

Abstract
Neutrophil arrest and migration on inflamed endothelium involves a conformational shift in CD11a/CD18 (LFA-1) to a high affinity and clustered state that determines the strength and lifetime of bond formation with ICAM-1. Cytoskeletal adaptor proteins Kindlin-3 and Talin-1 anchor clustered LFA-1 to the cytoskeleton and facilitate the transition from neutrophil rolling to arrest. We recently reported that tensile force acts on LFA-1 bonds inducing their colocalization with Orai1, the predominant membrane store operated Ca2+ channel that co-operates with the endoplasmic reticulum to elicit cytosolic flux. Since Kindlin-3 was recently reported to initiate LFA-1 clustering in lymphocytes, we hypothesized that it cooperates with Orai1 and LFA-1 in signaling local Ca2+ flux necessary for shear resistant neutrophil arrest. Employing microfluidic flow channels combined with total internal reflection fluorescence microscopy we applied defined shear stress to low or high affinity LFA-1 and imaged the spatiotemporal regulation of bond formation with Kindlin-3 recruitment and Ca2+ influx. Orai1 and Kindlin-3 genes were silenced in neutrophil-like HL-60 cells in order to assess their respective roles in this process. Kindlin-3 was enriched within focal clusters of high affinity LFA-1, which promoted physical linkage with Orai1. This macromolecular complex functioned to amplify inside-out Ca2+ signaling in response to IL-8 stimulation by catalyzing an increased density of Talin-1 and consolidating LFA-1 clusters within sites of contact with ICAM-1. In this manner, neutrophils utilize focal adhesions as mechanosensors that convert shear stress mediated tensile force into local bursts of Ca2+ influx that catalyze cytoskeletal engagement and an adhesion strengthened migratory phenotype.
Introduction
Leukocyte recruitment requires activation of β2-integrins that upshift their affinity state to form bonds with ICAM-1 on the endothelium and mediate shear resistant adhesion at sites of inflammation. Integrin activation is a bi-directional process with a common feature being the conversion to a high affinity ligand binding state. Inside-out signaling is elicited following ligation of G-protein coupled receptors (GPCR) and also in response to binding of glycosylated ligands of selectins that cluster and signal during leukocyte rolling on inflamed endothelium(1). Outside-in signaling is another process that involves force transmission through membrane clusters of high affinity integrins that is initiated after leukocytes roll and arrest under the hydrodynamic force of blood flow (2). Calcium flux is a common secondary messenger whose cytosolic levels are amplified in response to superposition of inside-out and outside-in signaling (3). Downstream of Ca2+ flux is activation of Src family kinases and cytosolic association of Talin-1, Kindlin-3 and α-actinin to the integrin cytoplasmic domain. These events are collectively associated with conversion of LFA-1 to a high affinity and clustered configuration that supports shear strengthening of arrested leukocytes (4-8).
Repulsive forces imposed by hydrodynamic shear stress are implicated in stabilizing LFA-1 in a high affinity conformation and in synchronizing the transition from leukocyte rolling to arrest and transmigration at sites of inflammation (9-11). The mechanism is not well understood, but appears to involve outside-in transduction of force from the I-domain on the α subunit and I-like domain on the β subunit transmembrane to the cytoplasmic domain of LFA-1 (12-14). We have recently reported that shear stress acting on durable high affinity LFA-1/ICAM-1 bonds elicits Ca2+ flux and F-actin polymerization, providing spatial cues to guide neutrophil migration (10, 15). However, the precise mechanism by which shear force elicits localized intracellular calcium release at the site of LFA-1 engagement has yet to be defined.
Conversion of LFA-1 to a high affinity state can be induced allosterically by the binding of divalent cations manganese (Mn2+) or magnesium (Mg2+) to the MIDAS domain, or through binding of antibodies such as 240Q that recognize the IDAS domain on CD18 resulting in a conformational upshift to high affinity (16, 17). Conversely, binding of TS1/18 or Lovastatin to CD18 induce an allosteric downshift in LFA-1 to a low affinity state (18). Following leukocyte signaling, LFA-1 receptor clustering is another event that promotes the formation of multivalent and durable bond formation with ICAM-1 (18-20). Intracellular calcium flux via GPCR activation plays a crucial role in converting integrins to a high affinity and clustered state (21, 22). Local calcium flux in leukocytes is regulated by communication between endoplasmic reticular (ER) stores and the membrane calcium channel Orai1 that mediates store operated calcium entry (SOCE) (23, 24). Orai1 is important for coordinating the transition from an arrested neutrophil to one projecting pseudopods and directionally migrating (10, 15, 25). This mechanism involves spatial regulation of calcium transients that occur proximal to integrin engagement and uptake of tensile forces imposed by shear stress (14, 22, 25). How Ca2+ flux is initiated by cooperation between high affinity LFA-1 and Orai1, and how it elicits membrane clustering required for shear strengthened neutrophil adhesion, is the focus of this study.
LFA-1 affinity upshift and clustering requires the activity of Src family kinases and the cytoskeletal adaptor proteins Talin-1 and Kindlin-3, which localize within membrane domains to alter LFA-1 function (4, 26). Talin-1 concentrates at focal adhesions along with paxillin to provide linkage of LFA-1 clusters to F-actin that in turn drives cell polarization (27). While Talin-1 is necessary for LFA-1 extension from a low to intermediate affinity state, Kindlin-3 functions to stabilize LFA-1 at high affinity and induce sub-micron size clusters. In T cells and neutrophils it has been demonstrated that Kindlin-3 promotes cell spreading and adhesion strengthening (6, 28, 29). Since Talin-1 and Kindlin-3 bind to distinct sites on the cytodomain of the β2-subunit they may physically interact to facilitate focal clustering of LFA-1 (30). Precisely how and in what sequence Kindlin-3 and Talin-1 interact with LFA-1 bonds as tension is taken up during cell deceleration to arrest and their role in initiating calcium flux via Orai1 remain key unanswered questions.
Here, we applied total internal reflection fluorescence (TIRF) to image the spatio-temporal events associated with LFA-1 conversion from low to high affinity and its association with Kindlin-3 and Orai1 under hydrodynamic shear stress during neutrophil adhesion strengthening. We hypothesized that high affinity LFA-1 bond formation and uptake of tensile force were necessary to catalyze downstream events required for neutrophil adhesion strengthening including Kindlin-3 recruitment and Orai1 mediated calcium flux. Tensile force facilitated physical association of Kindlin-3 with the cytodomain of LFA-1 and this step was necessary for local calcium influx and subsequent cytoskeletal assembly necessary for stable adhesion at stresses up to 40 dyne/cm2. Knockdown of Kindlin-3 abrogated colocalization of Orai1 with high affinity LFA-1, diminished Ca2+ flux, and the recruitment of Talin-1 to adhesive contacts. Knockdown of Orai1 or chelating intracellular Ca2+ flux eliminated the consolidation of LFA-1 into macroclusters, which correlated with diminished adhesion strengthening. These studies reveal how durable multivalent LFA-1/ICAM-1 bonds serve as mechanosensors that direct PMN cytoskeletal activity by transmission of tensile force to a supramolecular complex that triggers Ca2+ influx at sites of adhesive contact.
Materials and Methods
Antibodies, small molecules, and other reagents
Human and mouse ICAM-1-Fc and E-selectin-Fc were purchased from R&D systems (Minneapolis, Mn). Protein A/G was purchased from Pierce (Rockford, IL). 2-Aminoethoxydiphenyl borate (2-APB) was purchased from EMD Biosciences (San Diego, CA), resuspended in dry DMSO at concentration of 100 mM, and stored at -80°C under N2. Thapsigargin was purchased from Invitrogen (Carlsbad, CA), and resuspended to 1 mM in DMSO the same day as the experiment. Anti-CD18 327C, anti-LFA-1 TS2/4, anti-CD18 (240Q) were obtained as a generous gift from Eli Lilly Corp (Indianapollis IN). Anti-CD18 TS1/18, anti-CD45 and anti-Mac-1 ICRF44 were all purchased from Biolegend (San Diego, CA). Anti-CD18 IB4 purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Small molecule Lovastatin purchased from Calbiochem. Polyclonal anti-mouse Orai1, anti-human and anti-mouse Kindlin-3, anti-human and anti-mouse Talin antibodies were purchased from Abcam (Cambridge, MA). Goat anti rabbit-Alexa488 polyclonal secondary antibody and normal goat serum was purchased from Invitrogen (Carlsbad, CA) and Thermo-Fisher Scientific (Waltham, MA), respectively. Antibodies were used at a saturating concentration of 5μg/ml or per manufacturer's instructions. Human promyelocytic leukemia cells (HL-60 cells) were purchased from ATCC (Manassas, VA). Quantum simply cellular anti-mouse IgG beads were purchased from Bangs Laboratories (IN). Control and Orai1 siRNA were purchased from Qiagen (Valenica, CA).
