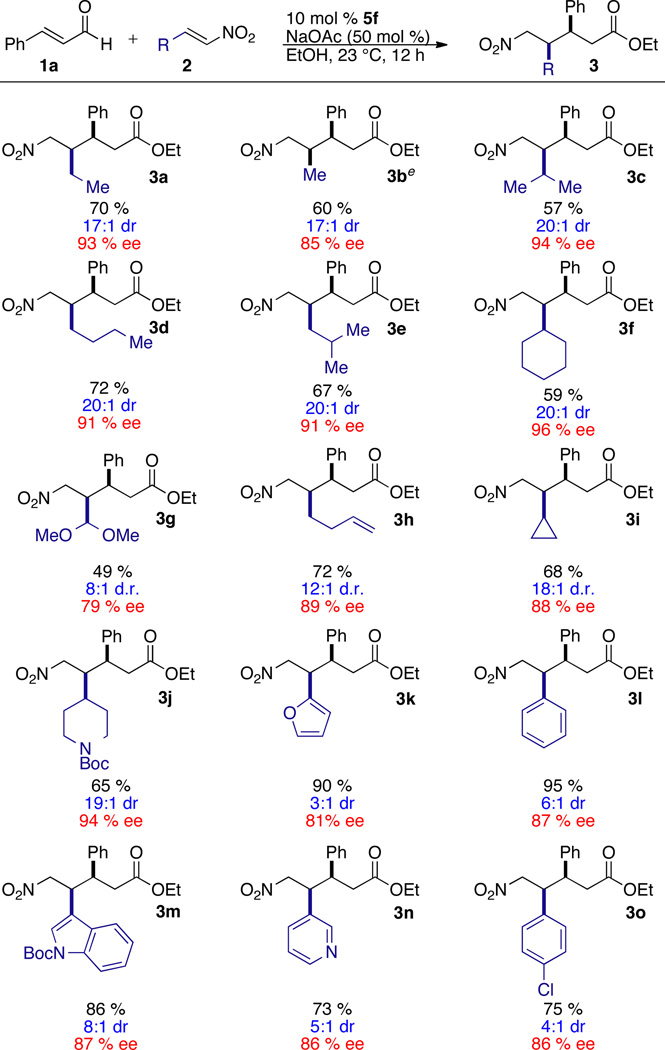
A variety of aliphatic and aryl nitroalkenes are competent in the reaction, Table 2. Aliphatic nitroalkenes typically provide product in excellent stereoselectivity, albeit with moderate yield (3a–3j). Sterically bulky nitroalkenes such as iso-propyl are tolerated but t-butyl does not participate (not shown). Notably, acetal 3g and terminal olefin 3h are formed with useful selectivities, and provide valuable handles for further manipulation. Enantio- and diastereoselectivities are typically lower for aryl nitroalkenes (3k–3o), but the reactions proceed in good yield. Heteroaromatic nitroalkenes participate as well (3k, 3m, 3n).
Table 2.
Nitroalkene Scope a–d
 |
See Table 1.
2.5 equiv. aldehyde 1a used.
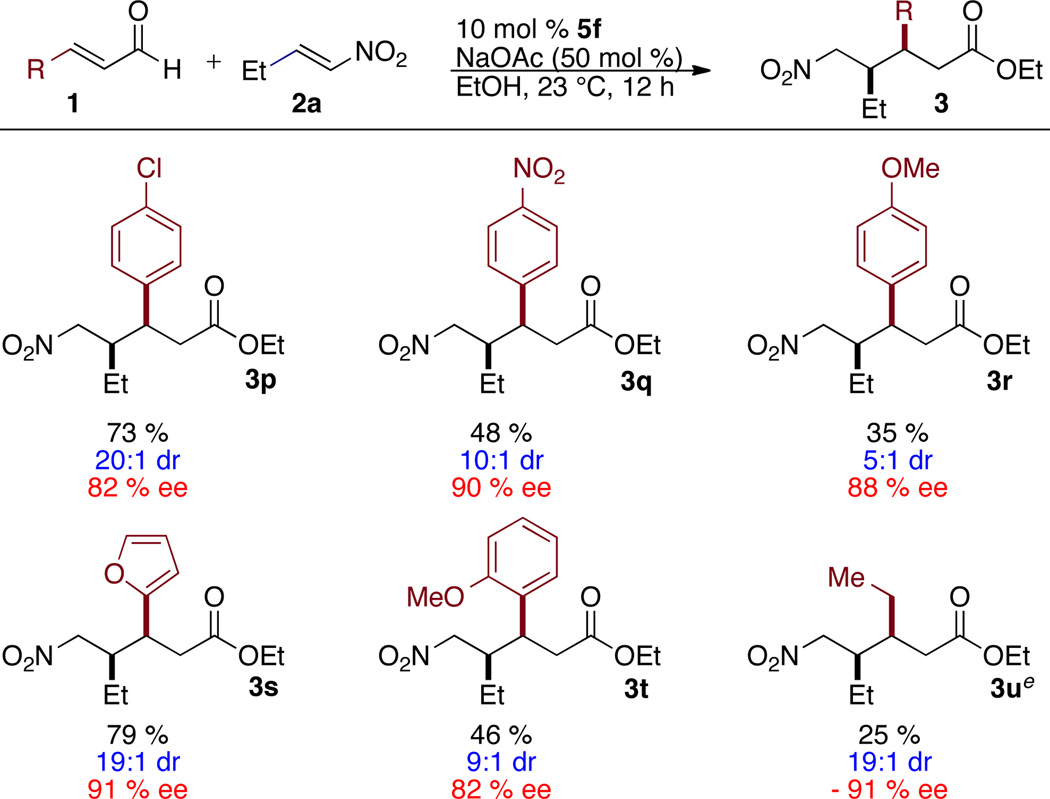
Electron withdrawing and electron rich aryl enals provide product in modest to good yield, Table 3. In the case of p-nitrocinnamaldehyde, the reaction delivers a modest yield of 3q, with the remainder of the aldehyde starting material converted to p-nitrodihydroethylcinnamate. In this highly electron withdrawing system, protonation of the homoenolate out-competes addition to the nitroalkene.11 Use of trans-2-pentenal in this reaction with catalyst 5f yields only the Stetter product This is unusual as it is the only substrate that gives Stetter product in greater than trace amounts. However, the use of fluorinated catalyst 5b at 50 °C delivers the desired product in 25% yield with the remainder of the mass balance the Stetter product 4.
