

Abstract
Purpose.
As in human populations, in which founder mutations have been identified in groups of families, a number of founder mutations have been observed across strains in mice. In this report, we provide a phenotype and genotype survey of three common eye diseases in the collection of JAX mice strains at The Jackson Laboratory (JAX). These eye diseases are retinal degeneration 1 (Pde6brd1), retinal degeneration 8 (Crb1rd8), and cone photoreceptor function loss 3 (Gnat2cpfl3).
Methods.
Ocular lesions for rd1 and rd8 were evaluated by fundus examination and fundus photography, and the abnormal retinal function observed in mice homozygous for cpfl3 was assessed by ERG. Genotyping protocols for rd1, rd8, and cpfl3 mutations were performed by PCR with appropriate primers.
Results.
We have actively screened retired breeders for surface dysmorphologies, and for intraocular defects by indirect ophthalmoscopy, slit-lamp biomicroscopy, and ERG to discover new spontaneous mutations in strains from the Genetic Resource Science (GRS) production colony. Through this process, we have found that of the strains screened, 99 strains carried the rd1 mutation, 85 strains carried the rd8 mutation, and 20 strains carried the cpfl3 mutation.
Conclusions.
Of the 1000 of strains screened during this study, 204 carried one of three founder mutations in Pde6b, Crb1, or Gnat2. Since these three retinal mutations occur commonly in various mouse strains, genotyping for these mutations, and/or avoiding mouse strains or stocks carrying these mutant alleles when studying new retinal disorders is recommended. The robust PCR genotyping protocols to test for these common alleles are described herein.
Keywords: retinal degeneration, fundus photography, spontaneous mutation, animal model, eye diseases
In this report, we provide a phenotype and genotype survey of three common eye diseases in the collection of JAX mice strains at The Jackson Laboratory. These eye diseases are retinal degeneration 1(Pde6brd1), retinal degeneration 8 (Crb1rd8), and cone photoreceptor function loss 3 (Gnat2cpfl3).
Introduction
Discovery of human retinal degenerations generally is identified by self-reports of visual problems with subsequent eye examinations. However, determining the etiology and the disease progression, elucidating the mechanisms underlying the disease, and exploring methods for delaying or ameliorating retinal degenerations can be impeded by ethical considerations of exploratory experimentation on humans. Also, human eye tissue (including biopsies) seldom is available for study because it is difficult to obtain tissue from the eye without risk of further damage to the patient's vision, and if samples are available, they usually are at the end stages of the disease process.
Animal models have advanced our understanding of eye diseases significantly as they allow for sampling of tissue throughout development and disease progression, for invasive studies, and for rapid genetic analysis. Use of mouse models, in particular, has increased through the past decades due to their ability to be modified genetically. In addition to manipulation by genetic engineering, a large number of spontaneous and chemically-induced mutations have been identified by screening mice using indirect ophthalmoscopy and ERG at The Jackson Laboratory (JAX; Bar Harbor, ME), where many mouse models of retinal degeneration and diseases have been discovered.1,2 These mouse models have increased significantly our understanding of the function of specific genes and how they lead to disease when disrupted. Studies of mouse models of human retinal degeneration have contributed to our understanding of the pathophysiology, as well as the etiology, of these diseases, and allowed for development of gene-based therapy to delay the progression of disease and increase visual function.3,4
Historically, the first retinal degeneration model was described by Clyde E. Keeler more than 80 years ago. It subsequently was identified as a disruption in phosphodiesterase 6 beta (Pde6brd1, formerly rd1, rd, identical to Keeler rodless retina, r).5–7 The second retinal degeneration model was discovered 50 years later, and named retinal degeneration-slow (Rds or Rd2).8 The third retinal degeneration was discovered in 1993 and named retinal degeneration 3, rd3.9 Subsequently, during the last 20 years, many additional mouse models of retinal degeneration have been discovered as the tools used commonly in human clinical eye examinations have been adapted for use with the smaller mouse eyes.10,11
Interestingly, through screening many retired breeders of different mouse strains at JAX, mutations in Pde6b, Crb1, and Gnat2 have been found to recur in high frequency. Because the strains examined all bore the same respective mutations within these particular genes, this suggested that they were founder mutations that had been propagated across multiple strains through the years of model development and inbreeding.12–14
As significant retinal pathology occurs in the presence of Pde6brd1 and Crb1rd8, and functional deficits occurs in the presence of Gnat2cpfl3, interpretation of disease phenotypes resulting from different gene/mutations and experimental studies where behavioral performance is an end point, respectively, may be confounded. Knowledge of the genotype for the common disease alleles in these experimental models will aid in the proper selection of mouse strains for use in research studies and interpretation of data generated from them.
Materials and Methods
Mice
The mice screened in this study were bred and maintained in standardized conditions at the Production and Research Animal Facilities at JAX. They were maintained on a NIH31 6% fat chow diet and provided acidified water, in a pathogen-free vivarium environment with a 14-hr light/10-hr dark cycle. One pair of retired breeders from the JAX Mouse Repository was screened for each strain. All experiments were approved by the Institutional Animal Care and Use Committee, and conducted in accordance with the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research.
Clinical Retinal Evaluation
Mouse eyes, dilated with 1% atropine (Alcon Laboratories, Inc., Fort Worth, TX), were evaluated by indirect ophthalmoscopy with a 90 diopter lens. If a mouse strain showed signs of early and rapid retinal degeneration, such as attenuation of retinal vessels or areas of depigmentation (Fig. 1B), the mouse strain was genotyped for the rd1 mutation. If a mouse strain in the C57BL/6 genetic background or related background showed a retinal spotting phenotype (Fig. 2A), the mouse strain was typed for the rd8 mutation. Fundus photographs were taken with the Micron III retinal imaging microscope from Phoenix Research Laboratories (Phoenix, AZ).
