

Preface
Collaboration is a cornerstone of many successful scientific research endeavors, which distributes risks and rewards to encourage progress in challenging areas. A striking illustration is the large-scale project that seeks to achieve a robust fundamental understanding of structure and function in the human G protein-coupled receptor superfamily. The GPCR Network was created to achieve this goal based on an active outreach program addressing an interdisciplinary community of scientists interested in GPCR structure, chemistry and biology.
Introduction
G protein-coupled receptors (GPCRs) are critical eukaryotic signal transduction gatekeepers, which have a common architecture of seven transmembrane helices and represent the largest protein family in the human proteome. There are more than 800 human GPCRs, which can be classified into five major classes and further divided into subfamilies based on sequence similarities (Figure 1). Located in the plasma membrane, GPCRs recognize an astonishing variety of widely different extracellular stimuli, including photons, ions, small molecules, peptides and proteins, and transmit the resulting extracellular signals 30 angstroms to elicit intracellular responses. Signal transmission occurs through coupling to different intracellular proteins (e.g., heterotrimeric G-proteins, arrestins and kinases)1, which then activate downstream effectors and trigger cascades of cellular and physiological responses. GPCR-mediated signaling pathways have been related to numerous human diseases, and GPCRs are the targets of an estimated 30–40% of all drugs currently on the market2. Consequently, understanding GPCR structure and function is of value to the basic science community interested in cell signaling and molecular recognition, as well as to the applied science community interested in drug discovery.
Figure 1.

Phylogenetic tree representation of the human GPCR superfamily constructed using sequence similarity within the seven-transmembrane region. Family members with determined structures are highlighted within the tree, and their binding pockets with the ligand, as captured in each of the distinct structures, are shown around the tree in the same orientation for ease of comparison. A2AAR (PDB code: 3EML), β1AR (2VT4), β2AR (2RH1), CXCR4 (3ODU), dopamine D3 (3PBL), δ-opioid (4EJ4), histamine H1 (3RZE), κ-opioid (4DJH), µ-opioid (4DKL), M2 muscarinic (3UON), M3 muscarinic (4DAJ), nociceptin/ophanin FQ peptide opioid (4EA3), rhodopsin (1GZM), sphingolipid S1P1 (3V2Y) receptors.
In this perspective we present a community-wide interdisciplinary infrastructure effort that was created to achieve a thorough understanding of GPCR structure–function relationships including, but not limited to, site specific mutagenesis of key residues and structure-activity relationships of each ligand-receptor structure determined. The receptors and their interactions are characterized using techniques of structural biology (X-ray and NMR), chemistry, biochemistry, biophysics and bioinformatics. An important element of the program is the active initiation of collaborations around the globe with fellow-scientists interested in specific GPCRs. Of key interest is access to an ever-widening range of ligands to support more detailed characterization of the rapidly expanding group of GPCRs that become accessible for structural biology.
The GPCR Network of PSI:Biology
The GPCR Network (http://gpcr.scripps.edu) was established as a collaborative effort funded by the U.S. National Institutes of Health Protein Structure Initiative (NIH/NIGMS PSI:Biology http://www.nigms.nih.gov/Research/FeaturedPrograms/PSI/psi_biology). The Center was established in 2010 with the goal to structurally characterize 15 to 25 representative GPCRs within a period of 5 years, with the vision to fully understand molecular recognition and signaling mediated by this membrane protein family. Full characterization includes that the receptors are studied in complexes with a wide range of different ligands, using x-ray crystallography, NMR and HDX. The arsenal of biophysical methods used will be extended to include, for example EPR experiments. In addition, computational methods of virtual ligand screening, conformational sampling of ligand-binding pockets and molecular dynamics simulations are used to explore an ever-widening ligand binding space. This work is then followed up by medicinal chemistry and tool compound development, whereby investigation of the biological significance of structural information is extensively conducted through collaborations with scientists who have long-standing individual interests in particular receptor systems. In the initial phase of the program, target selection is focused on GPCRs from different subfamilies with distant homology, to maximize the impact of each structure solved and expand with homology modeling. The 5-year goal, based on combination of experimentally solved structures and computationally predicted homology models of GPCRs, is to achieve 40% to 60% structural coverage of non-olfactory receptors (Figure 1). The 8 structures solved in the first two years of the GPCR Network cover about 80 modeled receptors when using a 35% sequence identity threshold for homology modeling (see below), which amounts to more than 20% structural coverage of non-olfactory receptors. Because of the potential scientific impact of peptide receptors, a few of these subfamilies, e.g., opioid, chemokine, class B and class C receptors, are tagged for more detailed coverage in order to experimentally characterize their structural variability and selectivity toward orthosteric and allosteric ligands. In the case of the class B (secretin) and C (glutamate) receptors, which have no representative structures determined to date, these receptors are being pursued for structural coverage by many different groups, including the GPCR Network. There do not seem to be any novel technical challenges to overcome for these subfamily members, except for the usual hurdle of finding the right ligand or ligands, which should in these classes of GPCRs stabilize the extracellular and transmembrane domains into a unique, compact conformation. Structures of class B and C receptors are anticipated within the next year or two.
