

Abstract
Previous studies related impaired myocardial microcirculation in diabetes to oxidative stress and endothelial dysfunction. Thus, this study was aimed to determine the effect of up-regulating pAMPK-pAKT signaling on coronary microvascular reactivity in the isolated heart of diabetic mice. We measured coronary resistance in wild-type and streptozotocin (STZ)-treated mice, during perfusion pressure changes. Glucose, insulin, and adiponectin levels in plasma and superoxide formation, NOx levels and heme oxygenase (HO) activity in myocardial tissue were determined. In addition, the expression of HO-1, 3-nitrotyrosine, pLKB1, pAMPK, pAKT, and peNOS proteins in control and diabetic hearts were measured. Coronary response to changes in perfusion pressure diverged from control in a time-dependent manner following STZ administration. The responses observed at 28 weeks of diabetes (the maximum time examined) were mimicked by L-NAME administration to control animals and were associated with a decrease in serum adiponectin and myocardial pLKB1, pAMPK, pAKT, and pGSK-3 expression. Cobalt protoporphyrin treatment to induce HO-1 expression reversed the microvascular reactivity seen in diabetes towards that of controls. Up-regulation of HO-1 was associated with an increase in adiponectin, pLKB1, pAKT, pAMPK, pGSK-3, and peNOS levels and a decrease in myocardial superoxide and 3-nitrotyrosine levels. In the present study we describe the time course of microvascular functional changes during the development of diabetes and the existence of a unique relationship between the levels of serum adiponectin, pLKB1, pAKT, and pAMPK activation in diabetic hearts. The restoration of microvascular function suggests a new therapeutic approach to even advanced cardiac microvascular derangement in diabetes.
Keywords: Coronary Microcirculation, Diabetic Cardiomyopathy, Heme-Oxygenase-1, Endothelial Dysfunction, Adiponectin
We have previously employed the isolated heart in Langendorff configuration to investigate coronary microvascular reactivity in rodents [Kusmic et al., 2006; L'Abbate et al., 2007]. In spite of the limitations of the isolated heart preparation (no blood perfusion, loss of basal coronary tone, uncertain equivalence with in vivo conditions), the vasoconstrictive response to transiently reduced perfusion pressure appears to be a simple, reliable way to examine coronary microvascular reactivity. Such a reaction, known as “paradoxical vasoconstriction,” as opposed to the expected vasodilation elicited by ischemia (low flow and lactate release), mimics the vasoconstrictive response observed downstream a severe stenosis in patients with chronic angina, during pacing-induced ischemia [Sambuceti et al., 1997, 2001, 2005]. Importantly, the active nature of this phenomenon (increased vascular tone as opposed to increased intramural vascular compression) was demonstrated in the mouse by its relief following papaverine injection [Kusmic et al., 2006]. In the wild-type mouse, “paradoxical vasoconstriction” appeared to be dependent upon endothelial NO production and reactive oxygen species (ROS). In particular, administration of L-NAME, an inhibitor of nitric oxide synthase (NOS), reversed “paradoxical” vasoconstriction to vasodilation and ROS scavengers suppressed the vasoconstrictive response suggesting that the combination of NO and ROS is essential for eliciting vasoconstriction during hypotension.
Microvascular reactivity was also examined during perfusion at “normal” pressure and coronary resistance remained constant in untreated heart while progressively increasing during L-NAME administration. We speculated that an unknown long-acting vasoconstrictor(s), accumulates with time [Kusmic et al., 2006] and showed in the isolated heart of wild-type mouse, L-NAME exerted a second apparently opposite role of NO, namely mediating vasoconstriction in response to low perfusion pressure while contrasting vasoconstriction at “normal” perfusion pressure.
Adipose tissue plays an important role in insulin resistance through the secretion of a variety of proteins such as TNF-α, IL-6, leptin, and adiponectin [Berg and Scherer, 2005]. In particular, adiponectin has attracted considerable attention as it has insulin-sensitizing properties that reduce serum triglyceride levels, enhance fatty acid oxidation and glucose uptake in the liver [Berg et al., 2001; Kim et al., 2007]. It has been suggested that adiponectin acts through the AMPK signaling pathway in the vasculature [Chen et al., 2003]. Activated AMPK and AKT have been identified as regulators of endothelial cell nitric oxide synthase (eNOS) activation as well as a number of cellular responses [Fleming et al., 2003; Kovacic et al., 2003; Ouchi et al., 2004; Skurk et al., 2004; Sun et al., 2005; Reihill et al., 2007]. Because AMPK has been identified as a potential target for the treatment of type 2 diabetes [Musi and Goodyear, 2002] and we previously observed that an increase in cardiac HO-1 levels increases serum adiponectin and myocardial pAKT in both control and diabetic rats [L'Abbate et al., 2007], a central issue in the present study was to determine whether the diabetic state is associated with abnormalities in upstream and downstream AMPK signaling pathway.
We hypothesized that progressive loss of NO in the diabetic heart would modify coronary reactivity in a manner similar to L-NAME in control heart and that activation of heme oxygenase-1 (HO-1) would restore control vasoreactivity by enhancing NO and reducing ROS. We examined coronary reactivity in mice with streptozotocin (STZ)-induced non-insulin-dependent diabetes, with three main objectives: firstly, to define the time course of changes in coronary microvascular reactivity during development of diabetes, secondly, to investigate the molecular mechanisms involved in vasoreactivity, in particular NO production and availability, and oxidative stress; thirdly, to assess the efficacy of HO-1 induction in restoring normal microvascular response, HO-1 is down-regulated in diabetic heart and induction results in increased adiponectin levels and enhanced heart tolerance to ischemia [L'Abbate et al., 2007].
We report a progressive deviation from normal of coronary microvascular reactivity in the diabetic heart that developed with time irrespective of constant hyperglycemia, insulinemia, and of glucose intolerance. Both systemic and coronary alterations are prevented by the HO-1 mediated increase in myocardial pLKB1, pAMPK, pAKT, and peNOS levels.
Materials and Methods
Surgical procedures and experimental protocols were approved by the Animal Care Committee of the Italian Ministry of Health and conformed to the “Guiding Principles for Research Involving Animals and Human Beings” approved by the Council of the American Physiological Society. All mice were housed under controlled 12/12h light/dark cycle and given food and water ad libitum.
Animal Models
Male C57BL/6 6- to 8-week-old mice (Harlan, Italy) were administered an intraperitoneal (i.p.) low-dose injection of STZ to induce diabetes by adapting the protocol used by members of the Animal Models of Diabetic Complication Consortium (USA). Briefly, STZ was made fresh daily and dissolved in 0.05 mol/L citrate buffer (pH 4.4) at a concentration of 5 mg/ml and injected i.p. at the dose of 35 mg/kg body weight for five consecutive days to obtain a stable animal model of experimental, non-insulin-dependent diabetes.
Diabetic mice were studied at 28 weeks following STZ administration, a result of a preliminary study, carried out at 6, 9, and 28 weeks, aimed at assessing the time course of coronary changes during development of diabetes. At 28 weeks (about one-third of the normal life span of mice) microvascular reactivity maximally diverged from that of control animals. Moreover, out of the STZ-treated animals only those showing glucose plasma levels of 180–250 mg/dl and insulin plasma values of 0.7–1.1 μg/L which better mimic the systemic humoral picture of patients with type 2 diabetes.
According to the above criteria, 49 diabetic mice were studied of which 20 did not receive any other treatment following STZ administration. Seventeen mice were treated with cobalt protoporphyrin (CoPP) in order to test the effect of the pharmacological up-regulation of HO-1 expression and the remaining 12 received an infusion of L-NAME during perfusion in the Langendorff configuration in order to investigate the effect of NO deprivation on microvascular reactivity. Forty-seven control mice, age and gender matched to diabetic mice, were treated with citrate buffer solution and examined at the same time points as the diabetic animals.
The increased expression of HO-1 in diabetic and control groups was pharmacologically induced by administering a dose of 1.5 mg/100 g body weight of CoPP subcutaneously every five days for a total of three injections. Tin protoporphyrin IX (SnPPIX), a selective inhibitor of HO activity, was administered i.p. at the dose of 2.0 mg/100 g body weight to CoPP-treated animals, 24h before heart's explant, in order to distinguish the specific effect of increased HO activity on the parameters under investigation.
In all animals, blood samples were collected weekly from the tail vein following a 4 h period of fasting to measure glucose levels (Glucocard GT-1610, Menarini Diagnostic, Firenze, Italia). Insulin levels were determined by ELISA (Mouse Insulin ELISA, Mercodia, Uppsala, Sweden) in serum samples collected before STZ treatment (basal level) and at the time of sacrifice. Animals, randomly chosen from all groups, underwent a glucose tolerance test. The test was performed on awake mice that were fasted for 6 h and injected with a glucose solution. A 100 mg/ml glucose stock solution was prepared in distilled water and sterilized by passing through a 0.2 μm filter. Following baseline blood glucose measurement, each mouse was challenged with a 1 mg glucose/gram body weight. Post-injection blood glucose measurements were performed at 10, 20, 30, 60, and 120 min after glucose loading.