Neutrophil isolation
Whole blood was obtained from consenting donors under an approved University of California, Davis Institutional Review Board protocol and layered over PMN separation media purchased from Thermo-Fisher Scientific, as previously described (31). After centrifugation, PMN were extracted from the appropriate density layer and washed with HEPES buffered salt solution. Mice heterozygous for expression of Orai1 were a generous gift from the laboratory of Dr. Anjana Rao (La Jolla Institute for Allergy and Immunology, CA) (32). Mice were genotyped from tail clippings by PCR, and PMN were isolated from the bone marrow of littermate Orai1+/− and wild type ICR strain mice as previously described (15, 33).
siRNA transfection
HL-60 cells were transfected with control scrambled siRNA or with Orai1-specific siRNA (QIAGEN) by electroporation using the Amaxa Nucleofector 4D (Lonza) according to the manufacturer's instructions. HL-60 cells were given fresh media 24 hours before electroporation to maximize survival. Cells were pelleted from media, resususpended in high resistance nuclefection buffer (Lonza) in the presence of 100nM siRNA, and electroporated. Immediately after electroporation, cells were transferred to 37°C media containing 1.3% DMSO and differentiated for 3 days under the influence of siRNA.
Adhesion assays
Human and Mouse ICAM-1, and antibodies 240Q, TS1/18, CD45, were absorbed at 5μg/ml to coverslips cleaned with piranha solution (34) and coated with aminosilane. PMN were perfused through custom made microfluidic chambers over substrates at 4 dynes/cm2 and imaged for calcium flux or fixed and labeled for Kindlin-3, Talin-1 and high affinity CD18 (327C) as previously described (15). For adhesion strengthening experiments, control, Talin and Kindlin-3 shRNA, control and Orai1 siRNA transfected HL-60 cells were differentiated to a neutrophil phenotype over 3 days with 1.3% DMSO or human isolated PMN were allowed to settle over an ICAM-1+240Q substrate derivitized on the substrate at 1:1 ratio (5 μg/ml each) to activate and stabilize high affinity LFA-1 at adhesive contact sites. Adhesion strengthening experiments performed on bone marrow derived PMN from Orai1+/+ and Orai1+/− mice were isolated and allowed to settle on ICAM-1 substrate in the presence of Mn2+. Cells were treated with Mac-1 excess blocking antibody in the media (ICRF44 for human and M1/70 for mouse) to ensure LFA-1 dependent adhesion. Shear was then ramped at 30 second intervals from 0, 4, 10, 20, 40 dynes/cm2 and the number of cells that remained adhered were measured over 5 separate fields of view at each shear level. In order to study the role of calcium flux in adhesion strengthening, PMN were treated with 50 μM BAPTA or 100 μM 2-APB prior to each experiment.
Real-time calcium imaging of PMN in microchannels
PMN (human and mouse) were suspended at a concentration of 2 × 106/mL in HEPES buffered salt solution and labeled with Fura-2 AM for 30 minutes at 37°C. Cells were then washed and resuspended in HEPES buffered salt solution. Labeled cells were perfused into a microfluidic flow chamber and imaged as previously described.(9, 35) Briefly, cells were drawn into the microfluidic chamber at a calculated shear stress of 4 dynes/cm2 (i.e. venular magnitude of shear stress) and sequentially imaged during interaction with the substrate with alternating excitation by 340nM and 380nM light generated by a mercury arc lamp attached to a filter wheel with 0.1 second frequency excitation. Images were acquired with an Orca-ER camera (Hamamatsu Corp) coupled to a Nikon 1200 microscope running Simple PCI 5.3 software. Image sequences were analyzed for the ratio between emission at 340nM and 380nM using custom macros written for Image Pro Plus 5.1. During analysis, the average intensity of each cell was identified in a confined area of interest around each cell for both the 340nM and 380nM exposure. This method of cell identification and local overlap accounted for any motion of rolling PMN during image acquisition.
TIRF in-plane microscopy of LFA-1, Kindlin-3 and Talin-1 expression
Human and mouse PMN were treated with or without Mn2+ or lovastatin, and perfused over ICAM-1, 240Q, TS1/18, or CD45 coated glass coverslips in a microfluidic flow chamber at a calculated shear stress of 4 dynes/cm2, fixed with 4% PFA, permeabilized with 0.1% Triton-X-100 and labeled with primary and secondary antibodies to specific proteins. Cells were imaged via total internal reflection fluorescence (TIRF) microscopy, which excites fluorophores within a focal height of ∼100 nm above the glass coverslip-cell membrane surface. High affinity CD18 receptor expression per cell was determined by comparing the MFI of bound 327C-Alexa-488 to Quantum Simply Cellular calibration bead set containing defined numbers of goat-anti-mouse binding sites on their surface. From this analysis, a linear relation between MFI and receptor expression was determined to convert TIRF obtained high affinity CD18 fluorescence to receptor # (see Supplementary Figure 2). An LFA-1 cluster was defined as an area of uniform fluorescence intensity registering two standard deviations above the mean fluorescence intensity of the cell. Once this threshold was set for each cell, cluster area was measured in μm2. Average cellular MFI and cluster MFI for each condition was converted into average receptor numbers per cell or per cluster using the calibration bead set as described above.
In vivo imaging of Orai1+/+ and Orai1−/− PMN recruitment to wound sites
EGFP-Orai1+/− and EGFP-Orai1−/− mice were generated by crossbreeding EGFP-lysozyme-knock in mice with Orai1+/− mice. Orai1+/+ and Orai1−/− mice were anesthetized with 2% and the back skin hair was then removed with a mechanical shaver. After sterilization with 10% wt/vol povidine-iodine and 70% alcohol, 6 mm in diameter circular full-thickness wound was made using a skin biopsy punch (Robbins Instruments, Chatham, NJ). The in vivo imaging of EGFP neutrophils appearing on the site of back skin wound was performed using the whole body small animal fluorescence imaging system (Xenogen IVIS 100 system, Xenogen) as described recently(36). Mice were anesthetized with 2% isoflurane and then put into the imaging chamber of the system. The EGFP-expressing neutrophils within the wound area were visualized using a GFP filter (excitation at 445–490 nm and emission at 515–575 nm) at an exposure time of 1 s. Live Image Pro. 2.5 software (Caliper Life Sciences), and fluorescence intensities expressed as average radiance (photons per cm2 per steradian) were measured by drawing a circular region of interest over the entire wound area
Immunoprecipitation of LFA-1 and CD45
Human PMN were isolated from whole blood or Control and Kindlin-3 shRNA transfected HL-60 cells that were differentiated for 3 days with 1.3% DMSO and allowed to settle on a 240Q, TS1/18, or CD45 antibody substrate in the microfluidic device in the presence or absence of shear stress. These cells bound to the microfluidic channel substrate were then sheared and lysed with IP lysis buffer containing protease inhibitor cocktail in the channel. 1mg protein lysates from cells bound to the substrate were collected from the microfluidic channel and incubated with anti-LFA-1 antibody TS2/4 or anti-CD45 antibody for 2 hours on ice. Protein A/G agarose resin from Pierce classic IP kit was used to pull down LFA-1 or CD45 and the resin was washed 4 times with IP lysis buffer (Pierce). Samples were boiled in SDS-PAGE sample buffer and equal concentrations of protein sample were subjected to electrophoresis. Polyvinylidene membranes were blocked with 5% Milk for 1 hour, probed with primary antibodies and HRP labeled secondary antibodies and developed using the ECL system.
Statistical Analysis
Data analysis was performed using GraphPad Prism version 5.0 software (GraphPad Software, San Diego, CA.). Differences between single pairs of conditions were analyzed for significance by two tailed unpaired Student t-test. For datasets derived from exclusively paired observations, p-values were obtained by two-tailed paired t-test. All error bars are mean +/− SEM based on the number of independent experiments indicated in the figure legends.
Results
Kindlin-3 is required for PMN adhesion strengthening through LFA-1
Deceleration of neutrophils during the transition to a stable arrested state involves a rapid shift in LFA-1 from an extended intermediate affinity state that supports rolling to a fully activated high affinity conformation (14, 37). While it is known that Kindlin-3 and Talin-1 are involved in LFA-1 activation (6, 28, 29, 38), their precise role in adhesion strengthening of PMN following arrest through high affinity LFA-1/ICAM-1 bond formation has not been elucidated. Kindlin-3 and Talin-1 expression was knocked down by ∼80% in promyelocytic HL-60 cells using lentiviral delivery of shRNA (Supplementary Fig 1). These HL-60 cells were then induced to differentiate to PMN and assayed for their adhesive capacity. We endeavored to image LFA-1/ICAM-1 bond formation on PMN activated by substrate bound allosteric antibody 240Q, which initiates adhesion from the outside-in as CD18 is engaged and forms durable multivalent bonds (39). PMN were perfused through the microfluidic flow channel and allowed to settle on substrates coated at a 1:1 protein ratio with ICAM-1/Fc and 240Q. This surface mimics an inflammatory substrate by shifting CD18 to high affinity upon contact, and in the presence of a function blocking antibody to Mac-1 receptor, activated high affinity LFA-1 bond formation with ICAM-1 at levels commensurate with IL8 stimulation of CXCR (18). Following attachment, shear was incrementally stepped up from 0-40 dynes/cm2 and the fraction of PMN that remained adherent to the substrate was measured (Fig 1A). Kindlin-3 knockdown cells exhibited a 60% defect in adhesion strengthening as compared to control scrambled shRNA transfected HL-60. In comparison, Talin-1 knockdown cells bound robustly under high shear stress and exhibited only a ∼20% defect in adhesion strengthening.
Figure 1. Kindlin-3 is required for PMN adhesion strengthening via clustered high affinity LFA-1.