GPCR Pipeline and Infrastructure
The initial process for determining GPCR structures was created through a combination of technologies developed via support from the NIH Common Fund in Structural Biology to the Joint Center for Innovative Membrane Protein Technologies (JCIMPT; http://jcimpt.scripps.edu), and by integration and optimization of technologies developed by other research groups. Efficient structure determination of GPCRs, which are reputably difficult to work with, required the development of a robust approach, both to increase the likelihood of success and to provide a platform amenable to optimization and cost reduction based on accumulated experience. The approach used in the initial successful structure determinations of β2-adrenergic receptor (β2AR) and A2A adenosine receptor (A2AAR)3, led to a pipeline with feedback loops (Box I and Figure 2). This pipeline is being constantly optimized, using a “family learning approach” similar to the one employed by the Structure Genomics Consortium (SGC) and successfully applied to other protein families, most notably protein kinases4. With the family learning approach, similar protocols and reagents (e.g., expression strategies, assays, inhibitors and co-factors) are re-used, and lessons learned for one family member can often be applied to other members, making it more cost effective for studying large protein families than traditional NIH R01 funding (single investigator with 2 to 3 students). Given the need for a funding mechanism for collaborative work on several different types of receptors enabling the family learning approach, the GPCR Network was established through the NIH’s U54 mechanism as part of the NIGMS Protein Structure Initiative (PSI:Biology). In parallel to the research efforts by scientists of the GPCR Network, a constant flow of scientists from around the world passes through GPCR Network laboratories. These visitors are trained in the use of technologies that have been developed as part of the JCIMPT program, and then return to their home institutions with new methods and tools, encouraging an expansion of the number of active researchers studying GPCRs.
BOX I. The GPCR Network Structure Determination Pipeline.
In order to go from the nomination of a specific GPCR to the final three-dimensional structure determination, we have developed a stage-gate process over the past 20 years that is now being refined to increase the efficacy and decrease the overall cost. This structure determination process relies on a set of family learning metrics through a pipeline with feedback loops (Figure 2). Once a structure is determined (step 1–6), follow up studies areinitiated to include complementary structure-function characterization (steps 7–9).
Gene Construct Design and Synthesis: An average of 75 constructs have been required to determine each structure to date. The cDNA for each of the target parent construct is codon-optimized and synthesized. Construct optimization includes placements of affinity tags, N- and C- termini truncations, and fusion protein considerations 48.
Expression Using Baculovirus Expression System: Receptor overexpression is carried out using a baculovirus expression system in insect cells at three different volume scales for rapid construct screening, ligand stability and biophysical characterization, and crystallization trials.
Purification: Solubilization in detergent/cholesterol mixtures is followed by metal affinity chromatography (IMAC)-based purification protocols.
Ligand Selection and Sample Characterization: Prior to any crystallization trials, ligand selection is carefully evaluated either during expression or at this stage. Purity, monodispersity, and ligand-binding affinity quality checks are followed by thermal stability analysis49, 50, and by receptor mobility in lipidic cubic phase (LCP) using Fluorescence Recovery After Photobleaching (LCP-FRAP)51.
Crystallization Studies: Crystallization trials are performed using the in meso (LCP)52 approaches. This crystallization approach was selected as the media most closely resembles the cell membrane and is important in aiding in the stabilization of detergent destabilized human GPCRs. Initial coarse screening is carried out using ∼400 conditions coupled with several host lipid matrices.
Data Collection, Processing, and Structure Determination: Diffraction data collection is done at synchrotron beamlines equipped with microbeam (e.g. GM/CA CAT at APS). Data collection, processing and structure solution are optimized based on experience of working with microcrystals53.
Structure Determination of GPCR–Ligand Complexes: For each receptor several receptor/ligand co-crystal structures are pursued to thoroughly map orthosteric and allosteric receptor binding sites.
Biophysical Characterization of GPCR–Ligand Complexes: Biophysical studies using NMR, and HDX are pursued to understand conformational equilibria and dynamic aspects of each receptor. The arsenal of techniques used will be further expanded, for example with EPR.
Computational Ligand Screening and Medicinal Chemistry: Ligand docking, virtual screening and homology modeling of close subtypes are conducted to further characterize and annotate each receptor structure. Medicinal chemistry is conducted through community outreach to yield an improved understanding of GPCR–ligand interactions.
Figure 2.

Process pipeline used by the GPCR Network to determine receptor structure and develop a deeper understanding of receptor dynamics and functional behavior. The stage-gate process relies on a set of metrics to advance target constructs for further processing. The bold black lines describe the process pathway; the gray lines are feedback loops where information at one stage can be used to repeat or adjust an earlier set of experiments. For example, if we find a construct is not stable enough or does not pass one of our pre-crystallization screens, we would return (gray lines) to construct design or search for more ligands that might help further stabilize the receptor.
Computational GPCR Studies
Experimental structure determination of all members of the human GPCR family, their complexes and associated multiple conformational states, however, is presently beyond practical possibilities. Therefore, an important activity of the GPCR Network is to leverage the impact of each experimental structure towards filling gaps in understanding the structures of related GPCRs, their specificity and other functional features. This goal is being achieved through comparative analysis, as well as homology-based modeling of structures and their complexes5, 6 (Figure 3), which has previously proven successful in applications to other major protein families7.