Isolated Heart Preparation
Animals were heparinized (500U i.m.) 10 min prior to anesthesia with pentobarbitone sodium (40mg/kg, i.p.). Hearts were excised and placed in ice-cold Krebs–Henseleit bicarbonate solution of the following composition (mmol/L): 118 NaCl, 24 NaHCO3, 4.7 KCl, 1.2 KH2PO4, 1.2 MgSO4, 2.5 CaCl2, 0.5 EDTA, and 5.5 glucose. The solution was pre-equilibrated with 95% O2 and 5% CO2 at pH 7.4. Extraneous tissues were removed, and the aorta was cannulated with a 20-gauge plastic cannula. The heart was then transferred to a modified non-recirculating Langendorff apparatus where it was allowed to beat spontaneously. Myocardial and buffer temperatures were kept constant at 37°C. Two side-arms in the perfusion line, located close to the heart inlet, allowed switching between two reservoirs set at “normal” (65mmHg) and “low” (30mmHg) pressure. Coronary flow was continuously measured with a flowmeter (model T106, Transonic System, Inc., Ithaca, NY) coupled to a in-line flow probe. The volume of effluent was also measured with a calibrated pipette as an additional estimate of coronary flow.
Coronary resistance was calculated as input pressure divided by coronary flow per gram of myocardial tissue (mmHg g min/ml). After an initial 10-min period of stabilization, not included in the analysis, two different protocols each of 70 min duration were followed:
Protocol 1—Control pressure
Preparations were maintained at 65mmHg input pressure during the entire 70 min experiment. A bolus of papaverine (50 μg) was used to examine the nature of coronary resistance by releasing vascular smooth muscle to document active vasoconstriction.
Protocol 2—Transient hypotension
Input pressure was reduced from 65 to 30 mmHg at the 20 min mark for a period of 20 min and then restored to 65 mmHg for the remaining 30 min.
In control animals (n = 12) as well as in diabetic animals (n = 12) L-NAME was added to the perfusion medium to a final concentration of 100 μmol/L at the time of aorta cannulation and remained in the preparation for the entire experiment in both protocol 1 and 2.
At the end of constant pressure experiments, hearts were rapidly frozen in liquid nitrogen and stored at −80°C.
Western Blot Analysis of Cardiac Tissue
Frozen hearts, obtained from animals perfused according to the constant pressure protocol, were pulverized under liquid nitrogen and placed in a homogenization buffer (in mmol/L: 10 phosphate buffer, 250 sucrose, 1 EDTA, 0.1 PMSF, and 0.1%, v/v, tergitol, pH 7.5). Homogenates were centrifuged at 27,000g for 10 min at 4°C, supernatant was isolated, and protein levels were visualized by immunoblotting with antibodies against HO-1, and HO-2 (Stressgen Biotechnologies Corp., Victoria, BC). Antibodies against LKB1, AKT, AMPK, pLKB1(Ser 428), pAMPK(Thr 172), pAKT(Ser 473), pGSK-3(Ser 9), 3-Nitrotyrosine (3-NT) (Cell Signaling Technology, Inc., Beverly, MA) eNOS, peNOS(Ser 1177), and β-actin (Santa Cruz Biotechnology, Santa Cruz, CA) were used. Myocardial β-actin expression was used as comparative protein. Antibodies were prepared in the following dilutions: HO-1, HO-2, and 3-NT 1:1000, eNOS, peNOS, LKB1, pLKB1, AMPK, pAMPK, AKT, pAKT, and pGSK-3 1:5000.
Briefly, 20 μg of heart tissue lysate supernatant was separated by 12% SDS/polyacrylamide gel electrophoresis and transferred to a nitrocellulose membrane. Immunoblotting was performed as previously described [Abraham et al., 2003]. Chemiluminescence detection was performed with the Amersham ECL detection kit (Amersham, Piscataway, NJ), according to the manufacturer's instructions.
Determination of HO Activity
Frozen hearts were pulverized under liquid nitrogen and placed in homogenization buffer (in mmol/L: 10 phosphate buffer, 250 sucrose, 1 EDTA, 0.1 PMSF, and 0.1%, v/v, tergitol, pH 7.5). Homogenates were centrifuged at 27,000g for 10 min at 4°C. Heme oxygenase activity was determined using a scanning double beam spectrophotometer (Lambda 17 UV/Vis; Perkin Elmer Cetus Instruments, Norfolk, CT) and expressed as nmol bilirubin/mg protein/h. Bilirubin formation was calculated using an extinction coefficient of 40mmol/L−1 cm−1 between 464 and 530 nm.
Measurement of Heart and NO Metabolites (NOx) Levels
Control and diabetic hearts were placed in plastic scintillation minivials containing 5 μmol/L lucigenin for detection of , in a final volume of 1 ml of air-equilibrated Krebs solution buffered with 10 mmol/L HEPES-NaOH (pH 7.4) as previously described [Abraham et al., 2003]. Lucigenin chemiluminescence was measured in a liquid scintillation counter (LS6000IC, Beckman Instruments, San Diego, CA). NOx levels were evaluated by measuring total nitrite and nitrate content in heart tissue homogenate using a NOx quantitation kit and following the manufacturer's instruction (Active Motif, Carlsbad, CA).
Serum Adiponectin Measurements
The high molecular weight form of adiponectin was determined using an ELISA assay (Pierce Biotechnology, Inc., Woburn, MA).
Drugs and Chemicals
STZ and L-NAME were purchased from Sigma (St. Louis, MO). Papaverine was purchased from Monico (Venezia, Italy). CoPP was purchased from Frontier Scientific (Logan, UT). Tin protoporphyrin IX was provided as a generous gift from A. Kappas, MD (Rockefeller University, New York, NY).
All the other chemicals used were purchased from Sigma and were of reagent grade.
Statistical Analysis
The data are presented as mean ± SE. Statistical significance of differences between groups was determined by ANOVA followed by post hoc Fisher's test or by paired and unpaired Student's t-test as appropriate. A probability (P) value of <0.05 was taken to indicate statistical significance.
Results
Time Course of Microvascular Reactivity Following STZ Administration
As shown in Figure 1, the coronary resistance response to both constant pressure and transient low pressure differed at different times (6, 9, and 28 weeks after STZ administration), progressively diverging from time-matched controls, with a maximum divergence at 28 weeks. At constant pressure (gray symbols) a progressive increase in resistance was observed during the 70min of perfusion. The magnitude of vasoconstriction significantly increased from 6 to 28 weeks. As already reported for control animals [Kusmic et al., 2006], a bolus administration of papaverine (50 μg), abolished the increase in resistance in diabetic mice (Fig. 1D, inset) proving its active nature and ruling out the intervention of extra-vascular compressive forces.
Fig. 1.
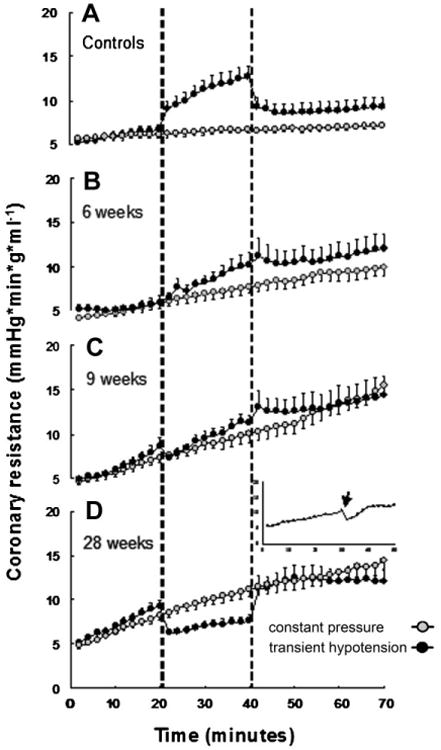
Coronary microvascular resistance in protocol 1 (perfusion at 65 mmHg constant pressure), gray circles, as compared to protocol 2 (transient 30 mmHg perfusion pressure), black circles, in controls and in diabetic animals at 6, 9, and 28 weeks following STZ or vehicle administration. Dashed vertical lines mark the period of low pressure in protocol 2. A: Control group (gray n = 11 and black n = 9). Controls have been pooled together as no differences were apparent at 6, 9, and 28 weeks. B: Six-week diabetic mice (gray n = 6 and black n = 5). C: Nine-week diabetic mice (gray n = 5 and black n = 6). D: Twenty-eight-week diabetic mice (gray n =11 and black n =10). Inset shows the effect of administration in bolus (arrow) of papaverine in 28-week diabetic mouse during protocol 1 (constant pressure perfusion).