Table 3.
Enal Scopea–d
 |
See Table 1.
Catalyst 5b was used and the reaction was run at 50 °C.
Due to the orthogonality of the two ends of products 3, we were motivated to explore manipulation of the nitro-ester product to deliver valuable synthons. We found we could generate the corresponding δ-lactams12 in good yield via a one-pot two-step process. Addition of zinc dust and acetic acid to the crude reaction mixture after 12 hours, followed by heating for an additional 4 hours provides an operationally simple protocol for the one-pot synthesis of δ-lactams.13 The δ-lactam product may be further reduced to the piperidine via the action of LiAlH4 (Scheme 1).14
Scheme 1.

Lactam Reduction
In conclusion we have developed a highly effective catalytic system for the asymmetric and diastereoselective generation of a diverse array of syn δ-nitro-esters. Key to our success was the development of catalyst 5f, which is highly selective for the homoenolate pathway over the established acyl anion (Stetter) pathway. Our system also allows the previously unreported coupling with aliphatic nitroalkenes and provides access to the syn diastereomer. We also report the operationally facile one-pot two-step reaction sequence to arrive at synthetically useful δ-lactams.
Supplementary Material
Table 4.
One-Pot Synthesis of δ-Lactamsa–d
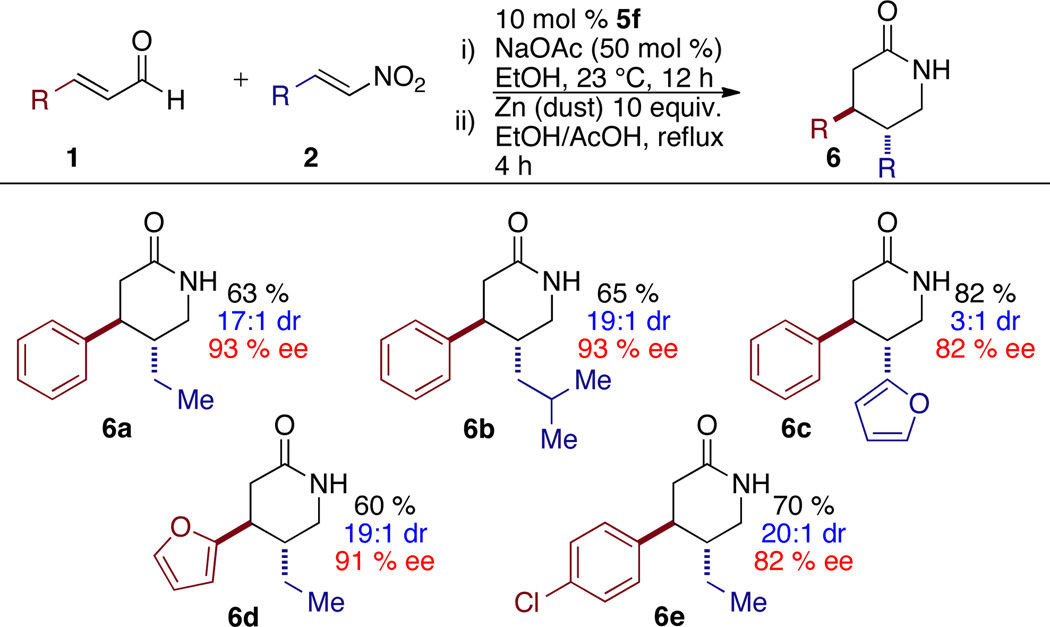 |
See Table 1.