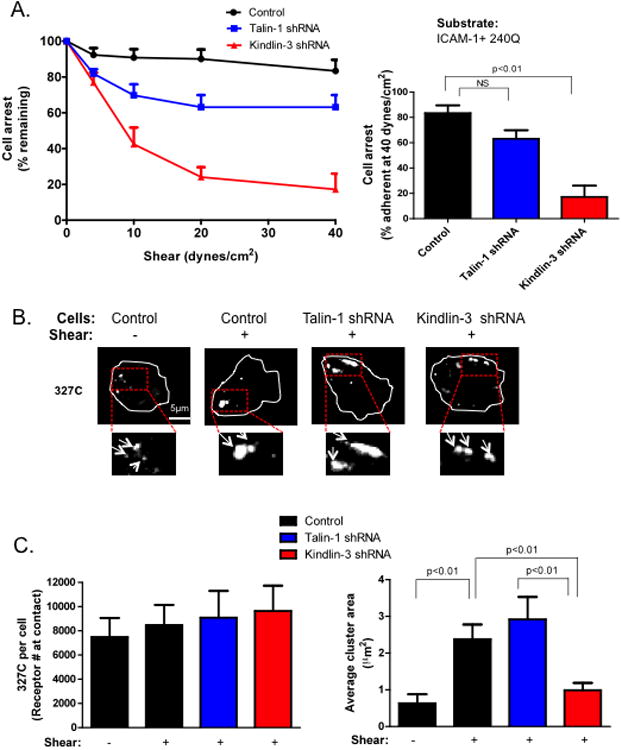
A) Control, Kindlin-3 and Talin-1 shRNA transfected HL-60 cells were differentiated into neutrophil-like cells by culture in DMSO for three days, perfused over a substrate coated with ICAM-1 and 240Q and shear stress was incremented from 0, 4, 10, 20, 40 dynes/cm2 in 30 second intervals. The fraction of cells remaining arrested at the end of each interval and at 40 dynes/cm2 was plotted. B) Control, Kindlin-3, and Talin-1 shRNA cells were pre-labeled with 327C at 10μg/ml and then perfused over the ICAM-1/240Q substrate at 4 dynes/cm2 for 2 minutes, fixed and imaged for measurement of high affinity CD18 expression and topography. Representative images are shown and boxed areas were magnified 2X. C) Mean fluorescence intensities of antibody binding were converted to receptor site number using Simply Cellular calibration beads as described in the Materials and Methods. Average cluster area in μm2 of MFI registering 2 S.D. above background was plotted. To ensure LFA-1 dependent adhesion, cells were pretreated with excess anti-Mac-1 ICRF44 in all experiments. Data shown is mean+/−SEM from 3 independent experiments.
To determine whether the topography of LFA-1 bonds influenced adhesion strengthening, high affinity CD18 was imaged by TIRF microscopy which limits fluorescence detection to within ∼0.1 μm of the plane of adhesive contact (Fig 1B). Activated CD18 receptors were imaged by labeling adherent PMN with 327C antibody and the number of binding sites were quantified using a bead set calibrated at defined receptor density (Supplementary Figure 2). HL-60 derived PMN sedimented to the substrate under static conditions were bound by ∼8000 high affinity CD18 sites. These receptors spontaneously coalesced into numerous clusters containing an average of ∼500 receptors (+/−100 receptors) of ∼0.5 μm2 in size (Fig 1B, C). With the application of 4 dynes/cm2 of shear stress for 2 minutes, PMN maintained an equivalent expression level of high affinity CD18. However, the smaller high affinity clusters coalesced into macromolecular clusters of ∼3000 receptors with an average area of ∼3μm2. These often redistributed to the PMN uropod as cells adopted an elongated shape. Kindlin-3 knockdown in HL-60 expressed equivalent expression of high affinity CD18, but exhibited impaired bond clustering in response to shear stress. In contrast, Talin-1 knockdown cells exhibited levels of bond clustering equivalent to control sheared cells, commensurate with the mild defect observed in adhesion strengthening. Thus, the consolidation of LFA-1 bond clusters within the plane of adhesion is dependent on application of shear stress and cytoplasmic association of Kindlin-3, but not Talin-1.
Kindlin-3 associates with high affinity LFA-1 under tension
Kindlin-3 has previously been shown to associate with the β-subunit of LFA-1 and mediate clustering in response to activation through TCR signaling (29). Since tensile force acting on high affinity LFA-1 promoted bond clustering, we next determined whether this required physical association with Kindlin-3. PMN were allowed to sediment at low shear flow on a substrate coated with 240Q to activate high affinity, or TS1/18 to stabilize LFA-1 in a low affinity state (Fig 2A). An equivalent number of LFA-1 were engaged within the plane of contact in the absence as compared to the presence of shear stress, as confirmed by imaging with fluorescence antibody to LFA-1 and high affinity CD18 (327C). The static condition correlated with a low albeit significant baseline level of Kindlin-3 expression. Application of shear stress to PMN resulted in a 4-fold increase in high affinity LFA-1 that corresponded to a ∼10-fold increase in Kindlin-3 expression. PMN stabilized at low affinity via TS1/18 did not exhibit this increase in Kindlin-3 expression. To confirm the requirement for both high affinity and tensile force on LFA-1, we infused Mn2+ into the microchannel to allosterically activate LFA-1 bond formation (Fig 2A). Only in the presence of shear stress did Mn2+ elicit Kindlin-3 localization to adhesive contact sites. Addition of Lovastatin, to stabilize LFA-1 in a low affinity conformation, maintained Kindlin-3 at a low baseline level similar to the static condition.
Figure 2. Kindlin-3 enrichment at sites of high affinity LFA-1 requires presence of shear force.
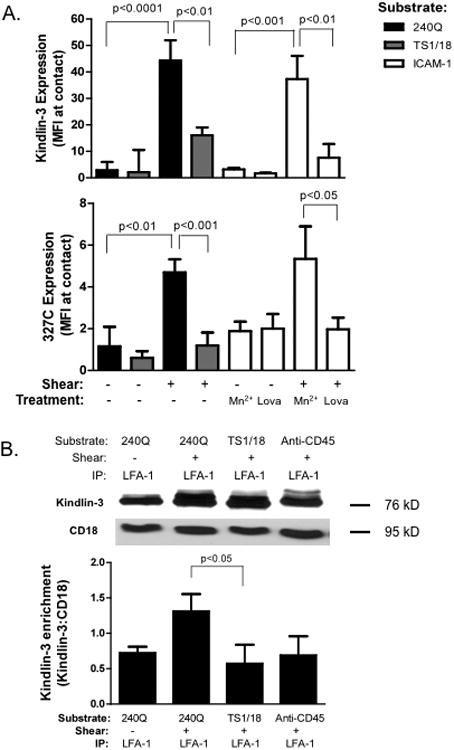
A) Human PMN, in the presence of 327C mAb were perfused over a substrate coated with 240Q or TS1/18 at a shear stress of 4 dynes/cm2, or sedimented on the substrate under static conditions. Samples on a substrate coated with ICAM-1/Fc were also stabilized in a high affinity state by treatment with Mn2+ or converted to low affinity by addition of Lovastatin in the presence of Mn2+ and exposed to shear stress, or maintained under static conditions. Cells were fixed, permeabilized and labeled for Kindlin-3. TIRF microscopy was used to detect expression of both Kindlin-3 and 327C labeled high affinity CD18 sites limited to the plane of adhesive contact. B) PMN were isolated from whole blood and perfused over a substrate of 240Q, TS1/18, or anti-CD45 substrate under static or 4 dynes/cm2 shear stress for 2 minutes. Cells remaining adherent to each substrate were then lysed and LFA-1 was immunoprecipitated and Western blots performed to detect Kindlin-3. Data depicts Kindlin-3 enrichment with CD18 as the density of each blot was normalized by CD18 expression harvested from each condition. Western blot is representative of 3 separate experiments. To ensure LFA-1 dependent adhesion, cells were pretreated with anti-Mac-1 ICRF44 for all experiments. Data shown is mean+/− SEM from 3 separate experiments.
We next examined if Kindlin-3 bound directly to the cytodomain of LFA-1 and its regulation by shifts in integrin conformation and the onset of shear stress. PMN were captured on a substrate of TS1/18, 240Q, or anti-CD45. Engagement of CD45 receptor served as a control to assess whether membrane tension alone elicited Kindlin-3 localization to sites of membrane contact under shear stress, since it is expressed at levels commensurate with CD18. In order to collect the membrane fraction of PMN tightly adherent to the indicated substrates, non-adherent and loosely bound PMN were sheared off the substrate prior to infusion of cell lysis buffer into the microchannels (Fig 2B). LFA-1 was isolated by immunoprecipitation on a protein A/G agarose column and the relative amount of Kindlin-3 pulled down on PMN bound to each substrate was detected by Western blot. Kindlin-3 was enriched by 2-fold on LFA-1 stabilized at high affinity on 240Q compared to low affinity on TS1/18, or on PMN isolated under static conditions. Kindlin-3 isolated from PMN captured on a substrate of anti-CD45 was not significantly different than those bound under static conditions. Taken together, these data indicate that Kindlin-3 rapidly recruits to the cytodomain of LFA-1 and that its association is amplified by transmission of tensile force acting on high affinity LFA-1.