During low-pressure perfusion (black symbols) the paradoxical vasoconstriction observed in control animals was blunted at 6 weeks and progressively reversed to vasodilation at 28 weeks (Fig. 1). On the basis of these results, the present studies employed diabetic animals 28 weeks after STZ treatment.
Effect of Chronic Diabetes on Body Weight, Plasma Glucose, and Insulin Levels
As seen in Table I, no significant difference in body weight and plasma insulin levels between control and diabetic mice was observed. Plasma glucose levels in diabetic mice were significantly higher than in control mice (P <0.001). In contrast treatment of diabetic mice with CoPP reduced glucose in plasma to the levels of the control group. CoPP treatment in control mice did not affect either fasting glucose or insulin levels.
Table I. General Characteristics of Experimental Animals.
| Group | n | Body weight (g) | Plasma glucose (mg/dl) | Plasma insulin (μg/L) |
|---|---|---|---|---|
| Control | 20 | 31 ± 2.4 | 131 ± 7.8 | 0.86 ± 0.1 |
| Diabetes | 20 | 33 ± 1.1 | 232 ± 27* | 0.83 ±0.3 |
| Effect of CoPP treatment | ||||
| Control + CoPP | 9 | 31 ± 4.0 | 135 ± 11 | 0.88 ±0.3 |
| Diabetes + CoPP | 17 | 34 ± 1.4 | 142 ± 5.0† | 0.77 ±0.1 |
Values are mean ± SE; n, number of animals tested. Animals were examined and treated with CoPP 28 weeks after citrate buffer (control) or STZ (diabetes) administration.
P< 0.001 versus control.
P< 0.001 versus untreated diabetes.
Effect of Chronic Diabetes on Glucose Tolerance Test
As expected, in control animals, glucose loading produced a marked increase in glucose levels that peaked at 20 min at around 300 mg/dl, thereafter progressively declining towards initial levels (Table II). However, in diabetic mice, glucose tolerance curves were typical of glucose intolerance, commencing at basal levels that were higher than in age-matched controls, glucose levels peaked 20–30 min after glucose infusion at values of 466 mg/dl and above and remained elevated (over 300 mg/dl) for the duration of the experiment. Glucose values in diabetic mice were statistically higher than in control animals (P < 0.001).
Table II. Plasma Glucose Level (mg/dl) During Glucose Tolerance Test.
| Group | n | 0 min | 20 min | 30min | 60 min | 120 min |
|---|---|---|---|---|---|---|
| Control | 20 | 131 ± 8.0 | 301 ± 15 | 287 ± 14 | 211 ± 16 | 160 ± 8.0 |
| Diabetes | 20 | 225 ±16* | 466 ± 21* | 461± 24* | 375 ± 29* | 303 ± 21* |
| Effect of CoPP treatment | ||||||
| Control + CoPP | 9 | 135 ± 11§ | 182 ± 10§ | 169 ± 8.0§ | 145 ± 8.0§ | 110 ± 5.0§ |
| Diabetes + CoPP | 17 | 142 ± 5.0† | 313 ± 15† | 276± 12† | 193 ±11† | 148 ± 6.0† |
Values are mean± SE; n, number of animals tested. Animals were examined and treated with CoPP 28 weeks after citrate buffer (control) or STZ (diabetes) administration.
P < 0.001 versus control.
P < 0.001 versus untreated diabetes.
P < 0.001 versus untreated control.
The effect of HO-1 expression on chronic diabetes and glucose tolerance, is shown in Table II. Diabetic mice treated with CoPP showed a marked decline in fasting plasma glucose levels to values no different from those seen in control animals. In addition, glucose levels during the glucose tolerance test were significantly reduced in diabetic mice following CoPP administration compared to untreated diabetic mice (P < 0.001). CoPP administered to control animals significantly impaired the glucose rise during the glucose tolerance test relative to untreated control animals (P < 0.001).
Effect of Diabetes on Coronary Resistance in Hearts Under Constant Pressure
Control hearts perfused at constant pressure exhibited relatively stable coronary resistance with only a small increase detectable over the study period (from 5.6 ± 0.4 to 7.1 ± 0.3 mmHg min g m−1 at 0 and 70 min, respectively; P < 0.05) (Fig. 1A and inset of Fig. 2A). In diabetic mice, the initial values of coronary resistance were similar to those in the control group. However, coronary resistance increased throughout the perfusion period in diabetic hearts, attaining values significantly higher (P < 0.05) than those observed in controls starting from 18min. At 70min coronary resistance achieved values of 14.4±0.3 mmHg min g ml−1 (P < 0.005 vs. controls) (Fig. 2A).
Fig. 2.
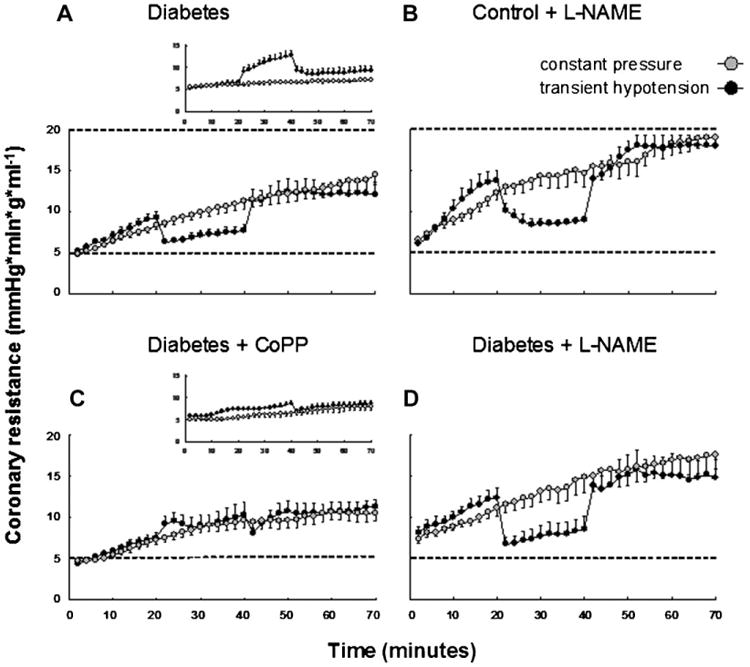
Coronary microvascular resistance in protocol 1 (perfusion at 65 mmHg constant pressure, gray circles) as compared to protocol 2 (transient 30 mmHg perfusion pressure, black circles). Dashed horizontal lines are set at 5 and 20 marks on the coronary resistance axis for a better comparison between panels. A: Twenty-eight-week diabetic mice (gray n = 11 and black n = 10) already shown in Figure 1A. Inset reproduces the response in control group (gray n = 11 and black n = 9) for comparison. B: The addition of the NOS synthase inhibitor L-NAME in controls (gray n = 6 and black n =6). C: The effect of CoPP treatment in diabetic mice (gray n = 6 and black n = 7). Inset reproduces the effect of CoPP treatment in controls (gray n = 6 and black n = 9) for comparison. D: The addition of L-NAME to diabetic mice (gray n = 6 and black n = 6).
Effect of Diabetes on Coronary Resistance in Hearts Under Low Flow ISCHEMIA
In control hearts hypotension caused a marked increase in coronary resistance (paradoxical vasoconstriction) (Fig. 1A and inset of Fig. 2A). The response was biphasic with a rapid initial increase followed by a slower but progressive increase. At the peak, coronary resistance rose about 1.9-fold over baseline (P < 0.001). The return to normal perfusion pressure caused a sudden fall in resistance to a steady value that was significantly higher than baseline (P < 0.01).
As shown in Figure 2A, 28 weeks of diabetes resulted in an early rise in coronary resistance during the first 20 min of the protocol not dissimilar from that reported in the constant pressure protocol at the same time points. However hypotension induced a marked drop of resistance, opposite to the vasoconstriction observed in controls (compare 2A vs. the inset or Fig. 1A). A subsequent return to normal perfusion pressure produced a new, steady increase in coronary resistance up to 1.3-fold of that of control (P < 0.05).
Effect of L-name on Coronary Resistance
In control mice, during constant pressure, a progressive rise in resistance was observed during infusion of L-NAME (Fig. 2B) confirming previous results [Kusmic et al., 2006]. This response closely resembled that recorded in long lasting diabetes (Fig. 2, compare B vs. A) although the increase in resistance following L-NAME was significantly higher (P < 0.05) when compared to the diabetic group. The addition of L-NAME to diabetic hearts further enhanced coronary resistance to values similar to L-NAME-treated controls (Fig. 2D).
