

Abstract
T cell receptors (TCRs) pair in millions of combinations to create complex and personally unique T cell repertoires. Using tetramers to analyze CD1b-reactive TCRs, we detected T cells with highly stereotyped TCR α chains present among genetically unrelated tuberculosis patients. These germline-encoded mycolyl-reactive (GEM) T cells were defined by CD4 expression and rearrangement of TRAV1-2 to TRAJ9 with few N-region additions. TCR analysis by high throughput sequencing, binding and crystallography showed linkage of TCR α sequence motifs to high affinity antigen recognition. Thus, the CD1-reactive TCR repertoire is composed of at least two compartments, high affinity GEM TCRs and more diverse TCRs with low affinity for CD1b-lipid complexes. These data demonstrate high inter-donor conservation of TCRs, which likely results from selection by a non-polymorphic antigen presenting molecule and an immunodominant antigen.
Studies of T cells in vaccines, autoimmunity and infection rely on the model that highly diverse TCRs recognize peptides bound to major histocompatibility complex (MHC) proteins. However, lipids presented by CD1 proteins expand the biochemical range of antigens for T cells 1. T cells recognize foreign lipid antigens from major pathogens like Mycobacterium tuberculosis, indicating that microbial lipid recognition occurs during the natural history of infectious diseases. Also, because CD1 and MHC proteins differ greatly in their rates of polymorphism in human populations, comparison of MHC and CD1-reactive TCRs provides an experimental system to study the role of host genetics in shaping human TCR repertoires. Currently, understanding of the complexity of the overall T cell repertoire is dominated by knowledge of MHC-restricted T cells, which form highly complex patterns based on pairing of rearranged TCR α and β chains that generate a unique TCR repertoire for each person. This diversity is generated through variable (V), diversity (D) and joining (J) segments, deletions and N-region additions. MHC class I and II proteins show corresponding complexity, as they are highly polymorphic in human populations, and any given protein sequence encoded at HLA-A, B, C, DR, DP, DQ loci can bind many peptides.
In contrast, CD1 polymorphisms are rare and are usually functionally silent 2. Thus, genetic polymorphism, which represents one major driver of MHC-reactive TCR repertoire diversity, is largely absent in the human CD1 system. To some extent, the non-polymorphic nature of CD1 antigen presenting molecules plays out as an apparently simplified TCR repertoire. The main example is invariant natural killer T cells (iNKT cells), which express TRAV10 and TRAJ18 (previously known as Vα24 and Jα18) and are found in most human donors 3, 4. However, most CD1-reactive T cells do not express this receptor. Other CD1d-reactive T cells (diverse NKT cells) 5, 6 and all known T cells recognizing CD1a, CD1b or CD1c utilize differing TCR V, D or J segments 7-10. Accordingly, ‘consensus’ classifications of the CD1 repertoire recognize three T cell types: iNKT cells, diverse NKT cells and diverse T cells recognizing CD1a, CD1b and CD1c 11.
CD1a, CD1b and CD1c, known as the group 1 CD1 proteins, are broadly retained during mammalian evolution 12, and they activate large numbers of T cells present in the blood and tissues of humans 8, 9. However, information about their biology and TCR diversity is extremely limited because no specific surface markers are known, and efficient methods to capture clones for TCR sequencing have not been developed. Taking advantage of CD1b tetramers as a new tool to study T cells in larger numbers, we identified highly stereotyped TCRs comprised of nearly identical TCR α chains. These TCR amino acid sequences derive from differing nucleotide sequences, showing that conserved TCRs arise through distinct rearrangement events within individual donors. Further, such TCRs are expressed on cell populations that are readily detectable among genetically unrelated donors ex vivo. These finding identify a previously unknown type of TCR-conserved αβ T cell type and point to inter-donor conservation of CD1b-reactive T cell responses.
RESULTS
T cells with high and low avidity for CD1b
To study lipid-reactive T cells in a disease setting, we used lipid extracts of Mycobacterium tuberculosis to activate T cells from tuberculosis patients (Supplementary Fig. 1). By screening a panel of clones for responses to mycobacterial lipids and CD1b, we detected clone 18 (Fig. 1a). We identified the stimulating antigen as free mycolic acid (MA) (Fig. 1b), an essential long-chain mycobacterial lipid (Supplementary Fig. 2). Comparing its TCR α and β chains to five previously known TCRs that recognize mycolyl lipids, we did not observe conservation of TCR sequence or V or J gene segment usage (Table 1) 1, 7, 13-16. Thus, initial results based on conventional cloning methods supported the widespread view that TCRs in the group 1 CD1 system are diverse, similar to those of MHC-restricted T cells 7, 8, 10, 17 and different from iNKT and mucosal-associated invariant T (MAIT) cells, which express highly conserved TCRs 17, 18.
Figure 1. CD1b-restricted T cell clones generated by conventional methods.
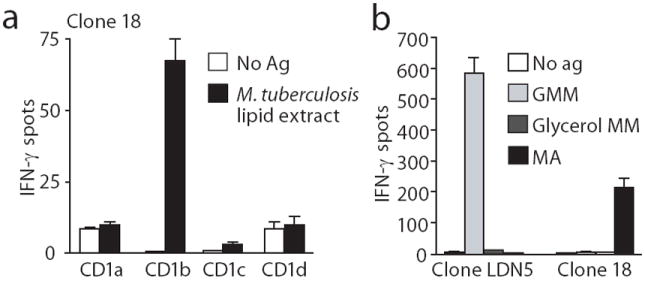
(a) ELISPOT assay of IFN-γ production by clone 18 after stimulation with K562 cells transfected with CD1a, CD1b, CD1c or CD1d in the presence or absence of M. tuberculosis lipid extract. (b) ELISPOT assay of IFN-γ production by clone LDN5 and clone 18 stimulated with glucose monomycolate (GMM), mycolic acid (MA) or glycerol monomycoalte (glycerol MM) and K562 cells transfected with CD1b. Panels a and b show standard deviation of triplicate measurements and are representative of three experiments
Table 1.