Calcium flux via CRAC is required for adhesion strengthening
Intracellular calcium flux initiated by membrane CRAC channels cooperates with GPCR signaling in synchronizing PMN arrest and shape polarization (15). We have recently reported that tensile force acting on LFA-1/ICAM-1 bonds clustered at a single site of adhesive contact activates Ca2+ influx by colocalizing with Orai1 (10). To assess the dependence of LFA-1 bond clustering and adhesion strengthening on intracellular Ca2+, PMN were depleted of calcium with BAPTA or treated with 2-APB to block CRAC channel mediated influx. Since we have previously reported that Orai1 is the predominant CRAC channel mediating Ca2+ influx in arrested PMNs (40), HL-60 cells were transfected with either a control scrambled siRNA or one specific to Orai1, and then differentiated into PMN. Cells were settled on a substrate of ICAM-1/Fc and 240Q at low shear and then stress was ramped in increments up to 40 dynes/cm2 and cell detachment was quantified. Orai1 knockdown HL-60 exhibited a ∼50% knockdown of Orai1 protein, which resulted in ∼50% defect in Ca2+ flux. These PMN weakly adhered to the substrate with only ∼40% cells remaining firmly bound as compared to ∼90% of the control siRNA transfected cells (Figure 3A and Supplementary Figure 3A and B). Similarly, PMN treated with BAPTA or 2-APB, displayed a 2-fold decrease in Ca2+ flux as compared to the control untreated PMN and bound weakly to the substrate, with only ∼20% remaining adherent as compared to 70% of control PMNs at 40 dyne/cm2 (Fig 3A and Supplementary Figure 3C). We next confirmed the defect in adhesion strengthening in PMN isolated from Orai1+/+ versus Orai1+/− mice. PMN were activated with Mn2+ and allowed to sediment on an ICAM-1 coated substrate and assayed for adhesion strengthening. Orai1+/− mice exhibited 30% less stably adherent PMN as compared to Orai1+/+ littermate controls at maximum shear stress (Supplementary Figure 4A) This defect was LFA-1 dependent since addition of a function blocking antibody to mouse β1-integrin abrogated the remaining 50% strongly adherent PMN (Data not shown). We conclude that Orai1 expression and its function in force mediated Ca2+ flux is required for durable adhesion at high shear stress in both human and mouse PMN.
Figure 3. Calcium flux via CRAC is required for adhesion strengthening.
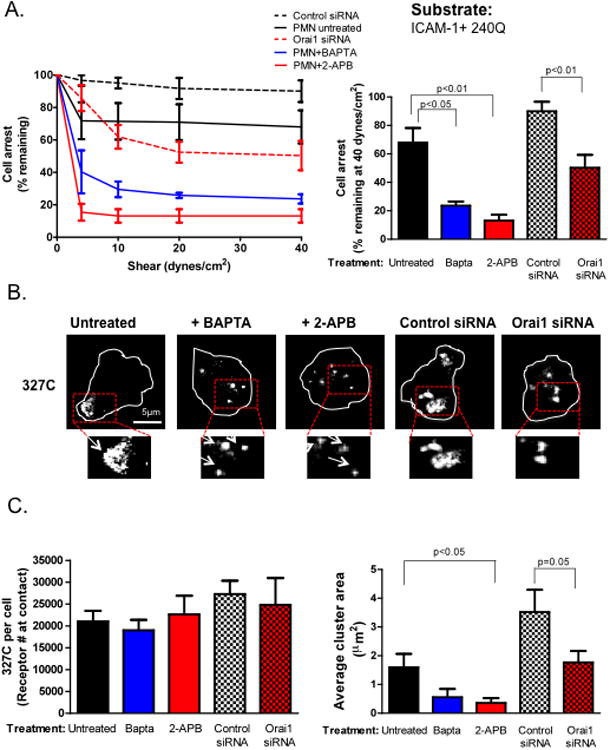
A) PMN were isolated from whole blood and treated with BAPTA or 2-APB, or HL-60 cells were transfected with control scrambled or Orai1 specific siRNA, differentiated over 3 days and perfused over an ICAM-1+240Q coated substrate. Shear was ramped from 0-40 dynes/cm2 at 30 second intervals and fraction of cells remaining arrested was measured over time. Percent of PMN remaining adherent at 40 relative to 0 dynes/cm2 is plotted. B) Cells for each condition were also labeled with 327C-488Alexa and then perfused over the ICAM-1/240Q substrate at 4 dynes/cm2 for 2 minutes, fixed and imaged for high affinity CD18 expression and topography. Representative images are shown and boxed regions were magnified 2X and shown below. C) 327C expression was quantified and the mean fluorescence intensities of antibody binding were converted to receptor site numbers using Simply Cellular calibration beads as described in the Materials and Methods. Average cluster area in μm2 of MFI registering 2 S.D. above background was plotted for each condition. To ensure LFA-1 dependent adhesion, cells were pretreated with anti-Mac-1 ICRF44 for all experiments. Data shown is mean+/− SEM from 3 separate experiments and representative images are shown.
We investigated whether the Orai1 mediated defect in adhesion strengthening translated into impaired PMN recruitment to a skin wound in a mouse model of acute inflammation. A full thickness (6 mm diameter) sterile wound was created on the flank of lys-EGFP knockin mice whose mature PMN upregulate lysozyme and express enhanced green fluorescence protein (EGFP-PMN)(40). Calcium levels measured in PMN from mice heterozygous or homozygous for Orai1 have previously been reported to express ∼50% that of wild-type littermates (10, 32). Lys-EGFP mice were crossbred with Orai1+/+ or Orai1−/− mice in order to non-invasively quantify PMN recruitment using whole animal fluorescence imaging of EGFP-PMN within the wound site (Supplementary Fig 4B and C). While PMN from both Orai1−/− and WT control mice demonstrated peak infiltration into wound at 24 hours, Orai1−/− exhibited ∼30% less PMN recruitment to the wound as compared to WT controls. PMN numbers in Orai1−/− mice rapidly declined in the wound following 48 hours, while the EGFP signal in Orai1+/+ were maintained significantly above Orai1−/− knockouts out to day 6 of injury. Spatial imaging of EGFP-PMN fluorescence within the wound revealed the highest concentrations in a circumferential zone just inside the margin of injury and PMN numbered significantly higher in the WT than Orai1−/− mice. These data clearly reveal a defect in the capacity of PMN to efficiently recruit into the wound margin and suggest that Orai1 participates in optimum PMN arrest and migration into a site of acute inflammatory injury.
To examine the mechanism by which Ca2+ flux played a role in mediating adhesion strengthening, we next examined the topography of high affinity LFA-1 induced by membrane contact with 240Q and ICAM-1/Fc. Allosteric induction of high affinity CD18 activated ∼20,000 sites per PMN, attributed to the processes of siRNA transfection and subsequent differentiation into PMN (Fig. 3B and 3C). As in Kindlin-3 or Talin-1 knockdown, this was not diminished by Ca2+ inhibition with BAPTA, 2-APB, or Orai1 knockdown. However, shear stress induced coalescence of LFA-1 bonds into macromolecular high affinity clusters measuring ∼3.5μm2 (∼9000 receptors) in isolated PMN, and ∼1.5μm2 (∼4000 receptors) in HL-60 controls. Coalescence of these clusters was abrogated by inhibition of Ca2+ flux and knockdown of Orai1, respectively. These data reveal that Ca2+ flux via Orai1 is necessary for subsequent consolidation of high affinity LFA-1 into macromolecular clusters and concomitant adhesion strengthening of PMN.
Kindlin-3, but not Talin-1, recruitment to adhesive contacts is independent of calcium flux
To define the intracellular mechanisms underlying impaired arrest strengthening, the relative capacity of Talin-1 and Kindlin-3 to recruit to adhesive sites and the dependence on Ca2+ influx through Orai1 was examined. PMN were exposed to shear flow, fixed/permeabilized, and then labeled with fluorescent antibodies to Talin-1 and Kindlin-3 before imaging by TIRF microscopy (Fig 4). Immunofluorescence of human PMN treated with BAPTA or 2-APB, and mouse PMN isolated from the bone marrow of Orai1−/− were compared with littermate WT controls. Activation was induced by 240Q on human PMN, and Mn2+ was added to mouse PMN to stabilize high affinity bond formation between LFA-1 and ICAM-1 on the substrate. Pharmacological inhibition with 2-APB or BAPTA each significantly diminished the enrichment of Talin-1 at sites of adhesive contact (Fig 4A). Likewise Talin-1 enrichment was also decreased by ∼1-fold in Orai1−/− PMN. In contrast, neither BAPTA, 2-APB, nor Orai1 knockout in mouse PMN reduced Kindlin-3 enrichment within sites of adhesive contact on sheared PMN (Fig 4B). Thus, Talin-1 recruitment to LFA-1 bonds under tension is responsive to Ca2+ influx via Orai1, whereas Kindlin-3 is independent of intracellular Ca2+ levels.
Figure 4. Dependence of Talin-1 and Kindlin-3 recruitment to sites of adhesive contact on calcium flux via Orai1.