Figure 3.

The strategy of the GPCR Network includes leveraging each experimentally determined receptor structure (e.g. human β2AR; center) towards the understanding of GPCR family diversity and receptor selectivity for ligands (right side) and individual GPCR structure-function and conformational selectivity by ligands (left side). By creating a complete data package for each receptor that includes complementary biophysical, structural, functional, and ligand data, the receptors can be more thoroughly understood in contrast to just solving the structure with limited follow up.
To assess the state-of-the-art in computer homology modeling for prediction of ligand–GPCR interactions, community-wide assessments of GPCR modeling and docking (GPCR Dock 2008 and 2010) were organized (Figure 4). In 2008, the goal was to assess the ability of existing protocols and programs to predict the structure of A2AAR bound to the antagonist ZM241385 (ref 8). A few groups, which employed the closest available β2AR/β1AR templates as well as extensive knowledge of mutagenesis and structure-activity relationships for the adenosine receptor subfamily, were able to predict the overall ligand orientation and about 40% of the ligand–receptor atomic contacts. However, accuracy of even the best models in the assessment was far below that of crystal structures, with regard to ligand–receptor contacts as well as ligand positioning (Figure 4A)8. In 2010, a second community-wide assessment included modeling of the dopamine D3 (Figure 4B) and chemokine CXCR4 receptors (Figure 4C,D) in complex with antagonists9. Analysis of these blind modeling results (Figure 4) suggests that prediction of GPCR interactions with small molecule ligands can attain useful reliability when homology models are based on more than 35% sequence identity to a structural template in the transmembrane domain, as in the case of the dopamine D3 receptor9. We currently use the 35% sequence identity threshold somewhat arbitrarily as a working hypothesis, while being aware that this threshold may vary between different families (e.g. opioid receptors) and also depend on specific interactions of particular ligands. To engage a wider community in this discussion and encourage additional progress, the results of each assessment are freely available (http://gpcr.scripps.edu). This effort is aided by the observation that the ligand binding pockets of GPCRs are ideal for structure based drug discovery given the deep and well defined binding pockets.
Figure 4.
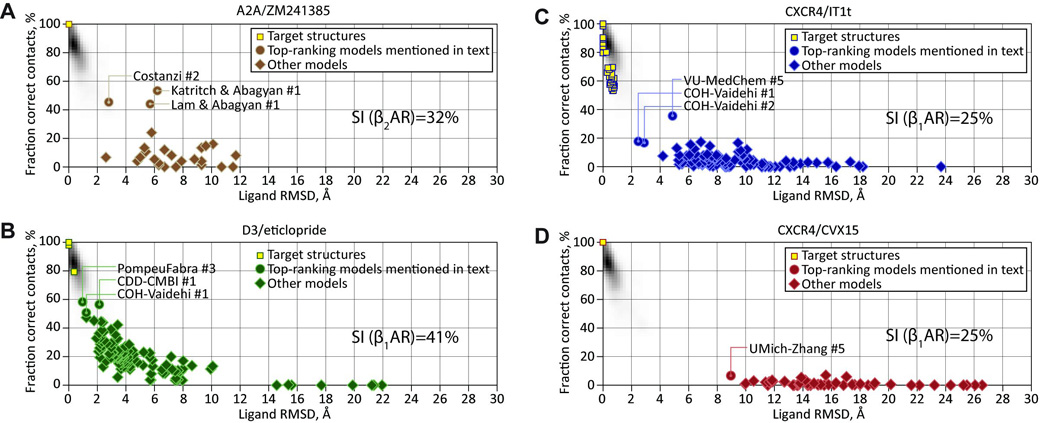
Community-wide prediction of GPCR-ligand docking and receptor interactions. In 2008 (A2AAR in complex with ZM241385) and 2010 (dopamine D3 in complex with eticlopride; CXCR4 chemokine in complex with IT1t and CVX15), two community wide assessments were conducted where the participants submitted prediction models of undisclosed GPCR ligand structures that were recently determined by the GPCR Network. The shaded plot background represents the distribution of the corresponding parameters for pairs of symmetry-related molecules in a subset of each PDB structure. (A) 2008 GPCR Dock8 with the assessment of the human A2AAR in complex with the antagonist ZM241385 highlighting limited success in receptor-ligand interactions. (B) 2010 GPCR Dock9 human dopamine D3 in complex with the antagonist eticlopride showing significant improvements in receptor-ligand docking, likely due to improved experimental models for the biogenic amine receptor subfamily. (C) 2010 GPCR Dock9 human CXCR4 receptor in complex with the small molecule IT1t. (D) 2010 GPCR Dock9 human CXCR4 receptor in complex with the peptide molecule CVX15. Panels A and B shows progress in the field based on available structural templates of related receptors, while panels C and D highlight the need for additional structural coverage needed for peptide receptors.