TCRs of mycolyl lipid specific T cell clones
| Clone | Phenotype | TRAV | TRAJ | TRBV | TRBJ | Antigen |
|---|---|---|---|---|---|---|
| 18 | CD4 | 1-2 | 9 | 6-2 | 2-2 | Mycolic acid |
| LDN5 | DN | 17 | 9 | 4-1 | 2-1 | Glucose monomycolate |
| DN1 | DN | 13-2 | 57 | 5-1 | 2-7 | Mycolic acid |
| DN.pott | DN | 3 | 31 | 7-3 | 2-1 | Mycolic acid |
| Z5B71 | CD4 | 35 | 52 | 3-1 | 2-5 | Glycerol monomycolate |
| Glucose monomycolate |
However, existing data 7, 8, 10 might inadequately represent the CD1b repertoire based on the small numbers of clones harvested to date and the need to extensively culture such clones to expand them. To bypass these problems and analyze T cells in the ex vivo setting among patients immunized by natural infection, we generated CD1b tetramers 19 and loaded them with glucose monomycolate (GMM; Supplementary Fig. 2). This mycobacterial glycolipid antigen is produced in vivo during infection 20 and potently activates CD1b-reactive T cells in humans and cows 15, 19, 21, 22. We sorted Tet+ T cells from three tuberculosis patients, cloned them at limiting dilution and screened for GMM-dependent functional response. Whereas Clone 18 and other T cell clones in the group 1 CD1 system were collected over many years (Table 1), tetramer binding to CD1b-reactive TCRs efficiently enriched for the relevant CD1b-reactive T cells, yielding multiple mycolyl lipid-reactive TCRs in all three patients studied (Fig. 2a). Thus, tetramer sorting represents an efficient method to sample the natural CD1b-GMM-reactive T cell repertoire ex vivo, demonstrating the existence of mycolyl lipid reactive clones among genetically unrelated donors.
Figure 2. Tetramer-based approaches efficiently generate CD1b-restricted T cell clones.
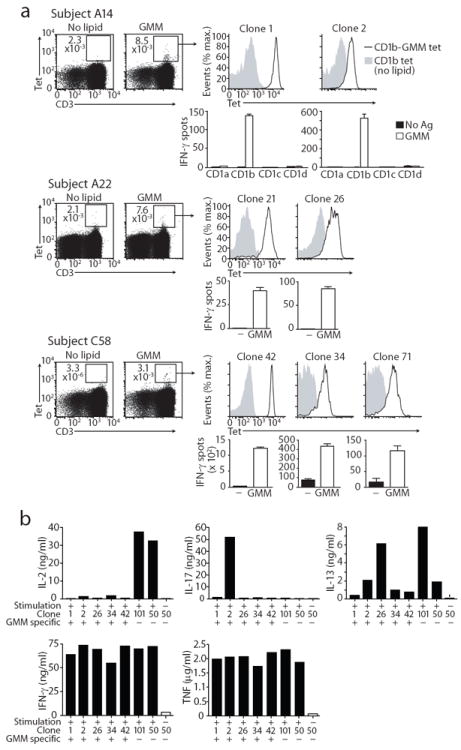
(a) Clones derived from PBMC of subjects A14, A22 and C58 were selected for staining with GMM-loaded CD1b tetramers. CD1b dependence and GMM reactivity of the Tet+ clones was confirmed by stimulation with K562 cells transfected with CD1a, CD1b, CD1c, or CD1d in the presence or absence of GMM. Tetramer staining was performed once on each clone, and ELISPOT experiments were performed twice on each clone. The SD of triplicate ELISPOT wells of one experiment is shown. (b) Supernatants of Tet+, GMM reactive clones 1, 2, 26, 34, 42, and Tet−, GMM non-reactive control clones 101 (CD4+TRAV1-2+) and 50 (CD8+TRBV4-1+), stimulated with or without PMA and ionomycin for 16 hours, were tested for cytokine content in a Luminex assay.
In all cases, clones that produced IFN-γ after GMM a ntigen stimulation also stained with GMM-loaded CD1b tetramers (Fig. 2). Based on staining intensity, the analyzed clones segregated into two groups detected with intermediate (Tetint) or high (Tethigh) staining. Tetint clones (clones 2, 26, 34, 71) showed diverse patterns of CD4 and CD8 expression, similar to clones generated by conventional methods (Table 1). In contrast, clone 18 and all Tethigh clones (clones 1, 21, 42) expressed CD4 but not CD8 (Fig. 2a and Fig. 3a). Both Tetint and Tethigh cells were isolated from each patient, suggesting that the differing avidity patterns were not patient-specific, but instead existed as distinct cell types present side by side in individual patients.
Figure 3. Conserved TCRs and CD4 define germline-encoded mycolyl-reactive (GEM) T cells.

(a) The CDR3 regions of CD1b-restricted GEM T cell clones are shown according to the their origin. GEM TCR sequences are compared to sequences from a non-GEM T cell clone recognizing CD1b (LDN5) and representative sequences from NKT (J3N.5) and MAIT cell clones (TRBV20). Light grey: germline-encoded V segment derived nucleotides; Dark grey: germline-encoded J segment derived nucleotides; Boxed: non-germline-encoded nucleotides in the α chain. TRBV6-2 and TRBV6-3 containing transcripts cannot be distinguished because the coding sequence of the two genes is identical. For simplicity, we refer to this shared sequence as TRBV6-2. (b) Human TCR conserved T cells types are illustrated.
GMM-specific T cells have antimicrobial effector functions
To gain insight into helper or effector functions of CD1b-reactive T cells we used a multiplex cytokine array to broadly profile cytokines produced by clones recognizing CD1b (1,2, 26, 34, 42) or control clones generated in the same way (101 and 50) (Fig. 2b). Our array design emphasized cytokines with defining roles in T helper cell function (TH1, TH17, TH2), such as interferon-γ (IFN-γ), IL-17 and IL-13. We also tested whether T cells produce two cytokines that are necessary for effective human anti-tubercular responses in vivo, TNF and IFN-γ 23, 24. In all cases, short-term T cell cultures produced IFN-γ and TNF, while helper cytokines like IL-2 and IL-17 were not consistently detected, suggesting an anti-microbial function of GMM-specific T cells 23, 24. As observed previously with clones cultured long-term in vitro 25, 26, these ex vivo studies confirmed that IFN-γ and TNF were reliably produced.