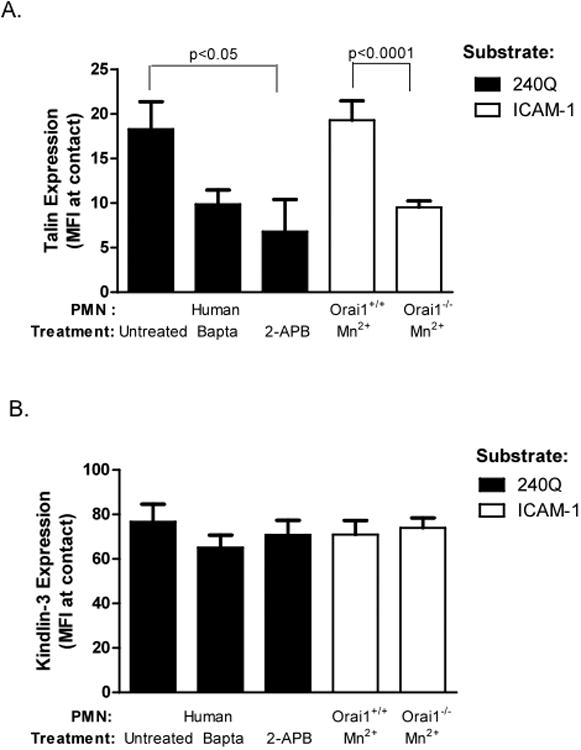
PMN were treated with BAPTA, 2-APB, and allowed to adhere on a substrate of 240Q antibody to stabilize high affinity LFA-1 bonds. Orai1+/+ and Orai1−/− PMN were isolated and LFA-1 was stabilized at high affinity by treatment with Mn2+ and perfused over an ICAM-1 substrate. Cells were fixed and labeled for A) Talin-1 and B) Kindlin-3 and their expression was imaged by TIRF. To ensure LFA-1 dependent adhesion, cells were pretreated with anti-Mac-1 ICRF44 or M1/70 for all experiments. Data shown is mean+/− SEM from 3 separate experiments.
Kindlin-3 is required for Talin/Orai1 recruitment and calcium influx at LFA-1 sites
Since Kindlin-3 redistribution to LFA-1 bonds is independent of intracellular Ca2+ flux, we hypothesized that physical association of Kindlin-3 with LFA-1 is required for Ca2+ influx during bond uptake of tensile force under shear flow. We first determined whether Ca2+ influx was defective in Kindlin-3 shRNA knockdown PMN. Calcium stores were depleted by incubation with 1μM Thapsigargin in Ca2+ free media and HL-60 derived PMN were then sheared on a substrate coated with TS1/18 or 240Q in order to stabilize LFA-1 binding sites at low or high affinity, respectively. To initiate the influx of Ca2+ through SOCE in arrested PMN, a bolus of 1.5mM Ca2+ buffer was perfused at 60 seconds (Fig 5A). PMN captured via high affinity LFA-1 registered ∼1000nM of Ca2+ flux, as compared to 500nM in PMN stabilized at low affinity. Kindlin-3 knockdown cells bound via high affinity LFA-1 did not exhibit a Ca2+ signal significantly different than cells bound via low affinity. In contrast, Talin-1 knockdown PMN exhibited levels of Ca2+ flux not significantly different from high affinity LFA-1 controls. These data suggest that Kindlin-3 is required for CRAC channel function induced by tensile force on high affinity LFA-1, while Talin-1 functions downstream of Ca2+ influx. This prompted examination of whether Kindlin-3 physically recruits Talin-1 and Orai1 to the cytodomain of LFA-1 in adherent and sheared PMN. Kindlin-3 knockdown or control differentiated HL-60 cells were bound to allosteric antibody substrates stabilized at a high or low affinity state. In the presence of shear stress, bound HL-60 were perfused with lysis buffer, then LFA-1 was isolated by immunoprecipitation for subsequent Western blot detection of Talin-1 and Orai1 (Fig 5B). Kindlin-3 knockdown cells contained ∼1-fold less Talin-1 and Orai1 as compared to control cells bound via high affinity LFA-1. Furthermore, immunoprecipitation with anti-CD45 pulled down an equivalent level of Talin-1 and Orai1 as PMN bound via low or high affinity LFA-1 under static conditions. These data indicate that shear acting on CD45 bonds is not sufficient to catalyze formation of a cytoskeletal complex of high affinity LFA-1, Orai1, and Talin-1. Rather, a sequence in which Kindlin-3 is enriched on high affinity LFA-1 under tension catalyzes localization with Orai1 mediated Ca2+ flux that in turn promotes Talin-1 binding and shear strengthened adhesion.
Figure 5. Kindlin-3 is required for calcium flux through SOCE and Talin1 and Orai1 recruitment to LFA-1 sites.
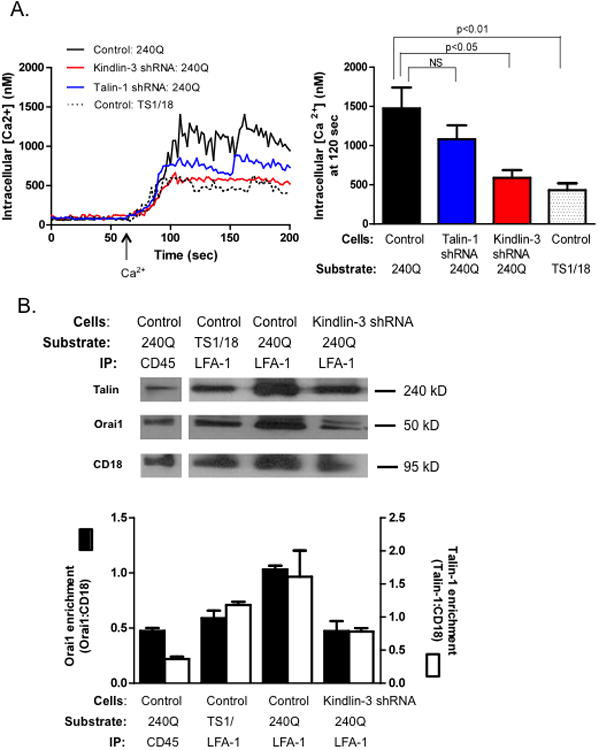
A) Control and Kindlin-3 shRNA transfected HL-60 cells, differentiated over 3 days were labeled with Fura-2 AM and treated with thapsigargin in presence of EGTA to deplete internal calcium stores and then perfused over a substrate of 240Q or TS1/18 at 4 dynes/cm2. Calcium buffer (1.5mM) was infused at 60 seconds and calcium flux was measured. Data shown is mean+/− SEM from 3 separate experiments B) Control and Kindlin-3 shRNA cells were perfused over a 240Q or TS1/18 substrate for 2 minutes, adherent cells were lysed and LFA-1 or CD45 was immunoprecipitated. Talin1 and Orai1 protein bound to LFA-1 and CD45 were detected by Western blot and data depicts Talin-1 and Orai1 association to CD18 as the density of each blot was normalized by CD18 expression measured for each condition. The two blot images for CD45 and LFA-1 IP are obtained from the same lane. Western blot is representative of n=3 separate observations. To ensure LFA-1 dependent adhesion, cells were pretreated with anti-Mac-1 ICRF44 for all experiments.
Chemokine amplification of calcium flux is mediated by LFA-1, Kindlin-3, and shear stress
Neutrophils rolling on E-selectin and arresting on ICAM-1 under shear flow exhibit a 100-fold increase in sensitivity to IL-8 signaling as compared to chemokine stimulation under static conditions as detected by the flux of intracellular Ca2+ on arrested cells (9). We investigated whether engagement of LFA-1/ICAM-1 bonds and Kindlin-3 function was necessary for this amplification in Ca2+ flux with increased dose of chemokine exposure of HL-60 arrested under shear stress. Differentiated control and Kindlin-3 shRNA transfected HL-60 cells were loaded with Fura-2 AM and perfused over an ICAM-1 substrate and the maximum level of intracellular Ca2+ flux was recorded following IL-8 perfusion (Fig 6A). Beginning at a baseline level of ∼150nM Ca2+ in adherent PMN stimulated at .01nM IL-8, a 10-fold increase up to ∼1300nM Ca2+ was detected at 1nM IL-8 and this increase required the presence of shear stress acting on high affinity LFA-1/ICAM-1 bonds. In contrast, Kindlin-3 knockdown cells barely registered an increase in Ca2+ flux under shear at maximum stimulation with 1nM IL-8 (Fig 6B). Talin-1 knockdown cells, however, exhibited equivalent levels of Ca2+ flux to the control cells at 1nM IL-8 under shear stress. A direct correlation was observed between the level of Ca2+ influx and the relative size of high affinity LFA-1 focal clusters supporting PMN arrest (Fig. 6B). These data suggest that IL-8 stimulated activation of LFA-1 involves not only an upshift to high affinity, but coalescence of focal clusters of LFA-1/ICAM-1 bonds in a process that is catalyzed by the level of chemokine stimulated Ca2+ influx. Shear stress imparted tensile force promotes Kindlin-3 association with LFA-1 and Orai1, which in turn mechano-transduced local Ca2+ influx that subsequently drives macromolecular clustering of LFA-1 bonds promoting durable adhesive contacts at sites of inflammation.
Figure 6. Shear stress, high affinity LFA-1, and Kindlin-3 amplify Ca2+ flux stimulated via IL-8.