GPCR crystal structures also provide a robust 3D structural framework for computational modeling of receptor dynamics and oligomerization state10, as well as ligand docking and virtual ligand screening (VLS). The growing number of structure-based VLS studies demonstrate encouragingly high hit rates (20 to 70 %) in identification of new ligand chemotypes as lead compounds for adenosine A2AAR 11, 12, chemokine CXCR4 13, dopamine D3 14, and histamine H1 15 receptors, as well as in lead optimization 16. Following successful applications to kinases, proteases and other protein target families, structure-based ligand screening technologies are now becoming an important part of the modern GPCR drug discovery process in pharmaceutical and biotech industries17, 18.
Outreach and Collaborations
As mentioned, the GPCR Network includes extensive efforts to involve a broadly-based scientific community. Each structure determined to date has been studied in collaboration with scientists who have intimate knowledge of the specific receptor system from a chemical and biological perspective (Table 1). For example, prior to our collaboration which resulted in the first detailed structure of the receptor at 2.4 Å resolution, Brian Kobilka worked for approximately 20 years to study details of β2AR and his insight into the biochemistry of the receptor was critical, including the protein engineering and understanding of receptor pharmacology. Adriaan IJzerman and Ken Jacobson had each spent over 20 years studying the adenosine receptors before the start of our collaborations, which yielded structures of A2AAR in complexes with an antagonist at 2.6 Å resolution5 and an agonist at 2.8 Å resolution19. Quickly following the publication of the first A2AAR structure, we worked with Ad Ijzerman, Laura Heitman and their colleagues on a careful site specific mutagenesis of the receptor binding site 20 and then with Ken Jacobson and colleagues at the NIH on the design and synthesis of novel ligands that bind the receptor 21. In the recent determination of the human κ-opioid receptor structure22, the laboratory of Bryan Roth, with 20 years of background in opioid receptors, was instrumental in selecting and providing the ligand for crystallization. Upon completion of the kappa opioid and NOP receptors, 28 and 5 mutants were made, respectively for the two different receptors to immediately probe receptor structure and function. Similar success resulted from the following collaborations: Tracy Handel and Pfizer on CXCR4 (ref. 23), Jonathan Javitch on dopamine D3 (ref. 24), So Iwata on histamine H1 (ref. 25), Mike Hanson and Receptos on sphingolipid S1P1 (ref. 26), and Girolamo Calo on nociceptin/orphanin FQ peptide opioid27 receptor. Given their long history of working with a specific receptor system, all these colleagues were ideal partners for conducting follow-up biochemical studies, including site-specific mutagenesis and/or structure–activity relationship analyses with numerous compounds. Community collaborations thus contribute to enabling structure determinations and to adding value to the structural insights once a structure is obtained, which is the heart of the mission at the GPCR Network.
Table 1.
Structures solved by the GPCR Network
| GPCR | Partner(s) | PDB Code | Method of crystallization |
Structure-Function Follow up studies |
|---|---|---|---|---|
|
β2-adrenergic receptor37, 38 |
B. Kobilka | 2RH1 | LCP with ICL3 T4L fusion |
Modeling full and partial agonists54 |
|
β2-adrenergic receptor41 |
3D4S | LCP with ICL3 T4L fusion |
Establishing specific cholesterol binding site41 |
|
|
β2-adrenergic receptor42 |
3NY8; 3NY9; 3NYA |
LCP with ICL3 T4L fusion |
Establishing conserved binding mode of β2AR42; ligand dependent perturbation by HDX28; biased signaling pathways by NMR47 |
|
|
A2A adenosine receptor3 |
A. IJzerman, L. Heitman |
3EML | LCP with ICL3 T4L fusion |
GPCR Dock 20088; Novel agonist chemotypes12; subtype selectivity analysis by mutations20 and modeling 55 |
|
A2A adenosine receptor19 |
K. Jacobson | 3QAK | LCP with ICL3 T4L fusion |
Assessment of activation-related changes in GPCRs, molecular modeling of agonist21; SBDD16 |
|
A2A adenosine receptor56 |
A. IJzerman, L. Heitman |
4EIY | LCP with ICL3 BRIL fusion |
Allosteric regulation by sodium ions56 |
|
Chemokine CXCR4 receptor23 |
A. Brooun, P. Wells, T. Handel; |
3ODU; 3OE6; 3OE8; 3OE9; 3OE0 |
LCP with ICL3 T4L fusion |
GPCR Dock 20109 |
|
Dopamine D3 receptor24 |
L. Shi, A.H. Newman, J. Javitch |
3PBL | LCP with ICL3 T4L fusion |
GPCR Dock 20109 |
|
Histamine H1 receptor25 |
S. Iwata, T. Kobayashi |
3RZE | LCP with ICL3 T4L fusion |
Virtual ligand screening15 |
|
Kappa opioid receptor22 |
B. Roth; F.I. Carroll; P.D. Mosier |
4DJH | LCP with ICL3 T4L fusion |
29 mutations studied for structure-function22 |
|
Nociceptin/orphan nin FQ peptide (opioid) receptor27 |
G. Calo; B. Roth |
4EA3 | LCP with N- term BRIL fusion |
8 mutations studied for structure- function27 |
|
Sphingolipid S1P1 receptor26 |
M. Hanson, H. Rosen |
3V2W; 3V2Y |
LCP with ICL3 T4L fusion |
5 mutations studied for structure- function26 |
To encourage and facilitate follow-up work on GPCRs with determined structures, the GPCR Network provides protein material to scientists who apply complementary biophysical methods. For example, hydrogen–deuterium exchange (HDX) experiments in the Griffin laboratory at TSRI-Florida were performed with GPCR protein material produced at the GPCR Network, and generated valuable insights into GPCR conformational equilibria in response to different pharmacological ligands28. Lastly, collaborative funding from the NIGMS PSI:Biology program is proving to be encouragingly successful in partnering experts in specialized biology areas with PSI:Biology Centers. As an illustration, the Handel lab at UCSD partnered with the GPCR Network through a NIH-funded collaboration on chemokine receptors. To assist in target selection and collaborator recruitment, PSI:Biology has also created the Community Nominated Targets program (CNT; http://sbkb.org/cnt), which enables fellow-scientists to suggest targets for close collaboration with the GPCR Network or another PSI:Biology Center.