Figure 1.
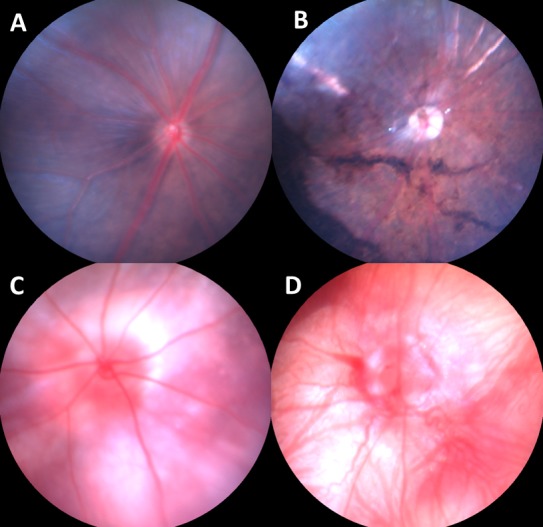
Fundus photographs of normal pigmented (A) and albino (C) mice as well as fundus photographs of mice bearing the Pdeb6rd1 mutation in pigmented (B) and albino (D) background.
Figure 2.
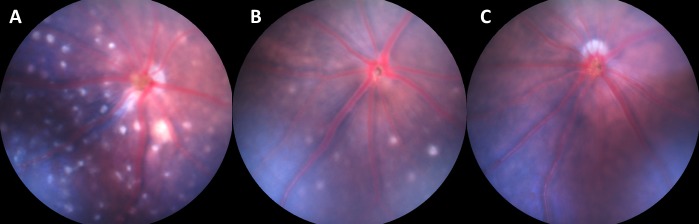
Fundus photographs of mice homozygous for the Crb1rd8 mutations with the typical pattern of white retinal spots (A), with a few white retinal spots (B), and with no retinal white spots (C).
Simplified Screening for Functional ERG Alterations
Attenuation of retinal vessels and disturbances in the retinal pigment epithelium are detected easily by indirect ophthalmoscopy. However, even if the retina has a normal appearance, retinal function still may be impaired. Therefore, all strains with a normal fundus appearance by indirect ophthalmoscopy were screened for functional deficits by ERG. Because the standard ERG test is time consuming, requiring approximately 30 to 60 minutes per mouse (not including the time necessary for dark adaption), we have developed a simple screening ERG test that takes less than 10 minutes per mouse to detect for functional deficits, such as retinal cone photoreceptor function loss (achromatopsia)14,15 and no b-wave (nob) mutations.16,17
Briefly, one eye per mouse was dilated with 1% cyclopentolate hydrochloride ophthalmic solution (Bausch & Lomb, Inc., Tampa, FL). A group of 5 to 10 mice could be dilated at the same time. Mice were weighed to determine the proper dosage of anesthesia (0.1 mL/20 g body weight of a mixture of 0.8 mL ketamine [Ketamine KCL injection USP, 100 mg/mL; Butler Schein Animal Health, Dublin, OH], 0.8 mL xylazine [Xylazine sterile solution, 20 mg/mL; Butler Schein Animal Health], and 3.4 mL 0.9% sodium chloride). Before administering the anesthetic via intraperitoneal injection, the eye to be tested was treated with cyclomydril (0.2% cyclopentolate hydrochloride, 1% phenylephrine hydrochloride ophthalmic solution, sterile; Alcon Laboratories, Inc.) to moisten the surface of the eye. The sedated mouse was placed on a 37°C heating pad to maintain the temperature of the animal during testing. A needle probe was inserted just under the skin at the base of the tail for grounding, a gold loop electrode was placed between the gum and cheek, and the active gold loop electrode was placed gently on the cornea slightly below the middle of the eye. A drop of 2.5% Gonioscopic Prism Solution (Wilson Ophthalmic, Mustang, OK) was placed on the cornea and electrode to assure a good contact and to prevent drying of the eye surface. In the simplified test, 10 flashes of the highest light intensity (light intensity = 1.9 log cd/m2) were averaged. If a mouse had a photopic b-wave amplitude at or above 100 μV and implicit times at approximately 50 ms, the mouse was considered normal (Fig. 3A), whereas a mouse with a b-wave amplitude at or below 50 μV and/or implicit times longer than 50 ms was considered abnormal (Fig. 3B). An abnormal ERG response was confirmed by the longer, standard ERG test with appropriate dark/light adaptations.18
Figure 3.

Representative ERG responses to a bright flash obtained from a mouse with normal retinal function (A) and a mouse with abnormal retinal function (B).
Genotyping Protocols
Genomic DNA for genotyping mice was prepared from tail tips by the rapid, hot sodium hydroxide and Tris (HotSHOT) procedure,19 and 1 μL of the DNA supernatant was used in a 10 μL PCR reaction. Amplicons were visualized with ethidium bromide after electrophoretic separation on a 4% agarose gel.
Genotyping for Pde6brd1
The genotyping for Pde6brd1 was done by three-primer PCR.20,21 A pair of primers, G1(5′-CCTGC ATGTGAACCCAGTATTCTATC) and G2 (5′-CTACAGCCCCTCTCCAAGGTTTAT AG) generated a product (240 base pair) from normal control DNA, while primer G2 paired with primer XMV (5′- AAGCTAGCTGCAGTAACGCCATTT), which primes within the virus sequence, generated a larger product (560 base pair) from the rd1 mutant allele. These primers were unique and were able to detect the mutant allele.