ACKNOWLEDGMENT
This paper is dedicated to Professor Teruaki Mukaiyama in celebration of the 40th anniversary of the Mukaiyama aldol reaction. We thank NIGMS for support (GM72586). TR thanks Amgen and Roche for unrestricted support. We thank Derek M. Dalton (CSU) for solving the structure of S3.
Footnotes
ASSOCIATED CONTENT
Supporting Information: Full experimental details, spectroscopic data for all new compounds, and crystallographic data (CIF). This material is available free of charge via the Internet at http:// pubs.acs.org.
REFERENCES
- 1.(a) Moore JL, Rovis T. Top. Curr. Chem. 2010;291:77. doi: 10.1007/978-3-642-02815-1_18. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Vora HU, Rovis T. Aldrichimica Acta. 2011;44:3. [PMC free article] [PubMed] [Google Scholar]; (c) Bugaut X, Glorius F. Chem. Soc. Rev. 2012;41:3511–3522. doi: 10.1039/c2cs15333e. [DOI] [PubMed] [Google Scholar]
- 2.(a) Nair V, Vellalath S, Babu BP. Chem. Soc. Rev. 2008;37:2691–2698. doi: 10.1039/b719083m. [DOI] [PubMed] [Google Scholar]; (b) Nair V, Menon RS, Biju AT, Sinu CR, Paul RR, Jose A, Sreekumar V. Chem. Soc. Rev. 2011;40:5336–5346. doi: 10.1039/c1cs15139h. [DOI] [PubMed] [Google Scholar]; (c) Chiang P-C, Bode JW. TCI MAIL. 2011;149:2–17. [Google Scholar]; (d) Cohen DT, Scheidt KA. Chem. Sci. 2012;3:53–57. doi: 10.1039/C1SC00621E. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Vora HU, Wheeler P, Rovis T. Adv. Synth. Catal. 2012;354:1617–1639. doi: 10.1002/adsc.201200031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.(a) Sohn SS, Rosen EL, Bode JW. J. Am. Chem. Soc. 2004;126:14370–14371. doi: 10.1021/ja044714b. [DOI] [PubMed] [Google Scholar]; (b) Burstein C, Glorius F. Angew. Chem. Int. Ed. 2004;43:6205–6208. doi: 10.1002/anie.200461572. [DOI] [PubMed] [Google Scholar]
- 4.(a) He M, Bode JW. Org. Lett. 2005;7:3131–3134. doi: 10.1021/ol051234w. [DOI] [PubMed] [Google Scholar]; (b) Rommel M, Fukuzumi T, Bode JW. J. Am. Chem. Soc. 2008;130:17266–17267. doi: 10.1021/ja807937m. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Raup DEA, Cardinal-David B, Holte D, Scheidt KA. Nat Chem. 2010;2:766–771. doi: 10.1038/nchem.727. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Zhao X, DiRocco DA, Rovis TJ. Am. Chem. Soc. 2011;133:12466–12469. doi: 10.1021/ja205714g. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Nair V, Vellalath S, Poonoth M, Suresh E. J. Am. Chem. Soc. 2006;128:8736–8737. doi: 10.1021/ja0625677. [DOI] [PubMed] [Google Scholar]; (f) Chiang PC, Kaeobamrung J, Bode JWJ. Am. Chem. Soc. 2007;129:3520–3521. doi: 10.1021/ja0705543. [DOI] [PubMed] [Google Scholar]; (g) Cardinal-David B, Raup DEA, Scheidt KA. J. Am. Chem. Soc. 2010:5345–5347. doi: 10.1021/ja910666n. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Nair V, Poonoth M, Vellalath S, Suresh E, Thirumalai R. J. Org. Chem. 2006;71:8964–8965. doi: 10.1021/jo0615706. [DOI] [PubMed] [Google Scholar]; (i) Phillips EM, Reynolds TE, Scheidt KA. J. Am. Chem. Soc. 2008;130:2416–2417. doi: 10.1021/ja710521m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.