Perhaps most critical to date, is the active participation of collaborating laboratories. This includes knowledge and access to compounds, or intuitive insight into specific receptor biochemistry. Of the collaborations that have not been successful to date, the primary reason has been a lack of commitment or passionate interest for structure. For example, requests are now being made on a regular basis for a specific receptor to be studied. Sadly, when we ask for assistance on the initial molecular biology or access to ligands, some collaborating labs reply that it would be nice to have the structure, but they do not have the resources to help or we do not get replies after asking for help. We have thus adopted a policy where active collaboration is required before we will commit our own resources which are also very limited.
Impact of new GPCR structures
As of July 9, 2012, the GPCR Network pipeline process has been used in the determination of 8 of the 9 presently available human GPCR structures, which represent the majority of the fourteen known unique GPCR structures. Specifically, the process has been used to determine the β2AR, A2AAR3, CXCR4 chemokine23, D3 dopamine24, H1 histamine25, S1P1 sphingolipid26, κ-opioid22, and nociceptin/orphanin FQ peptide27 receptors. Outside of the GPCR Network, several groups have contributed key structures, including the structures of bovine rhodopsin29, squid rhodopsin30, turkey β1 adrenergic receptor31, human M2 muscarinic receptor32, rat M3 muscarinic receptor33, mouse µ-opioid receptor34, and mouse δ-opioid receptor35. Additional structure reports include the characterization of the active and inactive states for rhodopsin/opsin29, 36, β2AR37–42 and A2AAR3, 19, 43, 44. A key breakthrough for the field was the structure determination of a complex of β2AR with the G-protein45, setting the stage for studies of GPCR complexes with their intracellular partner proteins. The ensemble of these structures illustrates that the seven-transmembrane protein architecture is ideally suited to recognize many different types of natural and synthetic ligands, and to transmit signals over a distance of 30 Å to the intracellular side of the membrane (Figure 1).
Receptor Structure Diversity and Modularity
The 14 distinct class A GPCR structures that have been determined to date confirm the overall structural conservation of the seven-transmembrane fold, with a Cα root mean squared deviation (RMSD) of < 3.0 Å calculated for the transmembrane-helices between any pair of GPCRs. In contrast, the orthosteric binding sites vary widely among the different structures, providing a rationale for the intricate molecular recognition potential in the GPCR family (Figure 1) that makes each receptor very unique46.
Variations in structure are especially pronounced on the extracellular side of receptors, reflecting a distinct evolutionary and functional modularity between extracellular (ligand-binding) and intracellular (downstream signaling) modules of GPCRs (Figure 5). Interestingly, although the protein backbone RMSD in the extracellular-transmembrane region calculated between different GPCR pairs is almost twofold larger than the RMSD value for the intracellular-transmembrane module, which reflects a larger variation in the extracellular half of the receptors, the intracellular side shows larger conformational changes of discrete molecular regions upon activation. A contrast in structural variability between the ligand-binding and the downstream signaling modules is also evident from comparison of the primary structures, which shows 6% sequence identity in the extracellular transmembrane region, as compared to 26% sequence identity in the intracellular transmembrane region.
Figure 5.

Receptor diversity in class A GPCRs, as illustrated by a simplified cartoon. Comparison of experimental GPCR structures reveals a larger structural diversity in the extracellular module (colored red) than in the intracellular module (colored blue), which seems to reflect the evolutionary pressure of recognizing hundreds of endogenous ligands while transferring their signals to only dozens of interacting partners (see the text for details).
For several GPCRs, structures have by now been determined in different functional states. This sets the stage for formulating hypotheses for follow-up work to the presently available data. There seems to be modularity also in the helix sub-bundles I to IV and V to VII. This leads to intriguing questions about splicing variants and receptor chimeras, as well as about the role of individual helices in receptor function beyond their involvement in ligand binding or receptor oligomerization. For example, the primary role of helix I, which is structurally the most diverse among the available GPCR structures, may be more involved in membrane insertion or oligomerization than previously proposed.