In the low-pressure protocol, L-NAME treatment in control mice converted the response to hypotension from vasoconstriction to vasodilation, once again mimicking the response observed in diabetic mice. However, as for the protocol that utilized constant pressure, during the initial and latter period of perfusion at 65 mmHg the relative amplitude of vasoconstriction was greater in L-NAME-treated hearts from control than from diabetic animals (Fig. 2B). The addition of L-NAME to diabetic hearts resulted in the response of coronary resistance to hypotension being similar to that observed in L-NAME-treated controls (Fig. 2D). The relative values of coronary resistance at 70 min in each protocol were: control L-NAME > diabetic L-NAME > diabetic > control.
Effect of CoPP on Coronary Resistance in Control and Diabetic Animals
Pre-treatment of control mice with CoPP did not significantly modify the coronary resistance response that occurred during constant pressure in the untreated control mice (Fig. 2C, inset). Conversely, in CoPP pre-treated diabetic mice, coronary resistance increased less than in untreated diabetic animals (P < 0.05 vs. untreated diabetics) (Fig. 2C vs. A).
During transient low pressure ischemia, pre-treatment with CoPP largely abolished the vasoconstrictive response to hypotension in control mice (inset Fig. 2C) while in diabetic mice it reverted the vasodilator response seen during hypotension into a vasoconstrictor response (Fig. 2C). A return to normal perfusion pressure induced a transient drop in coronary resistance followed by a new progressive increase.
Treatment with SnPPIX, in CoPP-treated diabetic mice 24h before heart explant, blunted the effect of CoPP on microvascular reactivity increasing resistance during constant pressure and restoring vasodilation during low pressure (results not shown).
Effects of CoPP on Heart HO-1 Gene Expression, HO Activity and Superoxide Levels
Western blot analysis of heart homogenates revealed that levels of HO-1 protein were significantly decreased in diabetes compared to control (Fig. 3A). In contrast, significant levels of HO-2 were detected in control hearts and were no different from HO-2 levels in diabetic hearts. Thus a significant decrease (P < 0.05) in the ratio of HO-1 to HO-2 in the hearts of diabetic mice compared to hearts from control mice was observed. CoPP administration increased heart HO-1 protein levels in diabetic (Fig. 3A) as well as control mice (P < 0.01 for both) but had no effect on HO-2 protein levels.
Fig. 3.
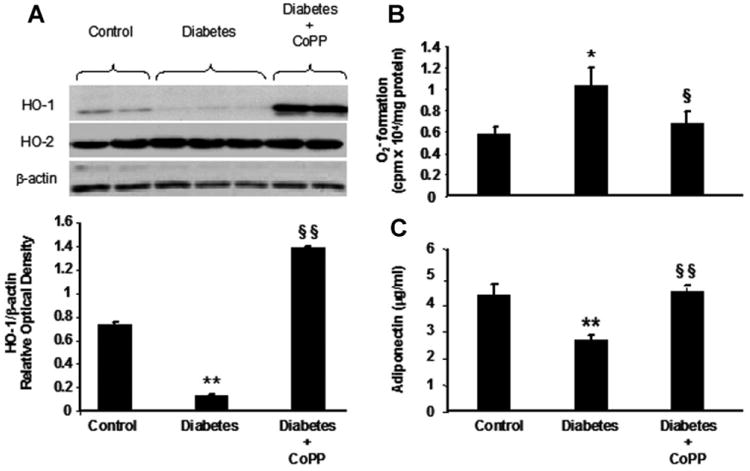
A: Western blot and densitometry analysis of HO-1, HO-2 and β-actin expression in myocardium of control and diabetic mice untreated or treated with CoPP. **P< 0.01 diabetes versus control, §§P<0.01 diabetes CoPP versus diabetes. Histograms are mean ±SE, n = 4 in each group. B: Myocardial production in control, diabetic mice, and diabetic mice treated with CoPP. *P< 0.05 diabetes versus control; §P<0.05 diabetes CoPP versus diabetes. Results are mean ±SE; n =4 in each group. C: Serum adiponectin levels in control and diabetic mice, untreated and treated with CoPP. Serum samples were obtained immediately prior to sacrifice. **P<0.01 diabetes versus control; §§P< 0.01 diabetes CoPP versus diabetes. Results are mean ±SE, n = 4 in each group.
HO activity was decreased in diabetic hearts compared to control hearts (0.43 ± 0.11 nmol bilirubin formed/mg protein/h in diabetes vs. 0.68 ± 0.12 nmol bilirubin formed/mg protein/h in control, P < 0.05). HO-1 induction resulted in increased HO activity in the hearts of diabetic mice following CoPP treatment, (1.51 ± 0.41 nmol bilirubin formed/mg protein/h), P < 0.05 versus untreated diabetic hearts.
Measurement of was performed at low concentrations of lucigenin (5 μmol/L). The decrease in cardiac HO-1 levels as a result of diabetes was associated with a significant reciprocal increase in ( formed in diabetic heart was 1.2 × 104 cpm/mg protein compared to 0.58 × 104 cpm/mg protein in control hearts, P < 0.05) (Fig. 3B). However, the increase in HO activity, as a result of CoPP administration, was associated with a significant decrease in in diabetic hearts to 0.67 × 104 cpm/mg protein (P < 0.05 treated vs. untreated diabetes) (Fig. 3B).
Effect of CoPP on Serum Adiponectin Levels
There was a significant decrease in serum adiponectin levels in diabetic mice compared to age-matched control mice, 2.61 ± 0.23 μg/ml versus 4.29 ±0.36 μg/ml (P < 0.01). CoPP administration resulted in a marked increase (P < 0.001) in the levels of serum adiponectin in diabetic mice as compared to diabetic untreated mice (4.4 ±0.89 μg/ml vs. 2.61 ±0.23 μg/ml). Moreover, the increase in adiponectin levels seen in diabetic mice following CoPP treatment attained a level similar to that found in control animals (Fig. 3C).
Effects of Perturbation of HO-1 Expression on Heart 3-Nitrotyrosine (3-NT) and No Metabolites (NOx) Levels
Densitometry analysis showed detectable levels of 3-NT in hearts of control mice (Fig. 4A). The 3-NT level increased significantly in diabetic hearts compared to control hearts (P < 0.05). Following CoPP treatment the level of 3-NT in diabetic mice was significantly reduced when compared to that in untreated diabetic animals (P < 0.05) (Fig. 4A).
Fig. 4.
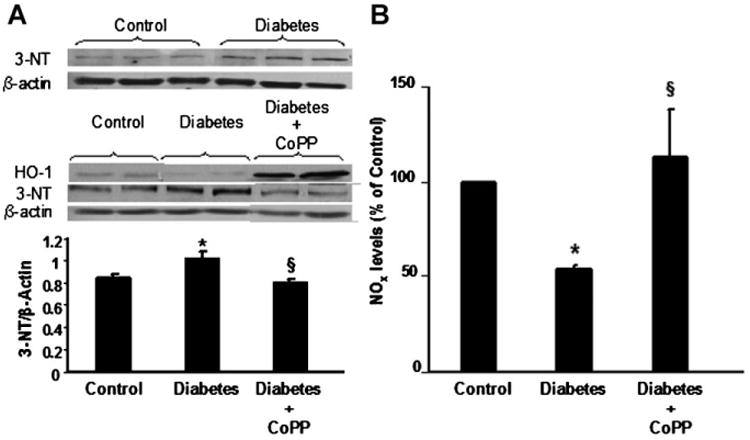
A: Western blot analysis showing 3-nitrotyrosine (3-NT), β-actin and HO-1 expression in hearts of control, diabetic mice untreated or treated with CoPP. Quantitative densitometry is expressed as ratio between 3-NT and the comparative protein β-actin. *P<0.05 diabetes versus control; §P<0.05 diabetes CoPP versus untreated diabetes. Results are expressed as mean ±SE, n = 5 in each group. B: Overall nitrates and nitrites (NOx) levels in heart tissues obtained from control and diabetic mice untreated or treated with CoPP. Results are means ±SE, n = 3 and they are expressed as percent of control (0.49 ±0.09 μmol/L). *P< 0.05 diabetes versus control and §P<0.05 diabetes CoPP versus diabetes.
As shown in Figure 4B, levels of NOx were significantly decreased in hearts from mice with diabetes, 54.5 ± 3.0% of control (P < 0.05). This decrease in NOx was prevented by treatment with CoPP, which restored levels of NO to 112.8 ± 24.8% of control (P < 0.05 vs. untreated diabetic mice) (Fig. 4B). Treatment of control mice with CoPP did not change levels of NOx in the heart as compared to untreated controls (92.7 ± 13.8% vs. 100% of control).