(a) Kathiravan MK, Salake AB, Chothe AS, Dudhe PB, Watode RP, Mukta MS, Gadhwe S. Bioorg. Med. Chem. 2012;20:5678–5698. doi: 10.1016/j.bmc.2012.04.045. [DOI] [PubMed] [Google Scholar]; (b) Khadem S, Maries RJ. Molecules. 2012;17:191–206. doi: 10.3390/molecules17010191. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Fattorusso E, Taglialatela-Scafati O, editors. Modern Alkaloids: Structure, Isolation, Synthesis and Biology. Weinheim, Germany: Wiley-VCH; 2008. [Google Scholar]; (d) Strunz GM, Findlay JA. In: The Alkaloids. Brossi A, editor. Vol. 26. New York: Academic Press; 1985. p. 89. [Google Scholar]; (e) O’Hagan D. Nat Prod. Rep. 2000;17:435. doi: 10.1039/a707613d. [DOI] [PubMed] [Google Scholar]; (f) Michael JP. Nat Prod. Rep. 2008;25:166–187. doi: 10.1039/b612168n. [DOI] [PubMed] [Google Scholar]
- 6.Nair V, Sinu CR, Babu BP, Varghese V, Jose A, Suresh E. Org. Lett. 2009;11:5570–5573. doi: 10.1021/ol901918x. [DOI] [PubMed] [Google Scholar]
- 7.Maji B, Ji L, Wang S, Vedachalam S, Rakesh G, Liu XW. Angew. Chem. Int. Ed. 2012;51:8276–8280. doi: 10.1002/anie.201203449. [DOI] [PubMed] [Google Scholar]
- 8. DiRocco DA, Oberg KM, Dalton DM, Rovis T. J. Am. Chem. Soc. 2009;131:10872–10874. doi: 10.1021/ja904375q. DiRocco DA, Rovis T. J. Am. Chem. Soc. 2011;133:10402–10405. doi: 10.1021/ja203810b. DiRocco DA, Noey EL, Houk KN, Rovis T. Angew. Chem. Int. Ed. 2012;51:2391–2394. doi: 10.1002/anie.201107597. (d) During review of this manuscript, Ye published an aza-benzoin between enals and ketimines; see: Sun LH, Liang ZQ, Jia WQ, Ye S. Angew. Chem. Int. Ed. 2013 doi: 10.1002/anie.201301304.
- 9.(a) Kerr MS, Read de Alaniz J, Rovis T. J. Am. Chem. Soc. 2002;124:10298–10299. doi: 10.1021/ja027411v. [DOI] [PubMed] [Google Scholar]; (b) Kerr MS, Rovis T. J. Am. Chem. Soc. 2004;226:8876–8877. doi: 10.1021/ja047644h. [DOI] [PubMed] [Google Scholar]
- 10.(a) Sun FG, Sun LH, Ye S. Adv. Synth. Catal. 2011;353:3134–3138. [Google Scholar]; (b) Enders D, Han J. Tetrahedron Asymmetry. 2008;19:1367–1371. [Google Scholar]; (c) He L, Lv H, Zhang YR, Ye S. J. Org. Chem. 2008;73:8101–8103. doi: 10.1021/jo801494f. [DOI] [PubMed] [Google Scholar]
- 11.(a) Chan A, Scheidt KA. Org. Lett. 2005;7:905. doi: 10.1021/ol050100f. [DOI] [PubMed] [Google Scholar]; (b) Sohn SS, Bode JW. Org. Lett. 2005;18:3873. doi: 10.1021/ol051269w. [DOI] [PubMed] [Google Scholar]
- 12.Bode has reported two complementary approaches for the direct asymmetric synthesis of δ-lactams (dihydropyridinones, specifically) via NHC catalysis from enals and unsaturated imines: He M, Struble JR, Bode JW. J. Am. Chem. Soc. 2006;128:8418–8420. doi: 10.1021/ja062707c. from enals and stable enamines: Wanner B, Mahatthananchai J, Bode JW. Org. Lett. 2011;13:5378–5381. doi: 10.1021/ol202272t.
- 13.Senkus M. Ind. Eng. Chem. 1948;40:506. [Google Scholar]
- 14.(a) Regan BM, Hayes FNF. J. Am. Chem. Soc. 1956;78:639–643. [Google Scholar]; (b) Hacksell U, Arvidsson LE, Svensson U, Nilsson JLG, Sanchez D, Wikstroem H, Lindberg P, Hjorth S, Carlsson A. J. Med. Chem. 1981;24:1475–1482. doi: 10.1021/jm00144a021. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.