Genotyping for Crb1rd8
Allele-specific PCR was used on genomic DNA to confirm the presence of the Crb1rd8 mutation22 using the primers: Crb1-mF1: 5′-GTGAAGAAGACAGCTACAGTTCTGATC; Crb1-mF2: 5′-GCCCCTGTTTGCATGGAGGAAACTTGGAAGACAGCTACAGTTCTTCTG; and Crb1-mR: 5′-GCCCCATTTGCACACTGAT. We made some sequence changes in each primer to improve the genotyping results.
Genotyping for Gnat2cpfl3
The Gnat2cpfl3 mutation resulted in a new MseI site that enabled us to genotype the cpfl3 mutation with a polymerase chain reaction and restriction fragment length polymorphism (PCR-RFLP) assay described previously.14
Results
JAX, which has the world's largest collection of mouse mutant stocks and genetically diverse inbred strains (available in the public domain at http://jaxmice.jax.org/index.html), has been an ideal place to discover and characterize models of heritable retinal disorders. By screening approximately 1000 strains of JAX mice from the JAX Mouse Repository, three highly prevalent retinal disorders have been detected.
Detection of the Pde6brd1Mutation
Mice homozygous for the Pde6brd1 mutation have a severe, early onset retinal degeneration associated with a murine viral insertion and a second nonsense mutation in exon 7 of the Pde6b gene, which encodes the beta subunit of cGMP-phosphodiesterase. Mice with the Pde6brd1 mutation can be detected easily by indirect ophthalmoscopy. Retinal vessels are attenuated (Figs. 1B, 1D) and areas of retinal depigmentation are observed (Fig. 1B). In this survey, once the rd1-like retinal degeneration phenotype (Figs. 1B, 1D) was detected, Pde6brd1 allele specific genotyping was done to confirm the rd1 mutation. By screening approximately 1000 mouse strains (the entire JAX mouse collection currently contains over 6000 strains) from JAX, 99 strains were confirmed to carry the Pde6brd1 mutation (Table 1).
Table 1.
JAX Mice With Retinal Degeneration 1 Mutation (Pde6brd1)
| # |
Strain Name |
Stock Number |
| 1 | STOCK Thtm1Srt/J | 008779 |
| 2 | STOCK Tg(Stra8-cre)1Reb/J | 008208 |
| 3 | STOCK Tg(Neurog1-cre)1Jejo/J | 012859 |
| 4 | STOCK Tg(Nefh-cre)12Kul/J | 009102 |
| 5 | STOCK Tg(Myh6-Prkca)1Jmk/J | 010579 |
| 6 | STOCK Tg(Myh6-Ppp3ca)37Eno/J | 009075 |
| 7 | STOCK Tg(mI56i-FLPe)39Fsh/J | 010815 |
| 8 | STOCK Tg(KRT5-rtTA)T2D6Sgkd/J | 017519 |
| 9 | STOCK Tg(I12b-cre/ERT2,-ALPP)37Fsh/J | 014600 |
| 10 | STOCK Tg(CAG-Venus)1Hadj/J | 011107 |
| 11 | STOCK Smn1tm1Msd Tg(H2-K1-tsA58)6Kio Tg(SMN2*delta7)4299Ahmb Tg(SMN2)89Ahmb/J |
006553 |
| 12 | STOCK Sirt6tm1.1Cxd/J | 017334 |
| 13 | STOCK Kcna2tm1Tem/J | 010744 |
| 14 | STOCK Fxntm1Mkn Tg(FXN)YG22Pook/J | 010963 |
| 15 | STOCK En1tm7(cre/ESR1)Alj/J | 007917 |
| 16 | STOCK En1tm2Alj/J | 007912 |
| 17 | STOCK En1tm2(cre)Wrst/J | 007916 |
| 18 | STOCK Blmtm4Ches/J | 008670 |
| 19 | STOCK Axltm1Grl/J | 011121 |
| 20 | Smn1tm3(SMN2/Smn1)MrphTg(SMN2*delta7)4299Ahmb | 008869 |
| 21 | SJL/J-Chr YB10/TeuJ | 016840 |
| 22 | SJL.129S2(C)-Cxcr2tm1Mwm/RmraJ | 013043 |
| 23 | RSV/LeJ | 000268 |
| 24 | HPG/BmJ | 000804 |
| 25 | FVB-Tg(Ttr-Igf1)1Sykr/J | 012662 |
| 26 | FVB-Tg(tetO-Ppargc1b)7Dpk/J | 012385 |