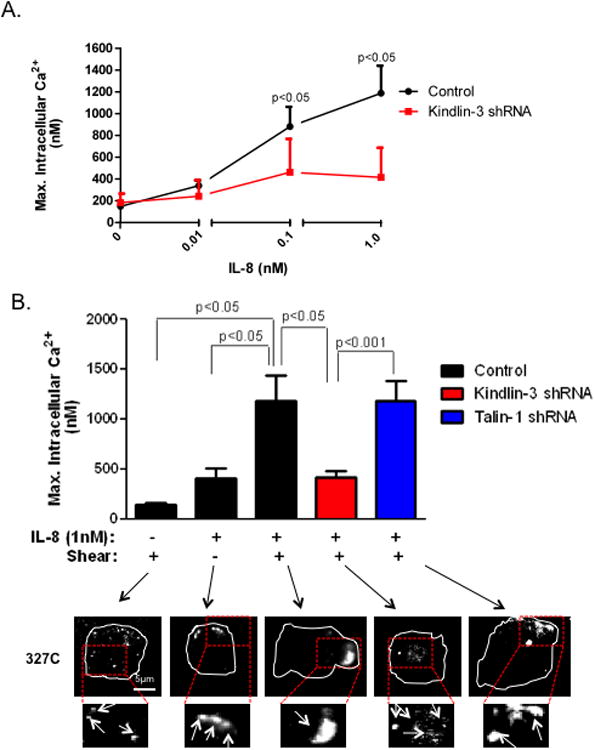
A) Control and Kindlin-3 shRNA HL-60 cells differentiated over 3 days, were labeled with Fura-2 AM and perfused over an ICAM-1 substrate. IL8 was perfused at indicated concentrations into the microfluidic channel to stimulate the cells and intracellular Ca2+ flux was measured over 2 minutes. Maximum Ca2+ flux elicited at each IL8 dose is plotted. B) Peak calcium values of control, Kindlin-3 and Talin1 shRNA differentiated HL-60 under different conditions were plotted and their representative 327C images detected within plane of adhesive contact using TIRF microscopy are shown. Representative images are shown and boxed regions were magnified 2X and shown below. To ensure LFA-1 dependent adhesion, cells were pretreated with anti-Mac-1 ICRF44 for all experiments. Data shown is mean+/− SEM from 3 separate experiments.
Discussion
Hemodynamic shear stress provides an ever-present repulsive force on leukocytes adherent at sites of inflammation. Leukocytes exploit this tensile force that is transmitted across the membrane via durable LFA-1/ICAM-1 bonds to generate integrin dependent outside-in signals that function to guide cytoskeletal assembly and cell migration (10, 11, 41, 42). Applying TIRF optics to image high affinity LFA-1 bonds and their interaction with adaptor proteins in the plasma membrane, we demonstrated that Kindlin-3 was central to conversion of mechanical force to intracellular signaling based upon the following criteria: 1) Kindlin-3 association with LFA-1 was significantly increased at sites of high affinity bond formation with ICAM-1 only in the presence of hydrodynamic shear stress. 2) Kindlin-3 and Orai1 mediated calcium flux were not required for LFA-1 conversion to high affinity, but mediated the coalescence of bond clusters with ICAM-1 that supported shear resistant adhesion. 3) Kindlin-3 association with LFA-1 was independent of cytosolic Ca2+, but necessary for shear mediated Ca2+ influx via formation of a focal complex with Orai1. 4) Influx of Ca2+ through Orai1 was essential for Talin-1 recruitment to LFA-1 bond clusters, which in turn linked the nascent focal complex to the actin cytoskeleton. Together, these data highlight the essential role of Kindlin-3 in transducing mechanical force necessary for the spatial regulation of Ca2+ influx and cytoskeletal assembly at focal sites where LFA-1 forms multivalent long-lived bonds with ICAM-1 that mediate leukocyte firm adhesion on inflamed endothelium.
Tensile force and LFA-1 conformation regulates outside-in signaling
An upshift in LFA-1 affinity is signaled by ligation of chemokine to GPCRs that rapidly induce microclustering of high affinity LFA-1 that are critical for PMN transition to stable arrest (2, 14, 20, 43). There is good evidence that stable bonds between high affinity LFA-1 and ICAM-1 serve as focal sites of intracellular signaling that elicit PMN shape polarization (4, 44). We examined the nature of this outside-in signaling by capturing PMN on a substrate presenting the allosteric antibody 240Q that binds to the IDAS domain of CD18 and promotes subsequent bond formation to co-localized ICAM-1/Fc heterodimers. Using microfluidic flow channels, we precisely incremented shear stress and examined the role of tensile force acting on either high affinity bond clusters, those stabilized at low affinity with TS1/18, or tethered via non-integrin CD45 membrane receptors. We demonstrated that tensile force acting on high affinity LFA-1 attachments was requisite for Ca2+ mediated receptor clustering, since equivalent numbers of high affinity LFA-1 was detected in the contact region of PMN adherent to the ICAM-1/240Q substrate under static conditions. Shear stress was required to elicit coalescence of 0.5 μm2 microclusters into ∼3μm2 macroclusters in the absence of a significant increase in the density of LFA-1 measured at ∼3000-4000 sites/μm2. Moreover, LFA-1 clustering was impaired in Kindlin-3 or Ca2+ depleted PMN, despite the presence of equivalent numbers LFA-1/ICAM-1 bonds. These data suggest that Kindlin-3 requires mechanical force to gain access to the LFA-1 cytodomain and subsequently provide linkage to Orai1.
LFA-1 mechanosignals via Orai1 and Ca2+ influx
Cytoplasmic Ca2+ functions as a versatile signaling molecule that functions to synchronize the transition from PMN rolling to arrest and shape polarization during its recruitment to sites of insult. GPCR engagement is followed by an intracellular Ca2+ burst mediated through phospholipase-C that releases intracellular stores via activation of inositol 1,4,5-trisphosphate associated with the ER (45, 46). Recently we reported that cooperation between release of ER calcium stores and activation of Ca2+ influx in arrested PMN occurs predominantly via Orai1 at sites of focal adhesion under tension (10, 15). In the current study, we demonstrated that chelating intracellular Ca2+ with BAPTA, impairing Ca2+ entry via SOCE with 2-APB, or in Orai1 knockdown PMN, the size of LFA-1 clusters was diminished resulting in a defect in adhesion strengthening at shear stress above 4 dynes/cm2. We further demonstrated the importance of Orai1 function for efficient PMN recruitment during acute inflammation in a skin wound model. Comparing EGFP-PMN recruitment dynamics in Orai1 deficient mice with WT controls revealed that PMN emigration was significantly impaired and consistent with published findings that Orai1 defective PMN are unable to induce LFA-1–mediated calcium flux and undergo F-actin driven shape change and migration under shear flow (10). Together, this supports the concept that LFA-1 clustering and polarization is tightly regulated by release of intracellular Ca2+ and downstream linkage to cytoskeletal proteins including Talin-1, F-actin, and Vinculin (26, 47, 48).
LFA-1 bond clusters amplify chemokine signaling dependent on association with Kindlin-3 and Ca2+ flux
In the absence of shear stress, PMN failed to elicit significant Ca2+ flux at maximal IL-8 stimulation, despite activation of ∼100,000 high affinity CD18 per cell (42, 49, 50). The requirement of shear in activating calcium flux in HL-60 differentiated PMN is qualitatively similar to previous observations on freshly isolated human PMN, wherein a 100-fold higher dose of IL-8 is necessary to elicit an equivalent level of Ca2+ flux under static versus shear flow conditions (9). The data support a mechanism by which engagement of high affinity LFA-1 acts synergistically with chemokine receptor signaling to effectively amplify release of intracellular Ca2+. Kindlin-3 association with high affinity LFA-1 was necessary for this amplification in Ca2+ release in the presence of shear flow. We speculate that Kindlin-3 functions as a mechanosensor since its association with LFA-1 did not itself require upshift to a high affinity conformation or Ca2+ signaling, but rather was dependent upon tensile force applied to high affinity bond clusters. Silencing expression of Kindlin-3 impaired LFA-1 clustering at adhesive contacts, which correlated with a significant decrease in adhesion strengthening. In contrast, silencing expression of Talin-1 did not alter high affinity LFA-1 clustering and only slightly diminished stable adhesion at high shear stress. This suggests that Talin-1 may not play an eminent role in clustering of LFA-1, but may assist in reorganization of the actin cytoskeleton and reinforcement of LFA-1 attachment to the cortical cytoskeleton via adaptor proteins such as Paxillin and Viniculin that associate with the integrin cytodomain (47, 51, 52). Our data demonstrates that Kindlin-3 recruitment to the β2-cytodomain is necessary for optimum calcium flux by physically linking LFA-1 bond clusters and Orai1 at a point upstream of Talin-1 association. Recent evidence indicates that a β2-specific NPXF motif is essential for triggering calcium signaling in Jurkat cells and our data suggests that Kindlin-3 may function as the scaffold protein linking the β2-integrin cytodomain to Orai1 during intracellular calcium mobilization (53). There are a number of cytoplasmic proteins that Kindlin-3 may interact with to activate calcium influx including STIM1, an ER luminal calcium sensor that facilitates clustering and spatial recruitment of Orai1 proximal to the ER (54, 55). Both calcium influx through Orai1 and Kindlin-3 are necessary for immune surveillance involving LFA-1 mediated signaling (56-58). Our data suggests that all three molecules contribute to mechanosignaling in PMN. Future studies will focus on how Kindlin-3 facilitates this cooperation between PLC mediated calcium flux and Orai1 CRAC and which cytoskeletal proteins act as the conduit linking mechanical force to calcium flux.
In summary, we define a mechanism by which tensile force acting on LFA-1/ICAM-1 bonds mechanically catalyzes its association with Kindlin-3; a necessary step in linkage with Orai1 and initiation of Ca2+ influx at focal sites of adhesion. This latter process provides a feed-forward mechanism that is cooperative with chemokine signaling to promote coalescence of LFA-1 bond clusters and cytoskeletal reinforcement of adhesion via Talin-1 and F-actin. In this manner, durable multivalent bonds are anchored to the cytoskeleton and serve to adhesion strengthen and direct pseudopod projections that efficiently guide PMN during transendothelial migration.