GEM T cell clones express invariant TCR α chains
Similar to clones generated by conventional methods (Table 1), Tetint clones showed differing TCR α genes. In contrast, all Tethigh clones expressed TCRs that were highly similar to one another and the TCR expressed by clone 18 (Fig. 3a). The TCR α chains of the Tethigh clones were identical in length and used the same variable (TRAV1-2) and joining (TRAJ9) segments, with few N region additions, yielding the CDR3 consensus sequence CAVRNTGGFKTIF (Fig. 3a). Because these similar rearrangements yielded largely germline-encoded TCR α chains and mediate recognition of mycolyl lipids, we designated these clones germline-encoded mycolyl lipid-reactive (GEM) T cells. Although lacking strict sequence conservation, TCR β chains were apparently biased in variable region (TRBV) usage. TRBV6-2 and TRBV30 are expressed in 3.6 % and 1 % percent of human T cells 27, 28, yet were found in all of the original set of GEM clones (Fig. 2a and Fig. 3). In addition to these clones from patients A22, C58, C52 and A14, single cell sequencing of Tet+ cells from patients C40, C52, C58 returned three TRBV6-2, but no TRBV30 sequences (Supplementary Fig. 3). Thus, GEM T cells are characterized by nearly invariant TCR α chains and a TCR β variable region gene bias, a pattern similar to iNKT cells and MAIT cells (Fig. 3b).
GEM TCRs bind CD1b-GMM with high affinity
Because strict TCR α conservation was seen only among Tethigh clones, we hypothesized that high-affinity binding of GEM TCRs to CD1b-GMM mediates the expansion of GEM T cells 19. Binding of any TCR to CD1b had not been previously measured, so we developed a plasmon resonance assay for binding of transmembrane region-truncated, disulfide-linked 29. GEM TCRs to CD1b-GMM complexes. All three GEM TCRs showed dissociation constants (KD) near to the value of ~ 1 μM (Fig. 4). These values are at the high end of the range for TCRs recognizing natural peptide or lipid antigens, and they are comparable to measurements of human NKT TCRs binding to the superagonist α-galactosylceramide in complex with CD1d30. Thus, the bright staining by CD1b tetramers for GEM clones (Fig. 2a) is explained by high affinity ternary interactions of αβ TCR heterodimers with CD1b-GMM complexes.
Figure 4. GEM TCR affinity.
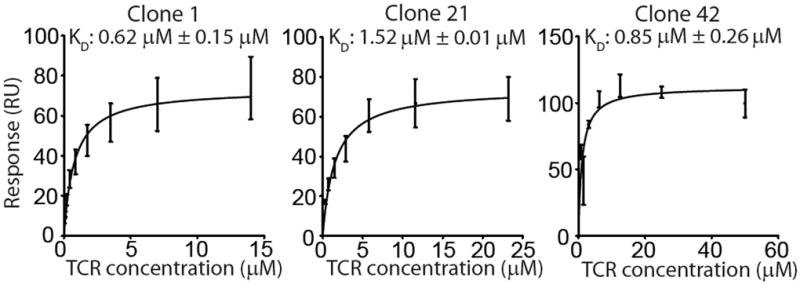
(a) Surface plasmon resonance measurements of the interaction between soluble TCRs of clones 1, 21 and 42 and immobilized CD1b-GMM complexes, and the equilibrium dissociation constants calculated from these measurements. Error bars represent SEM of two independent experiments.
GEM TCRs mediate antigen recognition
Next we determined the role of GEM TCRs in T cell activation by transferring TCRs into TCR β deficient JRT3-T3.5 cells. Untransfected cells were not activated by antigens, whereas expression of native GMM-reactive α and β TCRs (clones 42, 21, 1) in JRT3-T3.5 cells conferred specific recognition of GMM, but not MA, in all cases. TCR α and β chains from the MA-specific clone 18 reconstituted MA, but not GMM recognition (Fig. 5a). Thus, GEM TCRs are necessary for antigen-mediated activation, and the particular TCRs determine antigen fine specificity.
Figure 5. Antigen specificity of TCR transfectants.
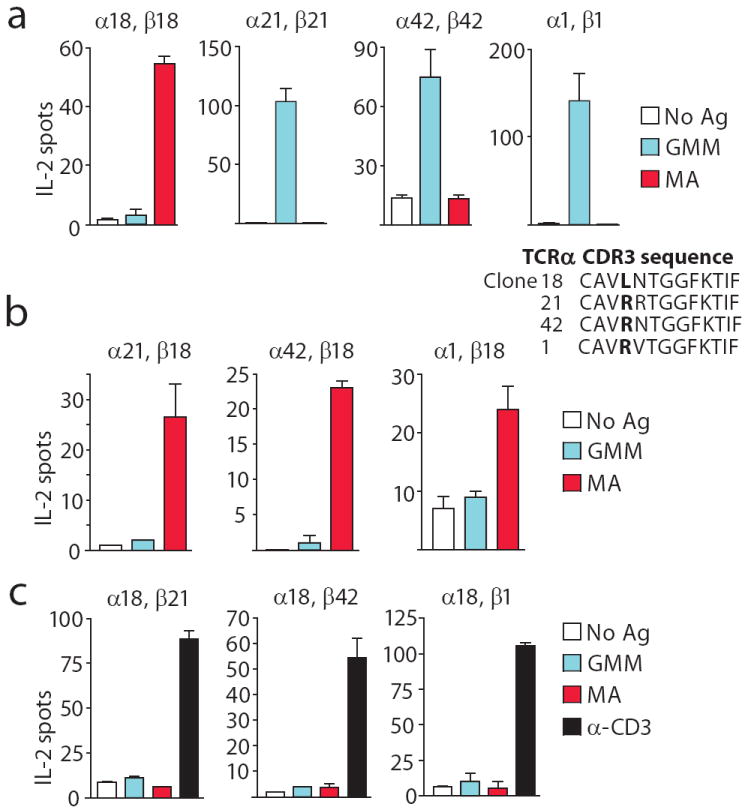
(a) JRT3-T3.5 cells were transfected with the α and β chains of the native TCRs of GEM clones 18, 21, 1 and 42 and analysed for recognition of GMM and MA presented by monocyte-derived dendritic cells. CDR3a sequences are shown on top with position 107 in bold. (b) To form chimeric TCRs, the TCR α chain of clone 18 was combined with the β chain of clones 21, 42, or 1 and (c) the α chains of clones 21, 42, or 1 were combined with the β chain of clone 18. Function of TCR reconstituted cells was determined with antigen or an activating α-CD3 (OKT3) antibody for cells refractory to antigen. The error bars represent the SD of triplicate wells, and results are representative of two independent transfection experiments.