| 27 | FVB-Tg(tetO-Ppargc1a)1Dpk/J | 012387 |
| 28 | FVB-Tg(tetO-MET)23Rwng/J | 008695 |
| 29 | FVB-Tg(tetO-Cib1)1Jmol/J | 013780 |
| 30 | FVB-Tg(tetO-Cacnb2)1Jmol/J | 013778 |
| 31 | FVB-Tg(Pbsn-IGF1*)5305Ng/J | 008874 |
| 32 | FVB-Tg(Pbsn-Ar*E231G)7353Ng/J | 008878 |
| 33 | FVB-Tg(NPEPPS)1Skar/J | 013732 |
| 34 | FVB-Tg(Myh6-TRPC3*)6.6Jmol/J | 016570 |
| 35 | FVB-Tg(Myh6-SOD2,Tyr)3Pne/J | 009438 |
| 36 | FVB-Tg(Myh6-cre)2182Mds/J | 011037 |
| 37 | FVB-Tg(Myh6/tetO-Itpr2)3.11Jmol/J | 014153 |
| 38 | FVB-Tg(Myh6/tetO-Itpr1)22.3Jmol/J | 014155 |
| 39 | FVB-Tg(Myh6/tetO-Gata6)2Jmol/J | 016571 |
| 40 | FVB-Tg(MECP2)1Hzo/J | 008679 |
| 41 | FVB-Tg(Lactb)74.2Lus/J | 008784 |
| 42 | FVB-Tg(Gstm5-EGFP)1Ilis/J | 010947 |
| 43 | FVB-Tg(GFAP-luc,GAPDH-rluc)172.9Mes/J | 009638 |
| 44 | FVB-Tg(GFAP-CRYAB)141.6Mes/J | 010676 |
| 45 | FVB-Tg(GAS7)63.2Lus/J | 010515 |
| 46 | FVB-Tg(Cdh5-tTA)D5Lbjn/J | 013585 |
| 47 | FVB-Tg(CAG-luc,-GFP)L2G85Chco/J | 008450 |
| 48 | FVB-Tg(C3-1-TAg)cJeg/JegJ | 013591 |
| 49 | FVB-Tg(ACTA1-PABPN1*A17)1Drub/DrubJ | 016193 |
| 50 | FVB;129S6-Sncatm1Nbm Tg(SNCA)1Nbm/J | 010710 |
| 51 | FVB/N-Tg(tetO-Fasl)BDepa/J | 014547 |
| 52 | FVB/N-Tg(Myh6-Gnaq)40Gwd/J | 012460 |
| 53 | FVB/N-Tg(Myh6*tetO-Capn1)L2Gwd/J | 012459 |
| 54 | FVB/N-Tg(LRRK2*R1441G)135Cjli/J | 009604 |
| 55 | FVB/N-Tg(LRRK2*G2019S)1Cjli/J | 009609 |
| 56 | FVB/N-Tg(LRRK2)1Cjli/J | 009610 |
| 57 | FVB/N-Tg(HTT*97Q)IXwy/J | 008197 |
| 58 | FVB/N-Tg(Hoxc13)61B1Awg/J | 011032 |
| 59 | FVB/N-Tg(GFAP-HTT*160Q)31Xjl/J | 012630 |
| 60 | FVB/N-Tg(Dazl-EGFP)10Rarp/J | 009354 |
| 61 | FVB/N-Tg(CAG-EGFP,-ALPP)2.6Ggc/J | 008200 |
| 62 | FVB/N-Tg(Acta2-RAC1*G12V)33Pjgc/J | 010634 |
| 63 | FVB/N-MidnTg(Tyr)2261EOve/J | 017438 |
| 64 | FVB/NJ-Tg(Slc6a3-PARK2*Q311X)AXwy/J | 009090 |
| 65 | FVB/NJ-Tg(Hspa1a-luc,-EGFP)2Chco/J | 012370 |
| 66 | FVB/N-Ckap5TgTn(sb-cHS4,Tyr)2320F-1Ove/J | 017437 |
| 67 | FVB.C-Prdm16csp1/J | 013100 |
| 68 | FVB.Cg-Tg(tetO-cre)1Jaw/J | 008244 |
| 69 | FVB.Cg-Smn1tm6(SMN2)Mrph/J | 009381 |
| 70 | FVB.Cg-Smn1tm1Msd Tg(SMN2)89Ahmb Tg(SMN2*A111G)591Ahmb/J | 009134 |
| 71 | FVB.Cg-Gt(ROSA)26Sortm1(CAG-lacZ,-EGFP)Glh/J | 012429 |
| 72 | FVB.B-WldS/UmonJ | 008820 |
| 73 | FVB.A-Irf6clft1/BeiJ | 012655 |
| 74 | FVB.129X1(B6)-Trfr2tm1Slu/J | 005215 |
| 75 | FVB.129S6-Fmn1tm2Made/J | 008665 |
| 76 | FVB.129S4-Ptpn1tm1Bbk/Mmjax | 032242 |
| 77 | FVB.129S4(B6)-Scn4atm1.1Ljh/J | 011033 |
| 78 | FVB.129-Fmr1tm1Rbd/J | 008909 |
| 79 | FVB.129(Cg)-Slc9a3tm1Ges/J | 012563 |
| 80 | FVB.129(B6)-Smn1tm5(Smn1/SMN2)Mrph/J | 008604 |
| 81 | FVB.129(B6)-Smn1tm4(SMN2)Mrph/J | 008713 |
| 82 | FVB.129(B6)-Igf1tm1Dlr/J | 012663 |
| 83 | FVB(Cg)-Tg(Ghrhr-cre)3242Lsk/J | 011034 |
| 84 | C3h-f | 009101 |
| 85 | C3H/HeOuJ-MaoaTg(H2-K1-Ifnb1)8Seif/J | 014132 |
| 86 | C3;B6-Tg(AAVS1)A1Xob/J | 009601 |
| 87 | C3.129S7(B6)-Ifngtm1Ts/J | 008228 |
| 88 | C3.129S4-Mtnr1btm1Drw/J | 010488 |
| 89 | C3.129S4(B6)-Mtnr1atm1Rep/J | 009681 |
| 90 | BXSB.129P2(Cg)-Tcratm1Mjo/TheoJ | 007848 |
| 91 | B6;SJL-Tg(Thy1-COP3/EYFP)8Gfng/J | 012348 |
| 92 | B6;SJL-Tg(tetO-Erbb2*)8-4Jek/J | 010577 |
| 93 | B6;SJL-Tg(MMTV-rtTA)4-1Jek/J | 010576 |