Effect of CoPP on heart pLKB1, pAMPK, pAKT, pGSK-3, AND peNOS Levels
Diabetes resulted in no significant changes in AKT and AMPK but significant decreases in both pAKT and pAMPK (Fig. 5). Up-regulation of HO-1 by CoPP administration increased the cardiac levels of both pAKT and pAMPK in diabetic mice (Fig. 5). pAKT/AKT ratio decreased significantly in diabetic animals compared to control mice (P < 0.01, Fig. 5A) and increased significantly in CoPP-treated diabetic animals compared to untreated diabetic mice (P < 0.05) (Fig. 5A). Similarly, a significant decrease in the heart pAMPK/AMPK ratio was seen in diabetic mice versus controls (P < 0.01, Fig. 5B), and CoPP treatment significantly increased the pAMPK/AMPK ratio in the diabetic heart (P < 0.01 vs. untreated diabetic mice, Fig. 5B).
Fig. 5.
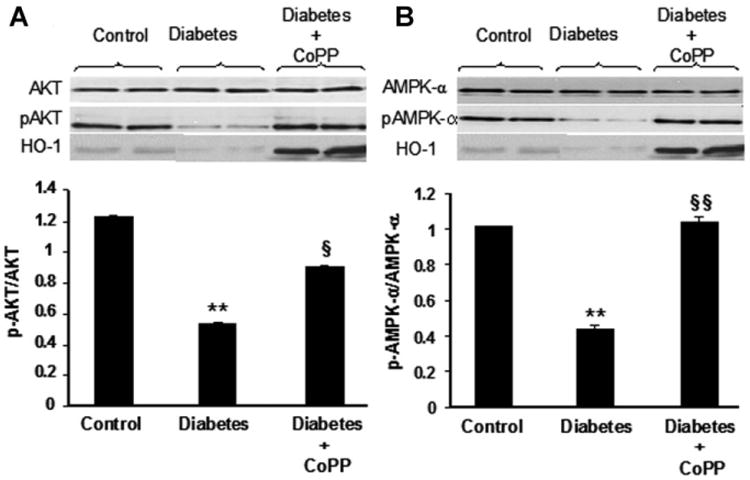
Western blot of total and phosphorylated (p−) forms of AKT (panel A), and AMPK (panel B) expression in hearts of control, diabetic mice untreated or treated with CoPP. Western blot bands of HO-1 are shown as control for CoPP efficacy in HO-1 over-expression. Quantitative densitometry for each enzyme is expressed as ratio between phosphorylated and total amount of protein. **P<0.01 diabetes versus control, §P< 0.05 and §§P<0.01 diabetes CoPP versus diabetes. Each bar represents mean ±SE of four experiments.
These changes in pAKT and pAMPK levels were paralleled by a decrease in both eNOS and peNOS(Ser 1179) levels in diabetic hearts (Fig. 6A). The quantitative densitometry expressed as ratio between phosphorylated and total eNOS showed significantly reduced values in diabetic mice compared to controls (P < 0.001, Fig. 6A). CoPP treatment enhanced both eNOS and peNOS levels, with a larger effect on the latter, this was manifest by a significant increase in the peNOS/eNOS ratio in CoPP-treated versus untreated diabetic mice (P < 0.01, Fig. 6A).
Fig. 6.
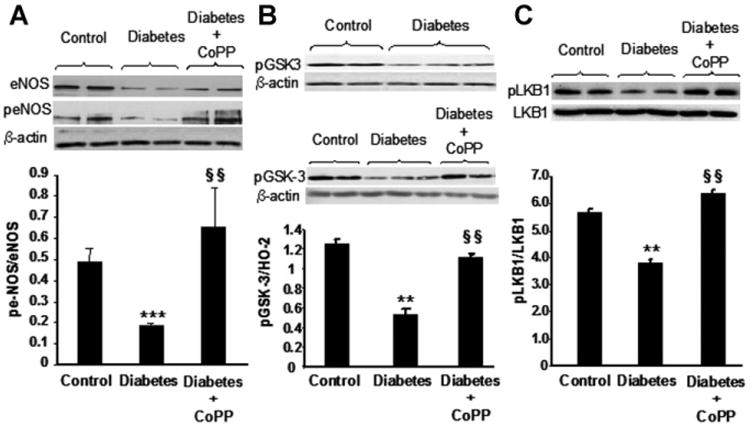
A: Western blot of total and phorphorylated (p−) forms of eNOS expression in hearts of control, diabetic mice untreated or treated with CoPP. Myocardial β-actin expression (comparative protein) is shown. Quantitative densitometry is expressed as ratio between phosphorylated and total amount of protein. ***P< 0.001 diabetes versus control, §§P<0.01 diabetes CoPP versus diabetes. Each bar represents mean ±SE of four experiments. B: pGSK-3(Ser 9) and β-actin expression in myocardium of control and diabetic mice untreated or treated with CoPP. Quantitative densitometry was determined. **P<0.01 diabetes versus control, §§P<0.01 diabetes CoPP versus diabetes. Each bar represents mean ± SE of four experiments in control and CoPP-treated diabetic mice and mean±SE of six experiments in diabetic group. C: Western blot of total and phosphorylated form of LKB1 in myocardium isolated from control, diabetic, and CoPP-treated diabetic mice. Quantitative densitometry is expressed as ratio between phosphorylated and total amount of protein. **P<0.01 diabetes versus control, §§P<0.01 diabetes CoPP versus diabetes. Each column represents mean ± SE of four experiments.
In addition, GSK-3, a substrate of pAKT and pAMPK activity, levels decreased in diabetic compared to control animals (P < 0.01, Fig. 6B). In contrast, hearts from CoPP-treated diabetic mice, exhibited a significant increase in pGSK-3 protein levels compared to hearts from untreated diabetic animals (P < 0.01). pLKB1 levels expressed as the ratio of pLKB1 to LKB1 decreased in diabetic hearts when compared to hearts isolated from control animals (P < 0.01). CoPP-treatment produced an increase in levels of pLKB1 in diabetic mice (P < 0.01 compared to untreated diabetic mice) to levels greater (P < 0.05) than in control animals (Fig. 6C).
Discussion
We examined coronary reactivity in the isolated heart of non-insulin-dependent diabetic mice for up to 28 weeks following STZ treatment. This is the first study to examine long-term changes in coronary reactivity in diabetes. The response of coronary resistance to different perfusion pressure regimens diverged from control in a time-dependent manner in spite of constant values of hyperglycemia and insulinemia. This finding might account for conflicting reports on type and/or magnitude of microvascular alterations in diabetic animal models. In agreement with our findings, Oltman et al. [2006] showed that Ach-induced response in Zucker rats was progressively attenuated in 16- to 24-week-old and 28- to 36-week-old rats. Moreover, Nagareddy et al. [2005] reported a gradual decreased expression of eNOS concomitant with increased nitrotyrosine and progressive depressed response to vasoactive agents in STZ-induced diabetes.
In the present study, vasoreactivity in control hearts was characterized by stable coronary resistance at constant pressure (65 mmHg) and “paradoxical” vasoconstriction at low pressure (30 mmHg). In contrast, 28-week diabetic hearts showed a progressive increase in coronary resistance at constant pressure, up to threefold basal value, while low-pressure perfusion resulted in vasodilation in place of vasoconstriction in control animals. Both responses at high and low pressure in diabetic hearts were reproduced by L-NAME infusion in healthy hearts.
“Paradoxical” vasoconstriction in hearts from different species of healthy animals, classifies this event as normal rather than a disturbance in vasomotor tone control [Frame and Powell, 1976; Canty and Klocke, 1985]. We hypothesized that, differently from a constant pressure condition where NO seems necessary to counteract the accumulation of an unknown vasoconstrictor, during low pressure the vasoconstrictor originates from the NO system itself, putatively the product of NO oxidation, peroxynitrite [Kozak et al., 2005; Schildknecht and Ullrich, 2009].
In support of NO shortage in chronic diabetic hearts we found decreased levels of eNOS compared to control hearts confirming previous reports [Nagareddy et al., 2005; L'Abbate et al., 2007; Peterson et al., 2007]. In addition, NOx levels were reduced while 3NT and levels increased in diabetic hearts compared to controls. This in spite of the likely increase in iNOS expression is already documented in the diabetic heart [Nagareddy et al., 2005; L'Abbate et al., 2007]. Hyperglycemia as well as ischemia enhances endothelial production leading to increased vascular formation of the potent vasoconstrictor ONOO− [Kossenjans et al., 2000]. Peroxynitrite, has been shown to oxidize the cofactor tetrahydrobiopterin into inactive molecules [Milstien and Katusic, 1999], resulting in a preferential increase in production over NO production by eNOS activity. Thus, in the hypothesis of peroxynitrite being responsible for “paradoxical” vasoconstriction in healthy animals, the reduction of NO production in the diabetic heart might explain the loss of vasoconstriction seen with the prevailing effect of ischemia-induced vasodilators such as adenosine. According to this interpretation, we predicted that restoration of NO production and reduction of oxidative stress in diabetic heart would normalize microvascular reactivity. To this end we administered CoPP, which enhanced eNOS expression, decreased and 3-NT levels, when compared to untreated diabetic animals, and abolished both vasoconstriction during constant pressure and vasodilation during low pressure. At low pressure, the incomplete restoration of “paradoxical” vasoconstriction in diabetic hearts after CoPP treatment (Fig. 2C vs. inset A) contrasted with the abolition of vasoconstriction in controls (inset Fig. 2C) with the result of almost identical resistance profiles in CoPP-treated control and CoPP-treated diabetic animals. This finding may be interpreted as the result of an abnormal reduction of with reduced production of peroxynitrite in CoPP-treated controls, as opposed to partially restored NO levels and thus increased production of peroxynitrite in CoPP-treated diabetic animals.