To formally evaluate the relative contribution of TCR α and β chains in antigen fine specificity, we transfected native, but mismatched, TCR α and β pairs from clones 1, 18, 21 and 42 to create chimeric TCRs and measured responses to MA and GMM antigens. In all cases, transfectants responded to antigen or anti-CD3 stimulation, suggesting that the chimeric TCRs were expressed and signaled into cells. Because all GEM T cell clones use nearly identical TCR α sequences (Fig. 5a), we tested if TCR α chains play a dominant role in determining antigen specificity. Pairing of α chains from three GMM-recognizing TCRs (clones 1, 21, 42) with the β chain from the MA-specific TCR (clone 18) reconstituted recognition of MA, but not GMM, in all cases (Fig. 5b). Thus, the TCR β chain determined the fine specificity for two homologous antigens.
Functional dominance of the β chain was unexpected, given that TCR α chains were more conserved in structure among GEM T cells. A converse set of TCR chain swaps, in which the α chain of clone 18 was paired with β chains from clones 1, 21 and 42, yielded three TCR chimeras, which did not respond to MA or GMM (Fig. 5c). Based on retained anti-CD3 response among transfectants, this result was not likely due to failure of transfection or signalling, but instead resulted from lack of antigen recognition by chimeric receptors. Thus, in contrast to preserved recognition of MA by chimeric TCRs (Fig. 5b), failure to recognize GMM in all chimeras suggested that the GMM glycolipid recognition was a more stringent interaction, dependent on both TCR chains. All four native TCR α chains (clones 1, 21, 42 and 18) are similar in sequence, but clone 18 differs only at position 107 from clone 42, with leucine substituted for arginine (Fig. 5a). Thus, position 107 in the CDR3 α sequence is crucial for determining MA antigen recognition.
GEM TCR structures identify a lateral push from the β chain
To address the structural basis of TCR α and β pairing and gain general insights into the molecular basis of antigen specificity of GEM TCRs, we expressed TCRs from clone 18 and 42 and solved their crystal structures at 2.2 Å resolution (Fig. 6a-c and Supplementary Fig. 4). By superimposing these two GEM TCRs with a human MAIT TCR 31, we could compare three TCRs that share TRAV1-2 use, but differentially recognize CD1b and MA, CD1b and GMM, or MR1 and ribityl-lumazine antigens 32 (Fig. 6d). Despite shared use of TRAV1-2, the MAIT and GEM TCRs differ in other aspects of their β chain and CDR3α conformations (Fig. 6d), which presumably mediate their non-cross-reactive recognition of CD1b and MR1 31, 33. However, this comparison provided specific insights into GEM TCR pairing that agreed with results from TCR swaps. Although similar in structure, the two GEM TCRs showed notable differences at the CDR3α-CDR3β interface, which derived from perturbations in the germline encoded CDR1α loop that arise mainly from the residue at position 107 in TCR β (Fig. 6). Whereas arginine 107 in the CDR3β of the MA-specific clone 18 packs against its CDR3α loop, the corresponding interaction is missing in the GMM-reactive clone, resulting in greater mobility in its CDR3α loop (Fig. 6d). This structural feature in the β chain, which likely determines MA versus GMM reactivity, is located nearby to the important TCR α residue 107, identified in TCR chain swap experiments (Fig. 6d). Thus, these structural data point to a specific mechanism for β chain influence on fine specificity, whereby the β chain pushes laterally on the invariant α chain at a defined point on the CDRα-CDRβ interface, analogous to fine-tuning NKT TCR specificity 34.
Figure 6. GEM TCR structures.
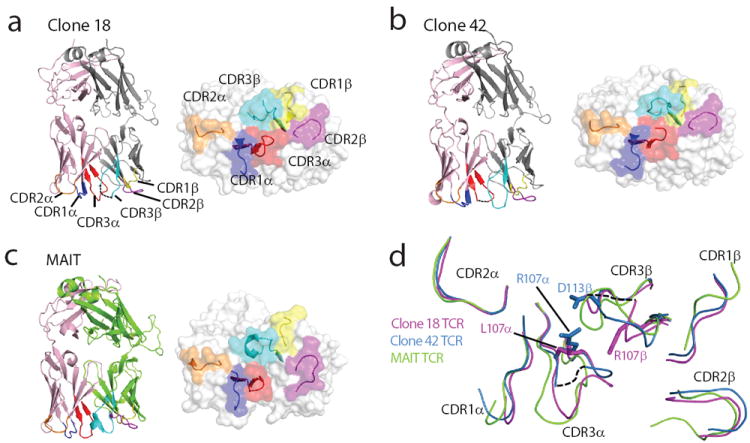
Structures of the TCRs of clone 18 (a), 42 (b) and a human MAIT clone 31 (c), each with a surface representation of the antigen binding face of the TCRs. Dashed lines represent residues missing from pdb coordinates. Pink: TRAV1-2 α chains; grey: TRBV6-2 β chains; green: TRBV20 β chain. (d) Overlay of CDR1-3α and CDR1-3β loops of GEM TCRs with the MAIT TCR. The structural superimpositions were performed over the Vα regions of the three TCRs. CDR loops of clone 18 (purple), clone 42 (blue) and the MAIT (green) TCRs are shown.