| 94 | B6;SJL-Tg(LRRK2)66Mjff/J | 013725 |
| 95 | B6;SJL-Tg(Gh1-rtTA)4-3Jek/J | 010574 |
| 96 | B6;129S2-Nfatc3tm1Glm/J | 010589 |
| 97 | B6.Cg-Tg(Tek-cre)1Ywa/J | 008863 |
| 98 | B6.Cg-Tg(Nlrp1b)1Die/DieJ | 006840 |
| 99 | B6.129S2-Seletm1Hyn Selptm1Hyn/J | 008437 |
Presence of the Crb1rd8Mutation
Mice homozygous for Crb1rd8 mutation exhibit large white retinal spots covering the inferior nasal quadrant of the retina (Fig. 2A) and a slow progressive retinal degeneration. It is caused by a single nucleotide deletion in the Crb1 gene. Because the degree of the spotting phenotype is highly variable (Figs. 2A–C) in mice homozygous for the rd8 mutation, it is difficult to determine whether a strain carries the rd8 mutation by clinical fundus examination alone. During the period between 2003 and 2011, 17 strains (Table 2) were found to carry the Crb1rd8 mutation by fundus examination and confirmed by rd8 genotyping. In the past year, the Crb1rd8 genotyping protocol was added to our Eye Mutant Resource screening program. By screening 302 strains, 66 strains were found to be homozygous for the Crb1rd8 mutation by genotyping (Table 2). However, only 6 of them had the retinal white spots phenotype (Table 2).
Table 2.
JAX Mice With the Retinal Degeneration 8 (Crb1rd8) Mutation
|
# |
Strain Name |
Stock Number |
Retinal Spots |
| 1 | STOCK Waptm2(rtTA)Kuw/J | 016116 | N |
| 2 | STOCK Trpa1tm2Kykw Tg(CAG-cre/Esr1*)5Amc/J | 008813 | Y |
| 3 | STOCK Tg(Tph2-icre/ERT2)6Gloss/J | 016584 | N |
| 4 | STOCK Ssttm2.1(cre)Zjh/J | 013044 | N |
| 5 | STOCK Nphp1tm1Jgg/J | 013169 | Y |
| 6 | STOCK Mov10l1tm1.1Eno/J | 014179 | N |
| 7 | STOCK Gt(ROSA)26Sortm6(Gli1)Amc/J | 013123 | N |
| 8 | STOCK Gad2tm1(cre/ERT2)Zjh/J | 010702 | N |
| 9 | STOCK Et(icre/ERT2)14602Rdav/J | 012692 | N |
| 10 | STOCK Epn3tm1.1Pdc/J | 014108 | N |
| 11 | STOCK Dclk2tm1Jgg/J | 013172 | N |
| 12 | STOCK Ccktm1.1(cre)Zjh/J | 012706 | N |
| 13 | CXB9/HiAJ | 001630 | N |
| 14 | CXB5/ByJ | 000355 | N |
| 15 | CXB3/ByJ | 000353 | N |
| 16 | CXB2/ByJ | 000352 | N |
| 17 | CXB12/HiAJ | 001633 | N |
| 18 | C57BL/6-Zbtb7btm1.2Litt/J | 008775 | N |
| 19 | C57BL/6-Tg(tetO-EDN1,-lacZ)9Mhus/J | 013729 | N |
| 20 | C57BL/6-Tg(LacZpl)60Vij/J | 002754 | Y |
| 21 | C57BL/6-Tg(Defa2-Myd88)1Lvh/J | 016133 | N |
| 22 | C57BL/6N-Tg(Slc6a3-icre/ERT2)2Gloss/J | 016583 | Y |
| 23 | C57BL/6N-Tg(Slc32a1-icre/ERT2)3Gloss/J | 016582 | N |
| 24 | C57BL/6N-Pawrtm1Rang/J | 015823 | N |
| 25 | C57BL/6N-Agtr1atm1Uky/J | 016211 | N |
| 26 | B6N;129-Egr1tm1Jmi/J | 012924 | N |
| 27 | B6N.Cg-Tg(UGT1A1*28)1Rhtu/J | 014170 | N |
| 28 | B6N.129-Ptpn5tm1Pjlo/J | 016556 | N |
| 29 | B6N.129-Ptch1tm1Hahn/J | 012457 | Y |
| 30 | B6N.129-Ptch1tm1Hahn/J | 011052 | N |
| 31 | B6N.129-Ptch1tm1Hahn/J | 012457 | N |
| 32 | B6N.129P2-Adora1tm1Bbf/J | 014161 | N |
| 33 | B6129S-Del(7Slx1b-Sept1)4Aam/J | 013128 | N |
| 34 | B6;DBA-Tg(Cited1-TagRFP)26Amc/J | 015853 | N |
| 35 | B6;D2-Tg(tetO-SNCA)1Cai/J | 012450 | N |
| 36 | B6;CBA-Tg(Thy1-spH)21Vnmu/J | 014651 | N |
| 37 | B6;C3-Tg(PDGFB-LRRK2*R1441C)574Djmo/J | 016576 | N |
| 38 | B6;129X1-Sncatm1Rosl/J | 003692 | N |