We have previously showed that HO-1 attenuates levels in diabetes and improves vascular function via an increase in extracellular superoxide dismutase [Kruger et al., 2005; Turkseven et al., 2005]. The effects of CoPP seen here may be due, in part, to the end-products of HO-1 activity, CO, and bilirubin. Bilirubin scavenges ROS and inhibits activation of NADPH oxidase and protein kinase-C, key signaling steps in oxidant-induced vascular injury [Abraham and Kappas, 2008]. CO has no antioxidant proprerties and its contribution to vasodilation and constriction of microvessels remains ill defined in our experimental model.
Additionally, we reported that increases in HO-1 increase myocardial levels of pLKB1, a serine/threonine protein kinase working in a complex with the “pseudokinase” STRAD and the protein MO25 which has been identified as an upstream activating kinase of AMPK [Hawley et al., 2003; Woods et al., 2003]. Therefore, HO-1 mediated increases of NO may involve pLKB1 and its down stream signaling. A major finding of the present paper is that STZ-induced diabetes was associated with decreased serum adiponectin and myocardial pLKB1, pAMPK, pAKT, and HO-1 levels. All the above alterations were reversed to levels observed in control animals by pre-treatment with CoPP. The HO-1 mediated decreases in ROS resulted in increased adiponectin and activation of pathways involving pLKB1, pAMPK, and pAKT (Fig. 7).
Fig. 7.
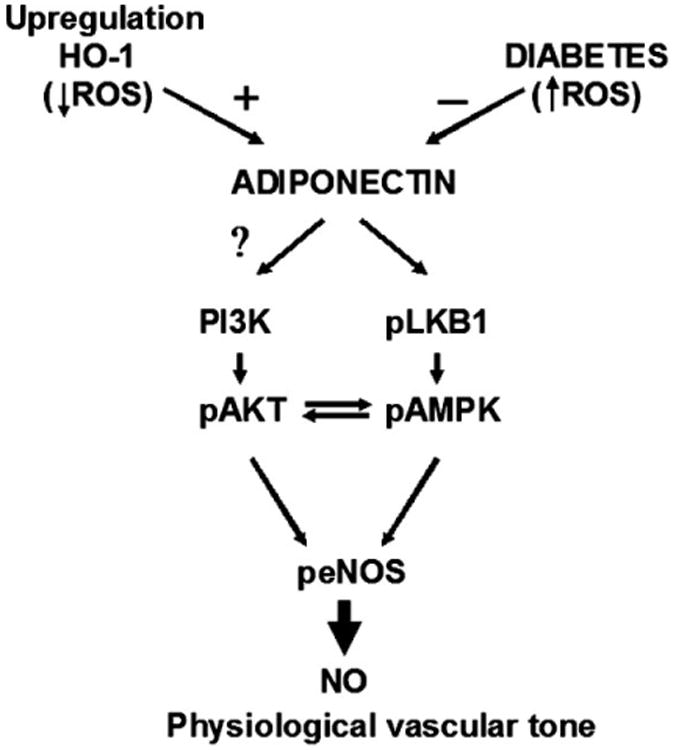
Hypothetical adiponectin-based mechanisms for coronary vasculature control. The scheme postulates the operation of adiponectin influence on LBK1-AMPK and PI3K/AKT pathways being negatively regulated by diabetic condition (increased ROS) and positively regulated by HO-1 expression.
Reduced plasma adiponectin levels have been reported in patients with coronary artery disease and diabetes, presumably as a result of increased ROS [Ohashi et al., 2006; Wang and Scherer, 2008]. Adiponectin is critical for endothelial cell survival and function via activation of eNOS and crosstalk between pAKT and pAMPK [Ouchi et al., 2004; Wang and Scherer, 2008]. Crosstalk between AMPK-AKT via pLKB1 is considered an important metabolic response necessary to attenuate ROS-mediated endothelial dysfunction [Schulz et al., 2008]. As both pAMPK and pAKT use eNOS as a substrate and enhance the levels of peNOS [Chen et al., 1999; Dimmeler et al., 1999; Abraham and Kappas, 2008], increased HO-1 expression becomes a key regulator of peNOS levels. Indeed when pAMPK and pAKT are increased peNOS is also increased [Fulton et al., 1999; Go et al., 2001]. We propose that the activated pLKB1, pAMPK-pAKT system observed in CoPP-treated diabetic hearts, could be reflected in the vessels by up-regulation of NO bioavailability and reduction of ROS (Fig. 7).
These results provide the first indication that increased LKB1 functions as a master upstream protein kinase regulating not only AMPK but also a subfamily of related kinases via an increase in HO-1. Activation of LKB1-AMPK is also important in cellular energy homeostasis via stimulation of glucose transport, switching off energy consumption by decreasing lipogenesis, increasing fatty acid oxidation and ATP [Tomas et al., 2002; Dyck and Lopaschuk, 2006]. Accordingly we measured pGSK-3 in our cardiac tissue as an internal control of AMPK and AKT activation; increased levels of pAMPK and pAKT were accompanied by increased pGSK-3 levels in treated diabetic mice.
Finally, glucose intolerance in our diabetic model deserves a comment. Increased insulin sensitivity which paralleled HO-1 over-expression may be related to several factors among which are the AKT mediated increase in glucose uptake and the reduction of oxidative stress. It should also be noted that, in addition to the reduced number of pancreatic β-cells, STZ affects glucose transport in skeletal muscle [Ramlal et al., 1989; Dimitrakoudis et al., 1992] through a decrease in the number of glucose transporters. These perturbations can be restored by increased expression of HO-1 and increased adiponectin levels. Moreover, Mosén et al. [2006] described a positive action of CO on glucose-stimulated insulin release in beta-cells, which might explain the lower glucose levels observed during the glucose tolerance test in CoPP-treated animals compared to untreated animals, in both control and diabetic mice.
Tissue assays in the present study was performed in hearts subjected to 80min Langendorff perfusion and the results could be biased, at least in part, by experimental conditions. Similarly, the absence of tissue assays in L-NAME-treated hearts prevented the clarification of the possible effect of NO via HO-1-mediated activation of pLKB1 and its downstream signal including pAMPK/pAKT axis. In addition, tissue assays were performed in heart tissue homogenate and the results cannot be assigned entirely to the vascular component. However, drugs which increase pAMPK in vivo, in muscle and myocytes [Al-Khalili et al., 2004; LeBrasseur et al., 2006] also increase pAMPK in the vascular system [Ouchi et al., 2004; Rodella et al., 2008].
A further limitation is the lack of measurement of iNOS expression in tissue samples. Under normal conditions, NO is produced by eNOS; however, iNOS is also expressed in cardiomyocytes from diabetic rats. Nagareddy et al. already reported decreased eNOS levels as opposed to increased iNOS levels in diabetic animals. We also showed the same pattern in the heart of mildly diabetic rats and its reversal to normal by HO-1 over-expression [L'Abbate et al., 2007]. In the present study, a putative increased expression of iNOS is compatible with the increased 3-nitrotyrosine levels observed in diabetic animals as well as with the overall reduction of NOx, which can be attributed to the scavenging of NO by superoxide as well as to peroxynitrite-induced deficiency of tetrahydrobiopterin. Further studies are required in order to understand the role of the eNOS/iNOS balance in coronary vasoconstriction by the use of specific inhibitors of iNOS as well as by tissue measurements timed for the different phases of the experiment (baseline, ischemia, reperfusion). The compartimentalization of NOS isoforms should also be considered as a further confounding factor in the use of tissue homogenates.
Finally, the meaning of the abolition of paradoxical vasoconstriction in chronic diabetes remains to be established. Paradoxical vasoconstriction has been interpreted as a mechanism to divert blood from less towards better perfused myocardium [Sambuceti et al., 2005], however, the effects of the loss of such a response has not been investigated.
Conclusion
In the present study the key event is HO-1 induction and increased HO activity that is associated with increases in adiponectin, pAMPK, pAKT, and peNOS levels. The study clearly demonstrates the existence of an HO-1-adiponectin regulatory axis that can be manipulated to enhance the crosstalk between pAKT-pAMPK signaling pathways. The consequent increase in both NO production and bioavailability is critical for microvascular function in our diabetic model. Thus, the effect of CoPP-treatment that we observed has to be considered to be the result of both local and systemic effects that results in the restoration of microvascular function in the hearts of diabetic mice.