GEM T cells expand in vivo
In vitro studies of clones identified key features of GEM T cells, such as high affinity TRAV1-2 TCRs and production of antimicrobial cytokines in response to mycobacterial lipids. To determine if GEM T cells are present in vivo and share these properties, we tested fresh polyclonal T cells from blood bank donors or tuberculosis patients in three types of experiments (Supplemental Fig. 1). We first used a monoclonal antibody that recognizes TCR α chains with TRAV1-2 sequences 35 to sort fresh PBMC into four populations based on TRAV1-2 and CD4 expression. CD1b- and GMM-reactive cells should fall in the TRAV1-2+CD4+ gate, and the majority of MAIT cells are expected in the TRAV1-2+CD4- gate 18, 33, 35. Both TRAV1-2- populations should contain lymphocytes that should be CD1b-independent. TRAV1-2-CD4- cells from a blood bank donor (BB2) showed antigen-independent IFN-γ responses, which might represent NK cell recognition of the MHC class Ilow target cell. The TRAV1-2-CD4+ control populations from all donors failed to recognize GMM. In two blood bank donors (BB12 and BB2) and one tuberculosis patient (C58), IFN-γ ELISPOT showed that TRAV1-2+CD4+ populations were activated by GMM or MA in a CD1b-dependent manner. Thus, the in vitro analysis of clones and ex vivo analysis of polyclonal T cells both agree that TRAV1-2 and CD4 expression are markers for a subset of cells in the repertoire that recognize CD1b and mycolyl lipids (Fig. 7a and Supplementary Fig. 1).
Figure 7. GEM T cells in polyclonal populations ex vivo.
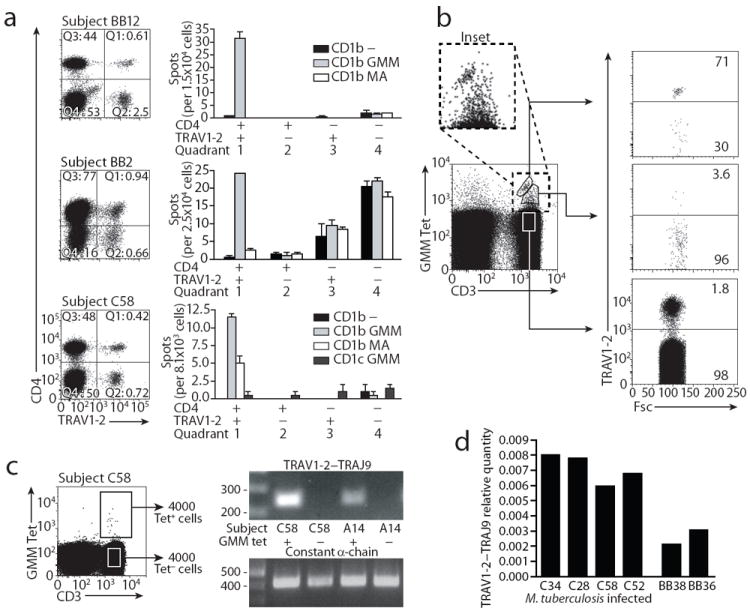
(a) Equal numbers of PBMC were sorted based on staining with antibodies against CD4 and TRAV1-2 and tested using K562 cells transfected with CD1b and CD1c in the absence or presence of GMM or MA. The experiment was performed twice in donors BB12 and BB2, and three times in donor C58, with similar results. (b) PBMC of subject C58 were stained with α-CD3, GMM-loaded CD1b tetramer and α-TRAV1-2. This experiment was performed twice with similar results. (c) The cDNA of 4000 sorted Tet+ and Tet− cells was used in a PCR with a TRAV1-2 and TRAJ9-specific primer pair. Cell sorting was performed once, and PCR was performed twice for each donor. (d) Quantity of TRAV1-2–TRAJ9 junctions relative to constant α chain was determined in cDNA from sorted CD4+TRAV1-2+ PBMC from blood bank donors and tuberculosis patients. Deep sequencing of the TCR repertoire of BB36 and BB38 revealed that 0.48 % and 0.90 % of the TRAV1-2+CD4+ T cells, or 0.0024 % and 0.0023 % of the total PBMC population, respectively, expressed typical GEM α chains.
Contrasts among GEM T cells, NKT cells and MAIT cells
Next we evaluated whether certain aspects of GEM T cell response initially observed in vitro, might also occur using more physiological APCs and apply to the ex vivo state. GEM T cell activation by CD1b transfection of transformed cells (Fig. 2 and Fig. 7a) also occurred in autologous monocyte-derived dendritic cells, which more closely resemble the main cell population to express CD1b in the periphery (Supplementary Fig. 5a). Similar to results obtained with clones (Fig. 2b), polyclonal GEM T cell responses from patient C58 to GMM lead to IFN-γ and TNF secretion when measured directly ex vivo (Supplementary Fig. 5b). Like clones, fresh TRAV1-2+CD4+ T cell populations showed detectable reactivity to both MA and GMM antigens, but they produced higher IFN-γ concentrations in response to GMM, suggesting that GMM is the immunodominant antigen (Figs. 1, 7a).
Ex vivo analysis of GEM T cells from tuberculosis patients highlights differences among GEM T cells, NKT cells and MAIT cells. NKT cells were discovered and named in part based on their expression of CD161 and other NK markers 11, and almost all MAIT cells express CD161 35. However, analysis of fresh CD1b Tethigh cells in three donors showed varying rates of CD161 expression, with two donors expressing this marker at low frequency, similar to that of total CD3+ cells (Supplementary Fig. 6a). NKT cells circulate in a pre-activated state and typically express the activation marker CD69 36. Prolonged T cell activation by antigens might additionally lead to the expression of the T cell exhaustion marker PD1 37. However, CD69 and PD1 were not expressed above background levels by Tethigh T cells in tuberculosis patients (Supplementary Fig. 6b-c). Overall, CD4 is a defining marker of GEM T cells that can further distinguish GEM T cells from most TRAV1-2+CD4- MAIT cells 18. CD69, CD161 or PD1 do not specifically mark GEM T cell populations ex vivo.