Supplementary Material
Acknowledgments
We are grateful to Dr. Anjana Rao for generously supplying Orai1+/− mice and to Ely Lilly for the gift of antibodies 327C and 240Q.
Grant Support: This work was supported by National Institutes of Health (NIH) grant A1472294 to S.I.S and German Research Foundation, AZ428/6-1 and SFB1009/1 to A.Z.
Abbreviations
- ICAM-1
intercellular adhesion molecule-1
- LFA-1
Leukocyte function antigen-1
- CD18
β2-integrin
- F-actin
filamentous-actin
- IL-8
Interleukin-8
- GPCR
G-protein coupled receptor
- CRAC
Calcium Release Activated Channels
- MFI
mean florescence intensity
- TIRF
total internal reflection fluorescence
- SOCE
Store operated calcium entry
- 2-APB
2-Aminoethoxydiphenyl borate
Literature Cited
- 1.Simon SI, Green CE. Molecular mechanics and dynamics of leukocyte recruitment during inflammation. Annu Rev Biomed Eng. 2005;7:151–185. doi: 10.1146/annurev.bioeng.7.060804.100423. [DOI] [PubMed] [Google Scholar]
- 2.Abram CL, Lowell CA. The ins and outs of leukocyte integrin signaling. Annu Rev Immunol. 2009;27:339–362. doi: 10.1146/annurev.immunol.021908.132554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dixit N, Simon SI. Chemokines, selectins and intracellular calcium flux: temporal and spatial cues for leukocyte arrest. Frontiers in immunology. 2012;3:188. doi: 10.3389/fimmu.2012.00188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sarantos MR, Zhang H, Schaff UY, Dixit N, Hayenga HN, Lowell CA, Simon SI. Transmigration of neutrophils across inflamed endothelium is signaled through LFA-1 and Src family kinase. J Immunol. 2008;181:8660–8669. doi: 10.4049/jimmunol.181.12.8660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Piccardoni P, Manarini S, Federico L, Bagoly Z, Pecce R, Martelli N, Piccoli A, Totani L, Cerletti C, Evangelista V. SRC-dependent outside-in signalling is a key step in the process of autoregulation of beta2 integrins in polymorphonuclear cells. Biochem J. 2004;380:57–65. doi: 10.1042/BJ20040151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lefort CT, Rossaint J, Moser M, Petrich BG, Zarbock A, Monkley SJ, Critchley DR, Ginsberg MH, Fassler R, Ley K. Distinct roles for talin-1 and kindlin-3 in LFA-1 extension and affinity regulation. Blood. 2012 doi: 10.1182/blood-2011-08-373118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Geng JG, Moore KL, Johnson AE, McEver RP. Neutrophil recognition requires a Ca(2+)-induced conformational change in the lectin domain of GMP-140. J Biol Chem. 1991;266:22313–22318. [PubMed] [Google Scholar]
- 8.Ginis I, Tauber AI. Activation mechanisms of adherent human neutrophils. Blood. 1990;76:1233–1239. [PubMed] [Google Scholar]
- 9.Schaff UY, Yamayoshi I, Tse T, Griffin D, Kibathi L, Simon SI. Calcium flux in neutrophils synchronizes beta2 integrin adhesive and signaling events that guide inflammatory recruitment. Ann Biomed Eng. 2008;36:632–646. doi: 10.1007/s10439-008-9453-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dixit N, Yamayoshi I, Nazarian A, Simon SI. Migrational guidance of neutrophils is mechanotransduced via high-affinity LFA-1 and calcium flux. J Immunol. 2011;187:472–481. doi: 10.4049/jimmunol.1004197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Alon R, Dustin ML. Force as a facilitator of integrin conformational changes during leukocyte arrest on blood vessels and antigen-presenting cells. Immunity. 2007;26:17–27. doi: 10.1016/j.immuni.2007.01.002. [DOI] [PubMed] [Google Scholar]
- 12.Lu C, Shimaoka M, Zang Q, Takagi J, Springer TA. Locking in alternate conformations of the integrin alphaLbeta2 I domain with disulfide bonds reveals functional relationships among integrin domains. Proc Natl Acad Sci U S A. 2001;98:2393–2398. doi: 10.1073/pnas.041618598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yang W, Shimaoka M, Chen J, Springer TA. Activation of integrin beta-subunit I-like domains by one-turn C-terminal alpha-helix deletions. Proc Natl Acad Sci U S A. 2004;101:2333–2338. doi: 10.1073/pnas.0307291101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Green CE, Schaff UY, Sarantos MR, Lum AF, Staunton DE, Simon SI. Dynamic shifts in LFA-1 affinity regulate neutrophil rolling, arrest, and transmigration on inflamed endothelium. Blood. 2006;107:2101–2111. doi: 10.1182/blood-2005-06-2303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schaff UY, Dixit N, Procyk E, Yamayoshi I, Tse T, Simon SI. Orai1 regulates intracellular calcium, arrest, and shape polarization during neutrophil recruitment in shear flow. Blood. 2009;115:657–666. doi: 10.1182/blood-2009-05-224659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Beals CR, Edwards AC, Gottschalk RJ, Kuijpers TW, Staunton DE. CD18 activation epitopes induced by leukocyte activation. J Immunol. 2001;167:6113–6122. doi: 10.4049/jimmunol.167.11.6113. [DOI] [PubMed] [Google Scholar]
- 17.Lu C, Shimaoka M, Salas A, Springer TA. The binding sites for competitive antagonistic, allosteric antagonistic, and agonistic antibodies to the I domain of integrin LFA-1. J Immunol. 2004;173:3972–3978. doi: 10.4049/jimmunol.173.6.3972. [DOI] [PubMed] [Google Scholar]
- 18.Sarantos MR, Raychaudhuri S, Lum AF, Staunton DE, Simon SI. Leukocyte function-associated antigen 1-mediated adhesion stability is dynamically regulated through affinity and valency during bond formation with intercellular adhesion molecule-1. J Biol Chem. 2005;280:28290–28298. doi: 10.1074/jbc.M501662200. [DOI] [PubMed] [Google Scholar]
- 19.Kinoshita K, Leung A, Simon S, Evans E. Long-lived, high-strength states of ICAM-1 bonds to beta2 integrin, II: lifetimes of LFA-1 bonds under force in leukocyte signaling. Biophys J. 98:1467–1475. doi: 10.1016/j.bpj.2009.12.4316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cambi A, Joosten B, Koopman M, de Lange F, Beeren I, Torensma R, Fransen JA, Garcia-Parajo M, van Leeuwen FN, Figdor CG. Organization of the integrin LFA-1 in nanoclusters regulates its activity. Mol Biol Cell. 2006;17:4270–4281. doi: 10.1091/mbc.E05-12-1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Altieri DC, Stamnes SJ, Gahmberg CG. Regulated Ca2+ signalling through leukocyte CD11b/CD18 integrin. Biochem J. 1992;288(Pt 2):465–473. doi: 10.1042/bj2880465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sa MD, Nelson WJ. Integrin-mediated calcium signaling and regulation of cell adhesion by intracellular calcium. Bioessays. 1996 doi: 10.1002/bies.950190109. [DOI] [PubMed] [Google Scholar]
- 23.Zhang SL, Yu Y, Roos J, Kozak JA, Deerinck TJ, Ellisman MH, Stauderman KA, Cahalan MD. STIM1 is a Ca2+ sensor that activates CRAC channels and migrates from the Ca2+ store to the plasma membrane. Nature. 2005;437:902–905. doi: 10.1038/nature04147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Navarro-Borelly L, Somasundaram A, Yamashita M, Ren D, Miller RJ, Prakriya M. STIM1-Orai1 interactions and Orai1 conformational changes revealed by live-cell FRET microscopy. J Physiol. 2008;586:5383–5401. doi: 10.1113/jphysiol.2008.162503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Krause KH, Campbell KP, Welsh MJ, Lew DP. The calcium signal and neutrophil activation. Clin Biochem. 1990;23:159–166. doi: 10.1016/0009-9120(90)80030-m. [DOI] [PubMed] [Google Scholar]
- 26.Puklin-Faucher E, Sheetz MP. The mechanical integrin cycle. J Cell Sci. 2009;122:179–186. doi: 10.1242/jcs.042127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Smith A, Carrasco YR, Stanley P, Kieffer N, Batista FD, Hogg N. A talin-dependent LFA-1 focal zone is formed by rapidly migrating T lymphocytes. J Cell Biol. 2005;170:141–151. doi: 10.1083/jcb.200412032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hyduk SJ, Rullo J, Cano AP, Xiao H, Chen M, Moser M, Cybulsky MI. Talin-1 and Kindlin-3 Regulate {alpha}4{beta}1 Integrin-Mediated Adhesion Stabilization, but Not G Protein-Coupled Receptor-Induced Affinity Upregulation. J Immunol. 187:4360–4368. doi: 10.4049/jimmunol.1003725. [DOI] [PubMed] [Google Scholar]