Receptor Conformational States and Dynamics
As a complement to the crystal structure data, HDX and NMR experiments are being used to understand receptor behavior. HDX is able to define the conformational flexibility in receptors at a global level, and through the use of selective labels in NMR one can ask specific questions about receptor conformational polymorphisms and dynamics. For example, using site-specific 19F-NMR labels to study β2AR complexes with various ligands, we observed that the cytoplasmic ends of helices VI and VII adopt two major conformational states47. Changes in the relative NMR signal intensities revealed that full-agonist binding shifts the equilibrium towards active states in both helix VI and helix VII, whereas β-arrestin–biased ligands predominantly impact the conformational states of helix VII. The selective effects of different ligands on the conformational equilibria involving helices VI and VII thus provided novel insights into the dynamic behavior of β2AR in full, partial and biased agonist signaling, which appears to relate with largely uncoupled conformational equilibria. Given the above observations on correlations between ligand structure and receptor response, this provides novel leads for correlating chemical structures of pharmacological ligands with their signaling pathways, acting either through G proteins or through β-arrestins.
Conclusions
Twelve years after the structure determination of bovine rhodopsin and five years after the first human GPCR structure with a diffusible ligand was obtained, a total of fourteen unique GPCR structures have now been reported. As a consequence, leveraging through modeling has become more reliable and will further improve as additional experimental structures are determined. As a current working practice, we use a 35% sequence identity cutoff for homology modeling, and estimate that up to 20% of the non-olfactory GPCRs could thus be reliably modeled based on the experimental structures of the aforementioned fourteen distinct receptors. For close to 100% coverage of the non-olfactory receptors, our current estimate indicates that ∼100 carefully selected, unique GPCR structures are needed. Beyond these considerations of “structural coverage”, the long-term focus of the GPCR Network is to obtain more and more detailed insights into structure–function correlations. In this regard, additional structure determinations of GPCR complexes with different ligands and with intracellular partner proteins, combined with biophysical characterization of these complexation interactions, will provide valuable input. As of July 9, 2012 seventy-two such structures have been deposited in the Protein Data Bank. Based on the currently available structures providing a basis for molecular replacement, it is expected that this number will grow rapidly as the GPCR Network and others pursue further improved understanding of GPCR structure and function. This ambitious project will continue to greatly benefit from close collaborations with fellow-scientists who have insights into specific receptor systems and access to GPCR-ligand libraries, with a common vision to continued growth of knowledge in this exciting and important area of biological and biomedical research.
With a more complete structure–function understanding of the GPCR superfamily, one can begin to investigate the next generation of questions about basic principles of molecular recognition, variation of receptor conformational states in disease, similarities and differences in the signaling mechanisms of GPCRs, rational design of more selective and desirable therapeutics, and evolution of GPCRs in the framework of the evolution of the human species.
Acknowledgements
The GPCR Network acknowledges support from NIH/NIGMS PSI:Biology grant U54 GM094618 and the NIH Common Fund grant P50 GM073197 to the JCIMPT for technology development. The authors are grateful to the Scientific Advisory Board Members (T.W. Schwarz, B.L. Roth, R.M. Stroud, G. Wagner, S.H. White and I.A. Wilson) and community collaborators including A. Brooun, G. Calo. R. Guerrini, T. Handel, M. Hanson, A. IJzerman, S. Iwata, K. Jacobson, J. Javitch, T. Kobayashi, B. Kobilka, P.D. Mosier, A.H. Newman, B.L. Roth, L. Shi, and P. Wells. We thank K. Kadyshevskaya and I. Kufareva for assistance with figure preparation; E. Abola and A. Walker for assistance with manuscript preparation.
Footnotes
Competing Interests Statement
R.C.S. is a founder, and H.R. is a founder and on the Scientific Advisory Board of Receptos, a GPCR structure–based drug discovery company.