TCR sequence conservation is linked to high avidity
CD1b-reactive clones with diverse TRAV1-2- TCRs or conserved TRAV1-2+ TCRs segregate into Tetint and Tethigh cells, respectively. Both clone types were present in each patient tested. This pattern suggested that GEM T cells and lower affinity CD1b-restricted T cells might exist within the same repertoire of any patient. Polyclonal T cell analysis of patient C58 yielded a broad range of tetramer staining intensity, extending from low to very high absolute signal (Fig. 7b). Further, this pattern of varying tetramer staining intensity was detected among other tuberculosis patients (C12, C32, C60 A14, A21, A22) (Supplementary Fig. 6). Next, we directly tested the relationship between tetramer staining intensity and TRAV1-2 expression. Fresh CD3+ T cells were analyzed for TRAV1-2+ T cell frequency within Tetneg, Tetint and Tethigh T cell populations. Whereas anti-TRAV1-2 stains 1.8 % of T cells and 3.6 % of Tetint cells, TRAV1-2 was expressed on 71 % of Tethigh cells (Fig. 7b). Thus, polyclonal T cells showed clear linkage of TRAV1-2 TCR expression with high-avidity binding to CD1b-GMM and indicated that GEM TCRs can dominate the high avidity repertoire.
GEM T cells clonally expand in vivo
We sought to detect GEM T cells in a tetramer-independent manner by PCR. TRAV1-2 is joined to TRAJ9 in all GEM clones, but TRAV1-2 is expressed in concert with other joining regions in MAIT cells and MHC-restricted T cells. We designed primers for TCR constant regions as a normalization control and primers that bind on both sides of the TRAV1-2–TRAJ9 junction. Standard PCR detected the GEM defining junction in Tet+, but not Tet− T cells from two tuberculosis patients (C58, A14) (Fig. 7c). Sequencing of the amplified rearrangements yielded new TCR α chain sequences that fit the TCR α sequence motif for GEM T cells (Fig. 3a). For example, rearrangements amplified from patient A14 contained a TCR α chain sequence identical to clone 1, which was also derived from patient A14, and two unique genetic rearrangements with 0 or 3 N nucleotides (Fig. 3a). Also, the rearrangements amplified from patient C58 were identical to the TCR α chain of clone 42, which was also derived from this subject. Furthermore, the same TCR α and β chain were identified more than once during single cell sequencing experiments (Supplementary Fig. 3). Isolation of identical TCR sequences from independent experiments using different blood samples from the same subject strongly suggests expansion of GEM clones in vivo.
GEM T cells exist in the naïve repertoire
Mycolyl lipids are foreign mycobacterial antigens that drive expansion of antigen-specific T cells during infection 16, 21, 22, 38, 39. After detecting GEM T cells in tuberculosis patients 19, we sought to determine if they also exist in the naïve repertoire. Sorted TRAV1-2+CD4+ T cells from two blood bank donors (BB2 and BB12) yielded detectable GMM- and CD1b-dependent responses (Fig. 7a, Supplementary Fig. 1). Based on this initial result, we quantitatively analyzed GEM-defining TCR sequences using real-time PCR (RT-PCR) and high-throughput sequencing. RT-PCRon TRAV1-2+CD4+ sorted cells from two blood bank donors, BB36 and BB38, and four tuberculosis patients (C28, C34, C52, C58) detected the TRAV1-2–TRAJ9 junction in all subjects tested but signals were significantly higher among the tuberculosis patients (Fig. 7d). These findings are consistent with the presence of a small GEM T cell population in the naive repertoire and expansion after tuberculosis infection. However, not every possible TRAV1-2–TRAJ9 junction corresponds to a GEM T cell sequence. To determine if the candidate GEM TCR α chains detected in blood bank donors actually met the length and sequence criteria for GEM T cells, we performed deep sequencing of the TCR α chains of the sorted TRAV1-2+CD4+ cells from donors BB36 and BB38 (Fig. 7d, Supplementary Fig. 7). Among 7,282 (BB36) and 6,861 (BB38) productive TRAV1-2 sequences analyzed, 0.48 % and 0.90 % encoded CAVRXTGGFKTIF or CAVLXTGGFKTIF, where X is any amino acid (Fig. 3, Supplementary Fig. 7). All eight clones identified used limited N region additions. Although deep sequencing did not provide information on the TCR β chain, the TCR α sequences proved that T cells bearing the defining GEM TCR α motif could be detected at low frequencies in the bloodstream of blood donors.
DISCUSSION
These data identify GEM T cells as one of three known TCR-conserved cell types in humans. GEM T cells, iNKT cells and MAIT cells show strict TCR α sequence conservation with biased selection of TCR β chains. Further, GEM T cells and MAIT cells are defined by the same TCR a variable region gene. TRAV1-2 is located at the most upstream position in the TRA-TRD locus, suggesting possibly shared mechanisms of TCR rearrangement or target binding to CD1b and MR1. However, clear differences in TCR structure and phenotype distinguish these three cell types. These data show that GEM T cells differ from MAIT cells in their CD4 expression, J segment usage, antigen binding surfaces and antigen specificity 18, 33. Contrasting with iNKT cells, GEM T cells have apparently lower baseline autoreactivity, lower precursor frequencies in the blood, low CD69 expression, suggesting that they are more dependent on infection to drive expansion 19. CD1b and CD1d differ in expression in the periphery, with CD1b expression largely restricted to DCs versus broad expression of CD1d on gastrointestinal epithelia, macrophages and B cells 40. Whereas iNKT cells are preactivated by self antigens and have a patrolling functions mediated by autoreactivity, these features point to a role for GEM T cells in the host response to infection.
We propose a model of the CD1b repertoire with at least two compartments: high affinity conserved TCRs and low affinity diverse TCRs. Because high affinity antigen recognition is linked to invariant TCR sequences, multiple TRAV1-2/TRAJ9 rearrangements create TCRs that likely drive expansion of GEM T cells through high affinity interactions with CD1b-lipid. Amino acid and nucleotide patterns suggest two mechanisms of expansion. First, identical TCR α and β chain nucleotide sequences were derived from different blood samples or culture wells from the same donors, suggesting that GEM T cells undergo clonal expansion in vivo prior to phlebotomy. Second, most sequences are distinct in ways that prove that GEM T cell populations in the blood derive from many independent TCR arrangement events. For example, three rearrangements with differing underlying nucleotide sequences give rise to identical CDR3 α amino acid sequences in donor BB36. Also, highly similar CDR3 α amino acid sequences in clones 1, 18, 21 and 42 derive from differing patterns of exonucleolytic trimming of the V and J regions. Autoreactivity to CD1b was not observed, and any self antigen that could support the thymic selection of GEM T cells is unknown. Therefore, the basis for emergence of GEM T cells among donors without known mycobacterial infection remains unexplained. However, it is possible that environmental or self antigens cause GEM T cell expansion in a way that is comparable to the recently described MHC-restricted, HIV-specific T cells in unexposed donors 41.