| 39 | B6;129X1-Dusp6tm1Jmol/J | 009069 | Y |
| 40 | B6;129X1-Dkkl1tm1.1Mldp/J | 012711 | N |
| 41 | B6;129S-Syn2tm1Sud/J | 002477 | Y |
| 42 | B6;129S-Seletm2Hyn Selltm4Hyn/J | 003806 | Y |
| 43 | B6;129S-Prkd1tm1Eno/J | 014181 | N |
| 44 | B6;129S-Nr1h2tm1Djm/J | 014635 | N |
| 45 | B6;129S4-Ighmtm1Che/J | 003751 | Y |
| 46 | B6;129S4-Col1a1tm1(tetO-Pou5f1,-Klf4,-Sox2,-Myc)Hoch/J | 011001 | Y |
| 47 | B6;129S2-Itgb3tm1Hyn/J | 004669 | N |
| 48 | B6;129S1-Dnm3tm1.1Pdc/J | 013543 | N |
| 49 | B6;129-Nrxn3tm4.1Sud/J | 016194 | N |
| 50 | B6;129-Mavstm1Zjc/J | 008634 | N |
| 51 | B6;129-Iis1tm3(CAG-Bgeo,-tdTomato/TEVP,-Dlg4,-GFP)Nat/J | 012587 | N |
| 52 | B6;129-Iis1tm2(CAG-Bgeo,-Ndp,-EGFP)Nat/J | 011077 | N |
| 53 | B6;129-Igf1rtm2Arge/J | 012251 | N |
| 54 | B6;129-Hprttm1Detl/J | 003183 | Y |
| 55 | B6;129-Crhr1tm1Klee/J | 004454 | Y |
| 56 | B6;129-Cd3etm1Lov/J | 004177 | Y |
| 57 | B6.SJL-Tg(Vil-cre)997Gum/J | 004586 | Y |
| 58 | B6.FVB-Tg(CMA1-cre)6Thhe/J | 014643 | N |
| 59 | B6.Cg-Tg(UBC-TVA)1Clc/J | 015808 | N |
| 60 | B6.Cg-Tg(tetO-Ifng)184Pop/J | 009344 | Y |
| 61 | B6.Cg-Tg(Itgax-TGFBR2)1Flv/J | 008378 | N |
| 62 | B6.Cg-Tg(Col1a1-cre/ERT2)1Crm/J | 016241 | N |
| 63 | B6.Cg-Tg(CMV-CASP3)17Edge/J | 016908 | N |
| 64 | B6.Cg-Selplgtm1Fur/J | 004201 | Y |
| 65 | B6.Cg-Pkd2l1tm1.1Yuni/J | 016853 | N |
| 66 | B6.Cg-Gt(ROSA)26Sortm1(rtTA*M2)Jae Col1A1tm6(tetO-MSI2)Jae/J | 014588 | N |
| 67 | B6.129S4-Mtortm1.2Koz/J | 011009 | N |
| 68 | B6.129S4(FVB)-Insrtm1Khn/J | 006955 | Y |
| 69 | B6.129S4(Cg)-Cdk5tm1.1Lht/J | 014156 | N |
| 70 | B6.129S2-Pak4tm1Amin/J | 015829 | N |
| 71 | B6.129S2-Mapk9tm1Flv/J | 004321 | Y |
| 72 | B6.129S2-Il10rbtm1Agt/J | 005027 | N |
| 73 | B6.129S2-H2-DMatm1Doi/J | 004513 | Y |
| 74 | B6.129S1-Nf1tm1Cbr/J | 007923 | Y |
| 75 | B6.129S1-Mapk10tm1Flv/J | 004322 | Y |
| 76 | B6.129S1(Cg)-Dnm2tm1.1Pdc/J | 013542 | N |
| 77 | B6.129S(Cg)-Id2tm1.1(cre/ERT2)Blh/ZhuJ | 016222 | N |
| 78 | B6.129S(C)-Batf3tm1Kmm/J | 013755 | N |
| 79 | B6.129P-Cx3cr1tm1Litt/J | 005582 | N |
| 80 | B6.129P2-Icostm1Mak/J | 004859 | N |
| 81 | B6.129P2-Axin2tm1Wbm/J | 009120 | N |
| 82 | B6.129-Bace1tm1Pcw/J | 004714 | Y |
| 83 | B6(Cg)-Il10tm1.1Karp/J | 014530 | N |
| 84 | B6(Cg)-Crhtm1(cre)Zjh/J | 012704 | N |
| 85 | B6(Cg)-Ccktm2.1(cre/ERT2)Zjh/J | 012710 | Y |
Frequency of the Gnat2cpfl3Mutation
Homozygous Gnat2cpfl3 mice exhibit reduced cone-mediated ERG responses as early as 3 weeks of age with normal rod-mediated ERG responses. The cpfl3 mutation originally was discovered in ALS/LtJ mice by routine ERG screening of retired breeders. Sequence analysis revealed a missense mutation due to a single base pair substitution in exon 6 of the Gnat2 gene. Subsequently, seven additional strains also were confirmed to carry the Gnat2cpfl3 mutation (Table 3).14 In our report, using the simple screening ERG method described above, once a strain showed an abnormal ERG response (Fig. 3B), it was genotyped using the Gnat2cpfl3 genotyping protocol. By genotyping the strains with abnormal ERG responses, 13 additional strains were confirmed to carry the Gnat2cpfl3 mutation (Table 3).
Table 3.