- 29.Feigelson SW, Grabovsky V, Manevich-Mendelson E, Pasvolsky R, Shulman Z, Shinder V, Klein E, Etzioni A, Aker M, Alon R. Kindlin-3 is required for the stabilization of TCR-stimulated LFA-1:ICAM-1 bonds critical for lymphocyte arrest and spreading on dendritic cells. Blood. 117:7042–7052. doi: 10.1182/blood-2010-12-322859. [DOI] [PubMed] [Google Scholar]
- 30.Moser M, Legate KR, Zent R, Fassler R. The tail of integrins, talin, and kindlins. Science. 2009;324:895–899. doi: 10.1126/science.1163865. [DOI] [PubMed] [Google Scholar]
- 31.Simon SI, Hu Y, Vestweber D, Smith CW. Neutrophil tethering on E-selectin activates beta 2 integrin binding to ICAM-1 through a mitogen-activated protein kinase signal transduction pathway. J Immunol. 2000;164:4348–4358. doi: 10.4049/jimmunol.164.8.4348. [DOI] [PubMed] [Google Scholar]
- 32.Feske S, Gwack Y, Prakriya M, Srikanth S, Puppel SH, Tanasa B, Hogan PG, Lewis RS, Daly M, Rao A. A mutation in Orai1 causes immune deficiency by abrogating CRAC channel function. Nature. 2006;441:179–185. doi: 10.1038/nature04702. [DOI] [PubMed] [Google Scholar]
- 33.Gwack Y, Srikanth S, Feske S, Cruz-Guilloty F, Oh-hora M, Neems DS, Hogan PG, Rao A. Biochemical and functional characterization of Orai proteins. J Biol Chem. 2007;282:16232–16243. doi: 10.1074/jbc.M609630200. [DOI] [PubMed] [Google Scholar]
- 34.Mitchon LN, White JM. Growth and analysis of octadecylsiloxane monolayers on Al2O3 (0001) Langmuir. 2006;22:6549–6554. doi: 10.1021/la0600189. [DOI] [PubMed] [Google Scholar]
- 35.Schaff UY, Xing MM, Lin KK, Pan N, Jeon NL, Simon SI. Vascular mimetics based on microfluidics for imaging the leukocyte-endothelial inflammatory response. Lab Chip. 2007;7:448–456. doi: 10.1039/b617915k. [DOI] [PubMed] [Google Scholar]
- 36.Kim MH, Liu W, Borjesson DL, Curry FR, Miller LS, Cheung AL, Liu FT, Isseroff RR, Simon SI. Dynamics of neutrophil infiltration during cutaneous wound healing and infection using fluorescence imaging. J Invest Dermatol. 2008;128:1812–1820. doi: 10.1038/sj.jid.5701223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chesnutt BC, Smith DF, Raffler NA, Smith ML, White EJ, Ley K. Induction of LFA-1-dependent neutrophil rolling on ICAM-1 by engagement of E-selectin. Microcirculation. 2006;13:99–109. doi: 10.1080/10739680500466376. [DOI] [PubMed] [Google Scholar]
- 38.Feng C, Li YF, Yau YH, Lee HS, Tang XY, Xue ZH, Zhou YC, Lim WM, Cornvik TC, Ruedl C, Shochat SG, Tan SM. Kindlin-3 mediates integrin alphaLbeta2 outside-in signaling and it interacts with the scaffold protein receptor for activated-C kinase 1 (RACK1) J Biol Chem. doi: 10.1074/jbc.M111.299594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Evans E, Kinoshita K, Simon S, Leung A. Long-lived, high-strength states of ICAM-1 bonds to beta2 integrin, I: lifetimes of bonds to recombinant alphaLbeta2 under force. Biophys J. 98:1458–1466. doi: 10.1016/j.bpj.2009.09.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schaff UY, Dixit N, Procyk E, Yamayoshi I, Tse T, Simon SI. Orai1 regulates intracellular calcium, arrest, and shape polarization during neutrophil recruitment in shear flow. Blood. 115:657–666. doi: 10.1182/blood-2009-05-224659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Astrof NS, Salas A, Shimaoka M, Chen J, Springer TA. Importance of force linkage in mechanochemistry of adhesion receptors. Biochemistry. 2006;45:15020–15028. doi: 10.1021/bi061566o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cinamon G, Shinder V, Alon R. Shear forces promote lymphocyte migration across vascular endothelium bearing apical chemokines. Nat Immunol. 2001;2:515–522. doi: 10.1038/88710. [DOI] [PubMed] [Google Scholar]
- 43.Constantin G, Majeed M, Giagulli C, Piccio L, Kim JY, Butcher EC, Laudanna C. Chemokines trigger immediate beta2 integrin affinity and mobility changes: differential regulation and roles in lymphocyte arrest under flow. Immunity. 2000;13:759–769. doi: 10.1016/s1074-7613(00)00074-1. [DOI] [PubMed] [Google Scholar]
- 44.Zhang H, Schaff UY, Green CE, Chen H, Sarantos MR, Hu Y, Wara D, Simon SI, Lowell CA. Impaired integrin-dependent function in Wiskott-Aldrich syndrome protein-deficient murine and human neutrophils. Immunity. 2006;25:285–295. doi: 10.1016/j.immuni.2006.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hellberg C, Molony L, Zheng L, Andersson T. Ca2+ signalling mechanisms of the beta 2 integrin on neutrophils: involvement of phospholipase C gamma 2 and Ins(1,4,5)P3. Biochem J. 1996;317(Pt 2):403–409. doi: 10.1042/bj3170403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Marks PW, Maxfield FR. Transient increases in cytosolic free calcium appear to be required for the migration of adherent human neutrophils. J Cell Biol. 1990;110:43–52. doi: 10.1083/jcb.110.1.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Franco SJ, Rodgers MA, Perrin BJ, Han J, Bennin DA, Critchley DR, Huttenlocher A. Calpain-mediated proteolysis of talin regulates adhesion dynamics. Nat Cell Biol. 2004;6:977–983. doi: 10.1038/ncb1175. [DOI] [PubMed] [Google Scholar]
- 48.Cairo CW, Mirchev R, Golan DE. Cytoskeletal regulation couples LFA-1 conformational changes to receptor lateral mobility and clustering. Immunity. 2006;25:297–308. doi: 10.1016/j.immuni.2006.06.012. [DOI] [PubMed] [Google Scholar]
- 49.DiVietro JA, Smith MJ, Smith BR, Petruzzelli L, Larson RS, Lawrence MB. Immobilized IL-8 triggers progressive activation of neutrophils rolling in vitro on P-selectin and intercellular adhesion molecule-1. J Immunol. 2001;167:4017–4025. doi: 10.4049/jimmunol.167.7.4017. [DOI] [PubMed] [Google Scholar]
- 50.Lum AF, Green CE, Lee GR, Staunton DE, Simon SI. Dynamic regulation of LFA-1 activation and neutrophil arrest on intercellular adhesion molecule 1 (ICAM-1) in shear flow. J Biol Chem. 2002;277:20660–20670. doi: 10.1074/jbc.M202223200. [DOI] [PubMed] [Google Scholar]
- 51.Ley K, Laudanna C, Cybulsky MI, Nourshargh S. Getting to the site of inflammation: the leukocyte adhesion cascade updated. Nature reviews Immunology. 2007;7:678–689. doi: 10.1038/nri2156. [DOI] [PubMed] [Google Scholar]
- 52.Emily M, Mace MAM, Marwali Muhammad Reza, Dreolini Lisa, Takei Fumio. LFA-1 binding to ligand induces Talin-mediated reorganization of the actin cytoskeleton in cytotoxic T cells. The Open Immunology Journal. 2008;1:51–61. [Google Scholar]
- 53.Sirim P, Zeitlmann L, Kellersch B, Falk CS, Schendel DJ, Kolanus W. Calcium signaling through the beta 2-cytoplasmic domain of LFA-1 requires intracellular elements of the T cell receptor complex. J Biol Chem. 2001;276:42945–42956. doi: 10.1074/jbc.M103224200. [DOI] [PubMed] [Google Scholar]
- 54.Kawasaki T, Lange I, Feske S. A minimal regulatory domain in the C terminus of STIM1 binds to and activates ORAI1 CRAC channels. Biochem Biophys Res Commun. 2009;385:49–54. doi: 10.1016/j.bbrc.2009.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Roos J, DiGregorio PJ, Yeromin AV, Ohlsen K, Lioudyno M, Zhang S, Safrina O, Kozak JA, Wagner SL, Cahalan MD, Velicelebi G, Stauderman KA. STIM1, an essential and conserved component of store-operated Ca2+ channel function. J Cell Biol. 2005;169:435–445. doi: 10.1083/jcb.200502019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Maul-Pavicic A, Chiang SC, Rensing-Ehl A, Jessen B, Fauriat C, Wood SM, Sjoqvist S, Hufnagel M, Schulze I, Bass T, Schamel WW, Fuchs S, Pircher H, McCarl CA, Mikoshiba K, Schwarz K, Feske S, Bryceson YT, Ehl S. ORAI1-mediated calcium influx is required for human cytotoxic lymphocyte degranulation and target cell lysis. Proc Natl Acad Sci U S A. 2011;108:3324–3329. doi: 10.1073/pnas.1013285108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Feng C, Li YF, Yau YH, Lee HS, Tang XY, Xue ZH, Zhou YC, Lim WM, Cornvik TC, Ruedl C, Shochat SG, Tan SM. Kindlin-3 mediates integrin alphaLbeta2 outside-in signaling and it interacts with the scaffold protein receptor for activated-C kinase 1 (RACK1) J Biol Chem. 2012 doi: 10.1074/jbc.M111.299594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Svensson L, Howarth K, McDowall A, Patzak I, Evans R, Ussar S, Moser M, Metin A, Fried M, Tomlinson I, Hogg N. Leukocyte adhesion deficiency-III is caused by mutations in KINDLIN3 affecting integrin activation. Nat Med. 2009;15:306–312. doi: 10.1038/nm.1931. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.