Current views emphasizing the diversity of group 1 CD1-reactive TCRs derive from small panels of T cell clones 7-10. We initially viewed clone 18 as further evidence of TCR diversity in the CD1b repertoire, but it was actually the prototype for a larger T cell population cells later identified in many unrelated donors. Based on reliability of higher throughput tetramer methods and the non-polymorphic nature of human CD1 molecules, we predict that inter-donor TCR conservation in the CD1 repertoire is more common than is currently appreciated. This hypothesis can be addressed through analysis of many clones using newly generated of human CD1a, CD1b 19 and CD1c 42 tetramers.
Inter-donor CDR3 diversity is the dominant pattern for MHC-restricted TCRs, with the notable exception of public TCRs detected in donors that share an MHC allele and specific antigenic exposure 43. The GEM TCR could be considered a kind of public TCR that recognizes CD1b. CD1b is expressed in an identical form in essentially all humans, which is not the case for even the most common MHC allele. In the MHC system, public TCRs are proposed to arise via convergent recombination, a process whereby multiple recombination events with few N nucleotide additions produce differing nucleotide sequences that encode the same amino acid sequence 44. GEM TCR α chain sequences display these hallmarks. Inter-donor TCR conservation might be of practical use in tracking of CD1-restricted T cells or possibly TCR-based immunodiagnosis. In the MHC system, particular TCR sequences do not normally predict the antigens recognized by genetically unrelated donors. Even the conserved iNKT TCR does not correspond to any single antigen of known biological function. Here we show that the GEM TCR α chain sequence is associated with recognition of lipids produced by a defined genus of pathogenic bacteria. Thus, CD1b tetramers, PCR primers or monoclonal antibodies for the GEM TCR α chain might now be evaluated as TCR-based tests for tuberculosis. GEM T cells are a plausible target for vaccination with mycobacterial lipids.
METHODS
Antigens
C32 GMM was purified from R. equi and C80 GMM was purified from M. phlei as described 20. MA was obtained from Sigma. M. tuberculosis lipid extract was prepared by extracting a pellet of 7H9 grown H37Rv with choroform/methanol 1:2 (V:V) for two hours at room temperature, followed by chloroform/methanol 2:1. Glycerol monomycolate was purified from lipid extract from M. bovis BCG grown in Sauton’smedium, followed by loading on a silica column and elution with chloroform.
Flow cytometry
CD1b monomers (NIH tetramer facility) were loaded with R. equi-derived GMM and assembled into tetramers 19. Tetramers were incubated for 30 minutes at room temperature prior to adding monoclonal antibodies for an additional 30 minutes on ice. Flow cytometry data were pre-gated for lymphocytes based on forward and side scatter. Antibodies are detailed in Table 1 Cells were sorted on an 11-color FACSAria (Becton Dickinson).
T cell cloning and T cell assays
Blood was obtained after informed consent from asymptomatic tuberculin positive subjects clinically assessed to have latent tuberculosis but with no clinical or radiographic evidence of active tuberculosis, from a patient with smear-positive pulmonary tuberculosis, and from blood bank donors, as approved by the institutional review boards of the Lemuel Shattuck Hospital and Partners Healthcare. Sorted GMM-loaded Tet+19 T cells were stored overnight in medium containing 0.2 ng/ml IL-15 and plated the next day at 1 cells/well in round bottom 96-well plates containing 2 ×105 irradiated allogeneic peripheral blood mononuclear cells, 4 ×104 irradiated Epstein-Barr virus transformed B cells, 30 ng/ml anti CD3 antibody OKT3 and 1 ng/ml of IL-2 which was added on day 2 of the culture. Alternatively, PBMC were stimulated with autologous monocyte-derived DC and 1 μg/ml total lipid extract of M. tuberculosis, and IFN-γ secreting cells isolated (Miltenyi Biotech) and cloned. After 3 weeks, the wells with visible growth were restimulated. Clones were tested for binding of GMM loaded tetramer by flow cytometry and for antigen specificity in an ELISPOT assay. For ELISPOT assays, cocultures of APCs and T cell were incubated for 16 h in a Multiscreen-IP filter plate (96 wells; Millipore) coated according to the manufacturer’s instructions (Mabtech). Antibodies are detailed in the online methods.
PCR and molecular cloning
From expanding T cell clones and sorted T cell populations, RNA was isolated with an RNeasy kit (Qiagen), and cDNA was synthesized with a Quantitect reverse transcription kit (Qiagen), including a genomic DNA–removal step. V segment usage was determined by PCR using primerset IPS000029 and IPS000030 as described at www.imgt.org in combination with TCR α constant region reverse primer 5’ GTGGTAGCAGCTTTCACCTCCTTGG 3’ and TCR β constant region reverse primer 5’ GGTGGCAGACAGGACCCCTTGC 3’. Taq polymerase was used in the supplied buffer (Denville) under the following cycling conditions: an initial denaturation of 5 min. at 95°C, followed by 35 cycles of 1 min. at 94°C, 1 min. at 59°C, 1 min. at 72°C, followed by a final elongation step of 7 min. at 72°C. For detection of TCRs using TRAJ9, a V segment specific forward primer was used in combination with TRAJ9 reverse primer 5’ GGTTCTGGATATTTGCTTTAACAAATAG 3’. For the cloning of the full length TCR α and β chains of clones 1, 18, 21, and 42 (Genbank accession numbers JQ778257-JQ778264) the following primers were used: FL_TRAV1-2_For_NheI: CTAGCTAGCGCAGCAGATGTGGGGAGTTTTCCTTC; FL_TRBV6-2_For_NotI: CTAGCGGCCGCCCTGCCATGAGCCTCGGGC; FL_TRBV30_For_NotI: CTAGCGGCCGCGGCTTGGATGATGCTCTGCTCTC; FL_TRAC_Rev_XhoI: CAGCTCGAGCAATCTTAATCATAAATTCGGGTAGGATCCCATC; FL_TRBC2_Rev_BamHI: TATGGATCCGGAGCTAGCCTCTGGAATCCTTTC. TCR α and β chains were cloned into pREP7 and pREP9 vectors, electroporated into JRT3-T3.5 cells and grown in medium containing 1.5 mg/ml Geneticin (Gibco) and 0.5 mg/ml hygromycin B (Invitrogen) for 4-6 weeks. Recognition of mycolyl lipids by transfectants was tested in an IL-2 ELISPOT assay in the presence of monocyte derived dendritic cells and 10 ng/ml phorbol 12-myristate 13-acetate.