In examining novel pharmacological strategies for the treatment of diabetes, it should be emphasized that the pharmacological targeting of HO-1 can be achieved by statins and apolipoproteins A1 mimetics [Abraham and Kappas, 2008] and the effect of CoPP can be mimicked by the CO releasing molecules [Rodella et al., 2008] suggesting that this approach merits further study.
Acknowledgments
This study was supported by Scuola Superiore Sant'Anna, CNR Medical Department and Cardiopulmonary project, Italy (A.L.); CNR Italy, contract grant number: RSTL2008 (C.K.) and FP7-ICT-2007 Art-TREAT project (grant agreement FP7-224297); National Institutes of Health grants USA, contract number HL55601, DK068134 and HL34300 (N.G.A.). We thank Dr. N. Vesentini for her helpful criticism and Martha Heck for her outstanding secretarial and editing assistance.
Grant sponsor: Scuola Superiore Sant'Anna; Grant sponsor: CNR Medical Department and Cardiopulmonary project, Italy; Grant numbers: RSTL2008, FP7-ICT-2007; Grant sponsor: National Institutes of Health grants USA; Grant numbers: HL55601, DK068134, HL34300.
References
- Abraham NG, Kappas A. Pharmacological and clinical aspects of heme oxygenase. Pharmacol Rev. 2008;60:79–127. doi: 10.1124/pr.107.07104. [DOI] [PubMed] [Google Scholar]
- Abraham NG, Kushida T, McClung J, Weiss M, Quan S, Lafaro R, Darzynkiewicz Z, Wolin M. Heme oxygenase-1 attenuates glucose-mediated cell growth arrest and apoptosis in human microvessel endothelial cells. Circ Res. 2003;93:507–514. doi: 10.1161/01.RES.0000091828.36599.34. [DOI] [PubMed] [Google Scholar]
- Al-Khalili L, Krook A, Zierath JR, Cartee GD. Prior serum- and AICAR-induced AMPK activation in primary human myocytes does not lead to subsequent increase in insulin-stimulated glucose uptake. Am J Physiol Endocrinol Metab. 2004;287:E553–E557. doi: 10.1152/ajpendo.00161.2004. [DOI] [PubMed] [Google Scholar]
- Berg AH, Scherer PE. Adipose tissue, inflammation, and cardiovascular disease. Circ Res. 2005;96:939–949. doi: 10.1161/01.RES.0000163635.62927.34. [DOI] [PubMed] [Google Scholar]
- Berg AH, Combs TP, Du X, Brownlee M, Scherer PE. The adipocyte-secreted protein Acrp30 enhances hepatic insulin action. Nat Med. 2001;7:947–953. doi: 10.1038/90992. [DOI] [PubMed] [Google Scholar]
- Canty JM, Jr, Klocke FJ. Reduced regional myocardial perfusion in the presence of pharmacologic vasodilator reserve. Circulation. 1985;71:370–377. doi: 10.1161/01.cir.71.2.370. [DOI] [PubMed] [Google Scholar]
- Chen H, Montagnani M, Funahashi T, Shimomura I, Quon MJ. Adiponectin stimulates production of nitric oxide in vascular endothelial cells. J Biol Chem. 2003;278:45021–45026. doi: 10.1074/jbc.M307878200. [DOI] [PubMed] [Google Scholar]
- Chen ZP, Mitchelhill KI, Michell BJ, Stapleton D, Rodriguez-Crespo I, Witters LA, Power DA, Ortiz De Montellano PR, Kemp BE. AMP-activated protein kinase phosphorylation of endothelial NO synthase. FEBS Lett. 1999;443:285–289. doi: 10.1016/s0014-5793(98)01705-0. [DOI] [PubMed] [Google Scholar]
- Dimitrakoudis D, Vranic M, Klip A. Effects of hyperglycemia on glucose transporters of the muscle: Use of the renal glucose reabsorption inhibitor phlorizin to control glycemia. J Am Soc Nephrol. 1992;3:1078–1091. doi: 10.1681/ASN.V351078. [DOI] [PubMed] [Google Scholar]
- Dimmeler S, Fleming I, Fisslthaler B, Hermann C, Busse R, Zeiher AM. Activation of nitric oxide synthase in endothelial cells by Akt-dependent phosphorylation. Nature. 1999;399:601–605. doi: 10.1038/21224. [DOI] [PubMed] [Google Scholar]
- Dyck JR, Lopaschuk GD. AMPK alterations in cardiac physiology and pathology: Enemy or ally? J Physiol. 2006;574:95–112. doi: 10.1113/jphysiol.2006.109389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleming I, Schulz C, Fichtlscherer B, Kemp BE, Fisslthaler B, Busse R. AMP-activated protein kinase (AMPK) regulates the insulin-induced activation of the nitric oxide synthase in human platelets. Thromb Haemost. 2003;90:863–871. doi: 10.1160/TH03-04-0228. [DOI] [PubMed] [Google Scholar]
- Frame LH, Powell WJ., Jr Progressive perfusion impairment during prolonged low flow myocardial ischemia in dogs. Circ Res. 1976;39:269–276. doi: 10.1161/01.res.39.2.269. [DOI] [PubMed] [Google Scholar]
- Fulton D, Gratton JP, McCabe TJ, Fontana J, Fujio Y, Walsh K, Franke TF, Papapetropoulos A, Sessa WC. Regulation of endothelium-derived nitric oxide production by the protein kinase Akt. Nature. 1999;399:597–601. doi: 10.1038/21218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Go YM, Boo YC, Park H, Maland MC, Patel R, Pritchard KA, Jr, Fujio Y, Walsh K, Darley-Usmar V, Jo H. Protein kinase B/Akt activates c-Jun NH(2)-terminal kinase by increasing NO production in response to shear stress. J Appl Physiol. 2001;91:1574–1581. doi: 10.1152/jappl.2001.91.4.1574. [DOI] [PubMed] [Google Scholar]
- Hawley SA, Boudeau J, Reid JL, Mustard KJ, Udd L, Makela TP, Alessi DR, Hardie DG. Complexes between the LKB1 tumor suppressor, STRADα/β and MO25α/β are upstream kinases in the AMP-activated protein kinase cascade. J Biol. 2003;2:28. doi: 10.1186/1475-4924-2-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JY, van de WE, Laplante M, Azzara A, Trujillo ME, Hofmann SM, Schraw T, Durand JL, Li H, Li G, Jelicks LA, Mehler MF, Hui DY, Deshaies Y, Shulman GI, Schwartz GJ, Scherer PE. Obesity-associated improvements in metabolic profile through expansion of adipose tissue. J Clin Invest. 2007;117:2621–2637. doi: 10.1172/JCI31021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kossenjans W, Eis A, Sahay R, Brockman D, Myatt L. Role of peroxynitrite in altered fetal-placental vascular reactivity in diabetes or preeclampsia. Am J Phyisiol Heart Circ Physiol. 2000;278:H1311–H1319. doi: 10.1152/ajpheart.2000.278.4.H1311. [DOI] [PubMed] [Google Scholar]
- Kovacic S, Soltys CL, Barr AJ, Shiojima I, Walsh K, Dyck JR. Akt activity negatively regulates phosphorylation of AMP-activated protein kinase in the heart. J Biol Chem. 2003;278:39422–39427. doi: 10.1074/jbc.M305371200. [DOI] [PubMed] [Google Scholar]
- Kozak AJ, Liu F, Funovics P, Jacoby A, Kubant R, Malinski T. Role of peroxynitrite in the process of vascular tone regulation by nitric oxide and prostanoids—A nanotechnological approach. Prostaglandins Leukot Essent Fatty Acids. 2005;72:105–113. doi: 10.1016/j.plefa.2004.10.007. [DOI] [PubMed] [Google Scholar]
- Kruger AL, Peterson S, Turkseven S, Kaminski PM, Zhang FF, Quan S, Wolin MS, Abraham NG. D-4F induces heme oxygenase-1 and extracellular superoxide dismutase, decreases endothelial cell sloughing, and improves vascular reactivity in rat model of diabetes. Circulation. 2005;111:3126–3134. doi: 10.1161/CIRCULATIONAHA.104.517102. [DOI] [PubMed] [Google Scholar]
- Kusmic C, Lazzerini G, Coceani F, Barsacchi R, L'Abbate A, Sambuceti G. Paradoxical coronary microcirculatory constriction during ischemia: A synergic function for nitric oxide and endothelin. Am J Physiol Heart Circ Physiol. 2006;291:H1814–H1821. doi: 10.1152/ajpheart.00220.2006. [DOI] [PubMed] [Google Scholar]