References
- 1.Rajagopal S, Rajagopal K, Lefkowitz RJ. Teaching old receptors new tricks: biasing seven-transmembrane receptors. Nat Rev Drug Discov. 2010;9:373–386. doi: 10.1038/nrd3024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wise A, Gearing K, Rees S. Target validation of G-protein coupled receptors. Drug Discov Today. 2002;7:235–246. doi: 10.1016/s1359-6446(01)02131-6. [DOI] [PubMed] [Google Scholar]
- 3.Jaakola VP, et al. The 2.6 angstrom crystal structure of a human A2A adenosine receptor bound to an antagonist. Science. 2008;322:1211–1217. doi: 10.1126/science.1164772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Weigelt J, McBroom-Cerajewski LD, Schapira M, Zhao Y, Arrowsmith CH. Structural genomics and drug discovery: all in the family. Curr Opin Chem Biol. 2008;12:32–39. doi: 10.1016/j.cbpa.2008.01.045. [DOI] [PubMed] [Google Scholar]
- 5.Reynolds K, Abagyan R, Katritch V. In: GPCR Molecular Pharmacology and Drug Targeting: Shifting Paradigms and New Directions. Gilchrist A, editor. Hoboken, NJ: Wiley & Sons, Inc; 2010. pp. 385–433. [Google Scholar]
- 6.Yarnitzky T, Levit A, Niv MY. Homology modeling of G-protein-coupled receptors with X-ray structures on the rise. Curr Opin Drug Discov Devel. 2010;13:317–325. [PubMed] [Google Scholar]
- 7.Cavasotto CN, Phatak SS. Homology modeling in drug discovery: current trends and applications. Drug Discov Today. 2009;14:676–683. doi: 10.1016/j.drudis.2009.04.006. [DOI] [PubMed] [Google Scholar]
- 8.Michino M, et al. Community-wide assessment of GPCR structure modelling and ligand docking: GPCR Dock 2008. Nat Rev Drug Discov. 2009;8:455–463. doi: 10.1038/nrd2877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kufareva I, Rueda M, Katritch V, Stevens RC, Abagyan R. Status of GPCR modeling and docking as reflected by community-wide GPCR Dock 2010 assessment. Structure. 2011;19:1108–1126. doi: 10.1016/j.str.2011.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Johnston JM, Filizola M. Showcasing modern molecular dynamics simulations of membrane proteins through G protein-coupled receptors. Curr Opin Struct Biol. 2011;21:552–558. doi: 10.1016/j.sbi.2011.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Carlsson J, et al. Structure-based discovery of A2A adenosine receptor ligands. J Med Chem. 2010;53:3748–3755. doi: 10.1021/jm100240h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Katritch V, et al. Structure-based discovery of novel chemotypes for adenosine A(2A) receptor antagonists. J Med Chem. 2010;53:1799–1809. doi: 10.1021/jm901647p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mysinger MM, et al. Structure-based ligand discovery for the protein-protein interface of chemokine receptor CXCR4. Proc Natl Acad Sci U S A. 2012;109:5517–5522. doi: 10.1073/pnas.1120431109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Carlsson J, et al. Ligand discovery from a dopamine D(3) receptor homology model and crystal structure. Nat Chem Biol. 2011 doi: 10.1038/nchembio.662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.de Graaf C, et al. Crystal structure-based virtual screening for fragment-like ligands of the human histamine H(1) receptor. J Med Chem. 2011;54:8195–8206. doi: 10.1021/jm2011589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tosh DK, et al. Optimization of adenosine 5'-carboxamide derivatives as adenosine receptor agonists using structure-based ligand design and fragment screening. J Med Chem. 2012;55:4297–4308. doi: 10.1021/jm300095s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Congreve M, Langmead C, Marshall FH. The use of GPCR structures in drug design. Adv Pharmacol. 2011;62:1–36. doi: 10.1016/B978-0-12-385952-5.00011-7. [DOI] [PubMed] [Google Scholar]
- 18.Shoichet BK, Kobilka BK. Structure-based drug screening for G-protein-coupled receptors. Trends Pharmacol Sci. 2012;33:268–272. doi: 10.1016/j.tips.2012.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xu F, et al. Structure of an agonist-bound human A2A adenosine receptor. Science. 2011;332:322–327. doi: 10.1126/science.1202793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jaakola VP, et al. Ligand binding and subtype selectivity of the human A(2A) adenosine receptor: identification and characterization of essential amino acid residues. J Biol Chem. 2010;285:13032–13044. doi: 10.1074/jbc.M109.096974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Deflorian F, et al. Evaluation of molecular modeling of agonist binding in light of the crystallographic structure of an agonist-bound A(2)A adenosine receptor. J Med Chem. 2012;55:538–552. doi: 10.1021/jm201461q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wu H, et al. Structure of the human k-opioid receptor in complex with JDTic. Nature. 2012 doi: 10.1038/nature10939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wu B, et al. Structures of the CXCR4 chemokine GPCR with small-molecule and cyclic peptide antagonists. Science. 2010;330:1066–1071. doi: 10.1126/science.1194396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chien EY, et al. Structure of the human dopamine D3 receptor in complex with a D2/D3 selective antagonist. Science. 2010;330:1091–1095. doi: 10.1126/science.1197410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shimamura T, et al. Structure of the human histamine H1 receptor complex with doxepin. Nature. 2011;475:65–70. doi: 10.1038/nature10236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hanson MA, et al. Crystal structure of a lipid G protein-coupled receptor. Science. 2012;335:851–855. doi: 10.1126/science.1215904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Thompson AA, et al. Structure of the Nociceptin/Orphanin FQ Receptor in Complex with a Peptide Mimetic. Nature. 2012 doi: 10.1038/nature11085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.West GM, et al. Ligand-dependent perturbation of the conformational ensemble for the GPCR beta2 adrenergic receptor revealed by HDX. Structure. 2011;19:1424–1432. doi: 10.1016/j.str.2011.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Palczewski K, et al. Crystal structure of rhodopsin: A G protein-coupled receptor. Science. 2000;289:739–745. doi: 10.1126/science.289.5480.739. [DOI] [PubMed] [Google Scholar]