JAX Mice With the Cone Photoreceptor Function Loss 3 (Gnat2cpfl3) Mutation
|
# |
Strain Name |
Stock Number |
| 1 | STOCK Zp2tm1Dean/J | 004129 |
| 2 | STOCK Tg(Ttr-RFP)1Hadj/J | 011108 |
| 3 | STOCK Tg(Trp53A135V)L3Ber/J | 003262 |
| 4 | STOCK Tg(tetO-Ipf1,EGFP)956.6Macd/J | 005699 |
| 5 | STOCK Tg(tetO-HIST1H2BJ/GFP)47Efu/J | 005104 |
| 6 | STOCK Tg(Stra8-cre)1Reb/J | 008208 |
| 7 | STOCK Tg(Neurog3-cre)C1Able/J | 005667 |
| 8 | STOCK Tg(Neurog1-cre)1Jejo/J | 012859 |
| 9 | STOCK Tg(Fos-lacZ)34Efu/J | 004623 |
| 10 | STOCK Tg(CAG-mRFP1)1F1Hadj/J | 005645 |
| 11 | STOCK Tg(CAG-KikGR)33Hadj/J | 013753 |
| 12 | STOCK Tg(CAG-EGFP)B5Nagy/J | 003115 |
| 13 | STOCK Tg(CAG-ECFP)CK6Nagy/J | 003773 |
| 14 | STOCK Sgk3fz-ica/McirJ | 006135 |
| 15 | STOCK Bcl2tm1Irt/J | 008882 |
| 16 | SENCARC/PtJ | 002748 |
| 17 | SENCARB/PtJ | 002747 |
| 18 | SENCARA/PtJ | 002746 |
| 19 | PN/nBSwUmabJ | 005052 |
| 20 | CD10/JlsJ | 006180 |
Discussion
Major advantages of using the mouse as a model system for ocular diseases include the physiologic and anatomic similarities of the mouse and human eye, the availability of clinical tools to characterize the models, and the well-developed resources available for data mining, for biochemical and physiologic studies, and for molecular manipulation of their genomes. For example, the ability to target and alter specific gene(s) is an important tool to produce mouse models with mutations in genes and tissues of choice. Inducing mutations by transgenesis or homologous recombination is termed “reverse genetics,” as opposed to “forward genetics” approaches whereby spontaneous/induced mutations are discovered as a result of overt phenotypes and the underlying gene/mutation subsequently is identified. Through “forward” and “reverse” genetic approaches, mouse models in >100 genes that underlie human retinal diseases have been studied.23
Examination of the mouse fundus by indirect ophthalmoscopy is a very powerful, noninvasive tool for evaluating the appearance of the mouse retina. It also is an effective approach for high throughput screening for mouse models of human retinal degeneration and diseases. Many examples of mouse models of retinal degeneration have been discovered by fundus examination.1,2 However, given the high frequency of the Pde6brd1 and Crb1rd8 mutations, it is important to avoid mouse strains or stocks carrying these mutant alleles, or to exclude these mutant alleles before studying new retinal models. Both of these founder mutations can be identified easily by genotyping. While mice with the Pde6brd1 mutation can be typed by fundus examination (Fig. 1) other mutations, such as Pde6anmf363, Pde6a282, and B6.129-tulp1−/−, are known to have similar disease characteristics, onset, and progression.24,25
The spontaneous Crb1rd8 mutation is likely to have occurred during the mid 1950s, when C57BL/6J mice were transferred to the National Institutes of Health (NIH; Bethesda, MD).13 During the intervening 60 years, this mutation has been widely disseminated, as indicated by the number of strains affected. Ocular models derived from the C57BL/6N strain background, which carries the Crb1rd8 mutation, should be genotyped even if a retinal spotting phenotype is not observed (Fig. 2, Table 2). Unfortunately, many embryonic stem cell lines that are used to generate genetically engineered models also carry the Crb1rd8 mutation as they were B6N derived.13 Despite the lack of retinal spots, Crb1rd8 mice all show a fragmented outer limiting membrane that may confound the interpretation of the disease characteristics of new retinal models being characterized.22
The simple screening ERG used in our study is another powerful, noninvasive and high throughput method to evaluate mouse retinal function, as mice with a normal fundus appearance still may be impaired functionally.14
The results from our survey provide information regarding the frequency of three commonly found mutations among different mouse strains. Strains that were positive for these three commonly found mutations will be listed in the JAX mice database (available in the public domain at http://jaxmice.jax.org/index.html). The genotyping protocols described should assist in identifying these founder mutations in new models of ocular diseases and for selection against these mutations to exclude their contributions to ocular phenotypes when they are found.
Acknowledgments
The authors thank Juergen Naggert and Mark Krebs for careful review of the manuscript, and the Genomic Resources Program at JAX for providing retired breeders for screening.
Supported by National Eye Institute Grants EY019943, EY011996, and EY016501.