- L'Abbate A, Neglia D, Vecoli C, Novelli M, Ottaviano V, Baldi S, Barsacchi R, Paolicchi A, Masiello P, Drummond GS, McClung JA, Abraham NG. Beneficial effect of heme oxygenase-1 expression on myocardial ischemia-reperfusion involves an increase in adiponectin in mildly diabetic rats. Am J Physiol Heart Circ Physiol. 2007;293:H3532–H3541. doi: 10.1152/ajpheart.00826.2007. [DOI] [PubMed] [Google Scholar]
- LeBrasseur NK, Kelly M, Tsao TS, Farmer SR, Saha AK, Ruderman NB, Tomas E. Thiazolidinediones can rapidly activate AMP-activated protein kinase in mammalian tissues. Am J Physiol Endocrinol Metab. 2006;291:E175–E181. doi: 10.1152/ajpendo.00453.2005. [DOI] [PubMed] [Google Scholar]
- Milstien S, Katusic Z. Oxidation of tetrahydrobiopterin by peroxynitrite: Implications for vascular endothelial function. Biochem Biophys Res Commun. 1999;263:681–684. doi: 10.1006/bbrc.1999.1422. [DOI] [PubMed] [Google Scholar]
- Mosén H, Salehi A, Henningsson R, Lundquist I. Nitric oxide inhibits, and carbon monoxide activates, islet acid α-glucoside hydrolase activities in parallel with glucose-stimulated insulin secretion. J Endocrinol. 2006;190:681–693. doi: 10.1677/joe.1.06890. [DOI] [PubMed] [Google Scholar]
- Musi N, Goodyear LJ. Targeting the AMP-activated protein kinase for the treatment of type 2 diabetes. Curr Drug Targets Immune Endocr Metabol Disord. 2002;2:119–127. [PubMed] [Google Scholar]
- Nagareddy PR, Xia Z, McNeill JH, MacLeod KM. Increased expression of iNOS is associated with endothelial dysfunction and impaired pressor responsiveness in streptozotocin-induced diabetes. Am J Physiol Heart Circ Physiol. 2005;289:H2144–H2152. doi: 10.1152/ajpheart.00591.2005. [DOI] [PubMed] [Google Scholar]
- Ohashi K, Kihara S, Ouchi N, Kumada M, Fujita K, Hiuge A, Hibuse T, Ryo M, Nishizawa H, Maeda N, Maeda K, Shibata R, Walsh K, Funahashi T, Shimomura I. Adiponectin replenishment ameliorates obesity-related hypertension. Hypertension. 2006;47:1108–1116. doi: 10.1161/01.HYP.0000222368.43759.a1. [DOI] [PubMed] [Google Scholar]
- Oltman CL, Richou LL, Davidson EP, Coppey LJ, Lund DD, Yorek MA. Progression of coronary and mesenteric vascular dysfunction in Zucker obese and Zucker diabetic fatty rats. Am J Physiol Heart Circ Physiol. 2006;291:H1780–H1787. doi: 10.1152/ajpheart.01297.2005. [DOI] [PubMed] [Google Scholar]
- Ouchi N, Kobayashi H, Kihara S, Kumada M, Sato K, Inoue T, Funahashi T, Walsh K. Adiponectin stimulates angiogenesis by promoting cross-talk between AMP-activated protein kinase and Akt signaling in endothelial cells. J Biol Chem. 2004;279:1304–1309. doi: 10.1074/jbc.M310389200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson SJ, Husney D, Kruger AL, Olszanecki R, Ricci F, Rodella LF, Stacchiotti A, Rezzani R, McClung JA, Aronow WS, Ikehara S, Abraham NG. Long-term treatment with the apolipoprotein A1 mimetic Peptide increases antioxidants and vascular repair in type I diabetic rats. J Pharmacol Exp Ther. 2007;322:514–520. doi: 10.1124/jpet.107.119479. [DOI] [PubMed] [Google Scholar]
- Ramlal T, Rastogi S, Vranic M, Klip A. Decrease in glucose transporter number in skeletal muscle of mildly diabetic (streptozotocin-treated) rats. Endocrinology. 1989;125:890–897. doi: 10.1210/endo-125-2-890. [DOI] [PubMed] [Google Scholar]
- Reihill JA, Ewart MA, Hardie DG, Salt IP. AMP-activated protein kinase mediates VEGF-stimulated endothelial NO production. Biochem Biophys Res Commun. 2007;354:1084–1088. doi: 10.1016/j.bbrc.2007.01.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodella LF, Peterson SJ, Drummond G, Rezzani R, Falck JR, Abraham NG. Heme oxygenase-derived carbon monoxide restores adiponectin levels and vascular function in type 1 diabetes. Drug Metab Lett. 2008;2:290–300. doi: 10.2174/187231208786734058. [DOI] [PubMed] [Google Scholar]
- Sambuceti G, Marzilli M, Marraccini P, Schneider-Eicke J, Gliozheni E, Parodi O, L'Abbate A. Coronary vasoconstriction during myocardial ischemia induced by rises in metabolic demand in patients with coronary artery disease. Circulation. 1997;95:2652–2659. doi: 10.1161/01.cir.95.12.2652. [DOI] [PubMed] [Google Scholar]
- Sambuceti G, Marzilli M, Fedele S, Marini C, L'Abbate A. Paradoxical increase in microvascular resistance during tachycardia downstream from a severe stenosis in patients with coronary artery disease: Reversal by angioplasty. Circulation. 2001;103:2352–2360. doi: 10.1161/01.cir.103.19.2352. [DOI] [PubMed] [Google Scholar]
- Sambuceti G, Marzilli M, Mari A, Marini C, Schluter M, Testa R, Papini M, Marraccini P, Ciriello G, Marzullo P, L'Abbate A. Coronary microcirculatory vasoconstriction is heterogeneously distributed in acutely ischemic myocardium. Am J Physiol Heart Circ Physiol. 2005;288:H2298–H2305. doi: 10.1152/ajpheart.00870.2004. [DOI] [PubMed] [Google Scholar]
- Schildknecht S, Ullrich V. Peroxynitrite as regulator of vascular prostanoid synthesis. Arch Biochem Biophys. 2009;484:183–189. doi: 10.1016/j.abb.2008.10.023. [DOI] [PubMed] [Google Scholar]
- Schulz E, Dopheide J, Schuhmacher S, Thomas SR, Chen K, Daiber A, Wenzel P, Munzel T, Keaney JF., Jr Suppression of the JNK pathway by induction of a metabolic stress response prevents vascular injury and dysfunction. Circulation. 2008;118:1347–1357. doi: 10.1161/CIRCULATIONAHA.108.784298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skurk C, Maatz H, Kim HS, Yang J, Abid MR, Aird WC, Walsh K. The Akt-regulated forkhead transcription factor FOXO3a controls endothelial cell viability through modulation of the caspase-8 inhibitor FLIP. J Biol Chem. 2004;279:1513–1525. doi: 10.1074/jbc.M304736200. [DOI] [PubMed] [Google Scholar]
- Sun JF, Phung T, Shiojima I, Felske T, Upalakalin JN, Feng D, Kornaga T, Dor T, Dvorak AM, Walsh K, Benjamin LE. Microvascular patterning is controlled by fine-tuning the Akt signal. Proc Natl Acad Sci USA. 2005;102:128–133. doi: 10.1073/pnas.0403198102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomas E, Tsao TS, Saha AK, Murrey HE, Zhang CC, Itani SI, Lodish HF, Ruderman NB. Enhanced muscle fat oxidation and glucose transport by ACRP30 globular domain: Acetyl-CoA carboxylase inhibition and AMP-activated protein kinase activation. Proc Natl Acad Sci USA. 2002;99:16309–16313. doi: 10.1073/pnas.222657499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turkseven S, Kruger A, Mingone CJ, Kaminski P, Inaba M, Rodella LF, Ikehara S, Wolin MS, Abraham NG. Antioxidant mechanism of heme oxygenase-1 involves an increase in superoxide dismutase and catalase in experimental diabetes. Am J Physiol Heart Circ Physiol. 2005;289:H701–H707. doi: 10.1152/ajpheart.00024.2005. [DOI] [PubMed] [Google Scholar]
- Wang ZV, Scherer PE. Adiponectin, cardiovascular function, and hypertension. Hypertension. 2008;51:8–14. doi: 10.1161/HYPERTENSIONAHA.107.099424. [DOI] [PubMed] [Google Scholar]
- Woods A, Johnstone SR, Dickerson K, Leiper FC, Fryer LG, Neumann D, Schlattner U, Wallimann T, Carlson M, Carling D. LKB1 is the upstream kinase in the AMP-activated protein kinase cascade. Curr Biol. 2003;13:2004–2008. doi: 10.1016/j.cub.2003.10.031. [DOI] [PubMed] [Google Scholar]