- 30.Murakami M, Kouyama T. Crystal structure of squid rhodopsin. Nature. 2008;453:363–367. doi: 10.1038/nature06925. [DOI] [PubMed] [Google Scholar]
- 31.Warne T, et al. Structure of a beta1-adrenergic G-protein-coupled receptor. Nature. 2008;454:486–491. doi: 10.1038/nature07101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Haga K, et al. Structure of the human M2 muscarinic acetylcholine receptor bound to an antagonist. Nature. 2012;482:547–551. doi: 10.1038/nature10753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kruse AC, et al. Structure and dynamics of the M3 muscarinic acetylcholine receptor. Nature. 2012;482:552–556. doi: 10.1038/nature10867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Manglik A, et al. Crystal structure of the mu-opioid receptor bound to a morphinan antagonist. Nature. 2012 doi: 10.1038/nature10954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Granier S, et al. Structure of the delta opioid receptor bound to naltrindole. Nature. 2012 doi: 10.1038/nature11111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Scheerer P, et al. Crystal structure of opsin in its G-protein-interacting conformation. Nature. 2008;455:497–502. doi: 10.1038/nature07330. [DOI] [PubMed] [Google Scholar]
- 37.Cherezov V, et al. High-resolution crystal structure of an engineered human beta2-adrenergic G protein-coupled receptor. Science. 2007;318:1258–1265. doi: 10.1126/science.1150577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rosenbaum DM, et al. GPCR engineering yields high-resolution structural insights into beta2-adrenergic receptor function. Science. 2007;318:1266–1273. doi: 10.1126/science.1150609. [DOI] [PubMed] [Google Scholar]
- 39.Rasmussen SG, et al. Crystal structure of the human beta2 adrenergic G-protein-coupled receptor. Nature. 2007;450:383–387. doi: 10.1038/nature06325. [DOI] [PubMed] [Google Scholar]
- 40.Rasmussen SG, et al. Structure of a nanobody-stabilized active state of the beta(2) adrenoceptor. Nature. 2011;469:175–180. doi: 10.1038/nature09648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hanson MA, et al. A specific cholesterol binding site is established by the 2.8 A structure of the human beta2-adrenergic receptor. Structure. 2008;16:897–905. doi: 10.1016/j.str.2008.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wacker D, et al. Conserved binding mode of human beta2 adrenergic receptor inverse agonists and antagonist revealed by X-ray crystallography. J Am Chem Soc. 2010;132:11443–11445. doi: 10.1021/ja105108q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lebon G, et al. Agonist-bound adenosine A2A receptor structures reveal common features of GPCR activation. Nature. 2011;474:521–525. doi: 10.1038/nature10136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dore AS, et al. Structure of the adenosine A(2A) receptor in complex with ZM241385 and the xanthines XAC and caffeine. Structure. 2011;19:1283–1293. doi: 10.1016/j.str.2011.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rasmussen SG, et al. Crystal structure of the beta2 adrenergic receptor-Gs protein complex. Nature. 2011;477:549–555. doi: 10.1038/nature10361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Katritch V, Cherezov V, Stevens RC. Diversity and modularity of G protein-coupled receptor structures. Trends Pharmacol Sci. 2012;33:17–27. doi: 10.1016/j.tips.2011.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liu JJ, Horst R, Katritch V, Stevens RC, Wuthrich K. Biased signaling pathways in beta2-adrenergic receptor characterized by 19F-NMR. Science. 2012;335:1106–1110. doi: 10.1126/science.1215802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chun E, et al. Fusion partner toolchest for the stabilization and crystallization of G protein-coupled receptors. Structure. 2012;20:967–976. doi: 10.1016/j.str.2012.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Liu W, Hanson MA, Stevens RC, Cherezov V. LCP-Tm: an assay to measure and understand stability of membrane proteins in a membrane environment. Biophys J. 2010;98:1539–1548. doi: 10.1016/j.bpj.2009.12.4296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Alexandrov AI, Mileni M, Chien EY, Hanson MA, Stevens RC. Microscale fluorescent thermal stability assay for membrane proteins. Structure. 2008;16:351–359. doi: 10.1016/j.str.2008.02.004. [DOI] [PubMed] [Google Scholar]
- 51.Xu F, Liu W, Hanson MA, Stevens RC, Cherezov V. Development of an Automated High Throughput LCP-FRAP Assay to Guide Membrane Protein Crystallization in Lipid Mesophases. Cryst Growth Des. 2011;11:1193–1201. doi: 10.1021/cg101385e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Caffrey M, Cherezov V. Crystallizing membrane proteins using lipidic mesophases. Nat Protoc. 2009;4:706–731. doi: 10.1038/nprot.2009.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cherezov V, et al. Rastering strategy for screening and centring of microcrystal samples of human membrane proteins with a sub-10 microm size X-ray synchrotron beam. J R Soc Interface. 2009;6(Suppl 5):S587–S597. doi: 10.1098/rsif.2009.0142.focus. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Katritch V, et al. Analysis of full and partial agonists binding to beta2-adrenergic receptor suggests a role of transmembrane helix V in agonist-specific conformational changes. J Mol Recognit. 2009;22:307–318. doi: 10.1002/jmr.949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Katritch V, Kufareva I, Abagyan R. Structure based prediction of subtype-selectivity for adenosine receptor antagonists. Neuropharmacology. 2011;60:108–115. doi: 10.1016/j.neuropharm.2010.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Liu W, et al. Structural basis for allosteric regulation of GPCRs by sodium ions. Science. 2012;337:232–236. doi: 10.1126/science.1219218. [DOI] [PMC free article] [PubMed] [Google Scholar]