Disclosure: B. Chang, None; R. Hurd, None; J. Wang, None; P. Nishina, None
References
- 1. Chang B, Hawes NL, Hurd RE, et al. Mouse models of ocular diseases. Vis Neurosci. 2005; 22: 587–593 [DOI] [PubMed] [Google Scholar]
- 2. Won J, Shi LY, Hicks W, et al. Mouse model resources for vision research. J Ophthalmol. 2011; 2011: 391384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Pang JJ, Chang B, Kumar A, et al. Gene therapy restores vision-dependent behavior as well as retinal structure and function in a mouse model of RPE65 Leber congenital amaurosis. Mol Ther. 2006; 13: 565–572 [DOI] [PubMed] [Google Scholar]
- 4. Alexander JJ, Umino Y, Everhart D, et al. Restoration of cone vision in a mouse model of achromatopsia. Nat Med. 2007; 13: 685–687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Keeler C. The inheritance of a retinal abnormality in white mice. Proc Natl Acad Sci U S A. 1924; 10: 329–350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Keeler C. Retinal degeneration in the mouse is rodless retina. J Hered. 1966; 57: 47–50 [DOI] [PubMed] [Google Scholar]
- 7. Pittler SJ, Keeler CE, Sidman RL, Baehr W. PCR analysis of DNA from 70-year-old sections of rodless retina demonstrates identity with the mouse rd defect. Proc Natl Acad Sci U S A. 1993; 90: 9616–9619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. van Nie R, Ivanyi D, Demant P. A new H-2-linked mutation, rds, causing retinal degeneration in the mouse. Tissue Antigens. 1978; 12: 106–108 [DOI] [PubMed] [Google Scholar]
- 9. Chang B, Heckenlively JR, Hawes NL, Roderick TH. New mouse primary retinal degeneration (rd-3). Genomics. 1993; 16: 45–49 [DOI] [PubMed] [Google Scholar]
- 10. Chang B, Hawes NL, Hurd RE, Davisson MT, Nusinowitz S, Heckenlively JR. Retinal degeneration mutants in the mouse. Vision Res. 2002; 42: 517–525 [DOI] [PubMed] [Google Scholar]
- 11. Chang B, Hawes N, Davisson M, Heckenlively J. Mouse models of RP. In: Tombran-Tink J, Barnstable C. eds Retinal Degenerations: Biology, Diagnostics, and Therapeutics. New York, NY: The Humana Press, Inc.; 2007: 149–164 [Google Scholar]
- 12. Bowes C, Li T, Frankel WN, et al. Localization of a retroviral element within the rd gene coding for the beta subunit of cGMP phosphodiesterase. Proc Natl Acad Sci U S A. 1993; 90: 2955–2959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Mattapallil MJ, Wawrousek EF, Chan CC, et al. The Rd8 mutation of the Crb1 gene is present in vendor lines of C57BL/6N mice and embryonic stem cells, and confounds ocular induced mutant phenotypes. Invest Ophthalmol Vis Sci. 2012; 53: 2921–2927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chang B, Dacey MS, Hawes NL, et al. Cone photoreceptor function loss-3, a novel mouse model of achromatopsia due to a mutation in Gnat2. Invest Ophthalmol Vis Sci. 2006; 47: 5017–5021 [DOI] [PubMed] [Google Scholar]
- 15. Chang B, Grau T, Dangel S, et al. A homologous genetic basis of the murine cpfl1 mutant and human achromatopsia linked to mutations in the PDE6C gene. Proc Natl Acad Sci U S A. 2009; 106: 19581–19586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Chang B, Heckenlively JR, Bayley PR, et al. The nob2 mouse, a null mutation in Cacna1f: anatomical and functional abnormalities in the outer retina and their consequences on ganglion cell visual responses. Vis Neurosci. 2006; 23: 11–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Maddox DM, Vessey KA, Yarbrough GL, et al. Allelic variance between GRM6 mutants, Grm6nob3 and Grm6nob4 results in differences in retinal ganglion cell visual responses. J Physiol. 2008; 586: 4409–4424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Nusinowitz S, Ridder WH III, Heckenlively JR. Electrophysiological testing of the mouse visual system. In: Smith RS. ed Systematic Evaluation of the Mouse Eye: Anatomy, Pathology, and Biomethods. Boca Raton, FL: CRC Press; 2002: 320–344 [Google Scholar]
- 19. Truett GE, Heeger P, Mynatt RL, Truett AA, Walker JA, Warman ML. Preparation of PCR-quality mouse genomic DNA with hot sodium hydroxide and tris (HotSHOT). Biotechniques. 2000; 29: 52–54 [DOI] [PubMed] [Google Scholar]
- 20. Qiao X, Pennesi M, Seong E, Gao H, Burmeister M, Wu SM. Photoreceptor degeneration and rd1 mutation in grizzled/mocha mouse strain. Vision Res. 2003; 43: 859–865 [DOI] [PubMed] [Google Scholar]
- 21. Giménez E, Montoliu L. A simple polymerase chain reaction assay for genotyping the retinal degeneration mutation (Pdeb(rd1)) in FVB/N-derived transgenic mice. Lab Anim. 2001; 35: 153–156 [DOI] [PubMed] [Google Scholar]
- 22. Mehalow AK, Kameya S, Smith RS, et al. CRB1 is essential for external limiting membrane integrity and photoreceptor morphogenesis in the mammalian retina. Hum Mol Genet. 2003; 12: 2179–2189 [DOI] [PubMed] [Google Scholar]
- 23. Samardzija M, Neuhauss SCF, Joly S, Kurz-Levin M, Grimm C. Animal models for retinal degeneration. In: Pang I, Clark AF. ed Animal Models for Retinal Diseases. New York, NY: The Humana Press, Inc.; 2010: 51–79 [Google Scholar]
- 24. Sakamoto K, McCluskey M, Wensel TG, Naggert JK, Nishina PM. New mouse models for recessive retinitis pigmentosa caused by mutations in the Pde6a gene. Hum Mol Genet. 2009; 18: 178–192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ikeda S, Shiva N, Ikeda A, et al. Retinal degeneration but not obesity is observed in null mutants of the tubby-like protein 1 gene. Hum Mol Genet. 2000; 9: 155–163 [DOI] [PubMed] [Google Scholar]