Single cell sequencing
For single cell TCR sequencing cells were sorted in wells of a PCR plate containing 2.5 μl cDNA synthesis reaction mix consisting of 0.1 % Triton X-100 (Sigma-Aldrich) and 0.2 μl iScript reverse transcriptase in 1x iScript reaction buffer, and incubated 25°C for 5 minutes, 42°C for 30 minutes, and 85°C for 5 minutes. Subsequently, a pre-amplification targeting TRAV1-2 and all TRBV segments was performed in GoTaq Flexi (Promega) reaction mixture containing 2.5 mM MgCl, 0.2 mM each dNTP, 0.025 U/μl GoTaq polymerase, 0.14 μM each of 26 TRBV-specific forward primers, 0.25 μM of constant β reverse primer 5’ GGTGGCAGACAGGACCCCTTGC 3’, 0.125 μM of TRAV1-2 specific forward primer 5’ GCAACATGCTGGCGAAGCACCCAC 3’, and 0.125 μM of constant α reverse primer 5’ GTGGTAGCAGCTTTCACCTCCTTGG 3’, under the following cycling conditions: an initial denaturation of 2 min. at 95°C, followed by 25 cycles of 30 sec. at 95°C, 1 min. at 59°C, 1 min. at 72°C, followed by a final elongation step of 5 min. at 72°C. The pre-amplification mixture was diluted to a total volume of 75 μl, and 5 μl was used in a RT-PCR reaction to determine TRAV1-2 message with 0.25 μM each of an internal TRAV1-2 primer 5’TCCTTAGTCGGTCTAAAGGGTACAG 3’ and internal constant α reverse primer 5’CATCAGAATCCTTACTTTGTGACACATTTG 3’using the Sybr green (BioRad) method 18. The TRAV1-2 PCR product of positive wells was isolated and sequenced and aliquots of the pre-amplified cDNA was amplified in individual PCR reactions with the V β specific forward primers and the constant β reverse primer 5’ ACACAGCGACCTCGGGTGGG 3’. Positive reactions were detected by agarose gel electrophoresis, and the amplicons were sequenced.
RT-qPCR, deep sequencing
Relative quantity of the TRAV1-2–TRAJ9 junction was determined relative to constant α chain message using the TaqMan (Applied Biosystems) method using the following primers and probes: TRAV1-2 forward primer: 5’TCCTTAGTCGGTCTAAAGGGTACAG 3’; TRAJ9 reverse primer: 5’TTGCTTTAACAAATAGTCTTGTTCCTGCTCC 3’; TRAV1-2 probe: 5’ FAM-CTCCAGATGAAAGACTCTGCCTCTTACCTCTGTGC-BHQ 3’; constant α forward primer: 5’CTGACCCTGCCGTGTACCAG 3’; constant α reverse primer: 5’ CATCAGAATCCTTACTTTGTGACACATTTG 3’; constant α probe: 5’FAM-CCAGTGACAAGTCTGTCTGCCTATTCACCGATTTTG-BHQ 3’. For deep sequencing of TRAV1-2 α chains, V segment-specific linear amplification and next-generation sequencing was performed as described previously 45, 46.
TCR affinity measurements
Soluble TCR proteins were expressed and purified as described 29 and floated in increasing concentrations over GMM-loaded CD1b coupled to research-grade streptavidin-coated chips in a Biacore 3000. The final response was calculated by subtraction of the response of the unloaded CD1b. BIAevaluation version 3.1 software (Biacore AB) was used to fit the data to the 1:1 Langmuir binding model and the equilibrium data were analyzed with the Prism program for biostatistics, curve fitting and scientific graphing (GraphPad).
TCR crystallisation, data collection and structure determination
Clone42 TCR crystals were grown by the vapour-diffusion method at 20°C with a protein/reservoir drop ratio 1:1 at a concentration 5mg/ml in 20 % PEG 3350, 0.05M Na-HEPES pH 7 and 1 % tryptone w/v. Clone18 TCR crystals were grown using the same technique with 25 % PEG1500 and 10 % Proprionate-cacodylate-bis tris propane pH7. TCR crystals were soaked in a cryoprotectant solution containing mother liquor solution with the PEG concentration increased to 25 % and then flash frozen in liquid nitrogen. Data were collected on the Micro-focus beamline (MX2) at the Australian Synchrotron with an ADSC-Q315r CCD detector at 100K. Data were processed as described previously 29 followed by maximum-likelihood refinement with the Buster program 47. The structures were determined by molecular replacement using the ELS4 TCR as model 48 with the PHSER program. Manual building was conducted with the COOT software followed by maximum-likelihood refinement with the Buster program. The final models were validated using the Protein Data Base validation website, and the final refinement statistics are summarized in Figure S4. The coordinates are available under the accession number: 4G8E for the clone18 TCR and 4G8F for the clone 42 TCR.
Supplementary Material
Table 2.
Antibodies used for cell sorting, flow cytometric analysis and T cell activation assays.
| Target molecule | Clone number | Fluorophore/tag | Manufacturer | Application |
|---|---|---|---|---|
| IFN-γ | 1-D1-K | none | Mabtech | ELISPOT |
| IFN-γ | 7-B6-1 | biotin | Mabtech | ELISPOT |
| IL-2 | IL-2-I/249 | none | Mabtech | ELISPOT |
| IL-2 | IL-2-II | biotin | Mabtech | ELISPOT |
| TNF | TNF3/4 | none | Mabtech | ELISPOT |
| TNF | TNF5 | biotin | Mabtech | ELISPOT |
| CD3 | SK7 | FITC | BD Bioscience | Flow cytometry |
| CD3 | OKT3 | none | In house | T cell activation |
| CD1b | BCD1b3.1 | none | In house | Neutralization |
| Isotype IgG1 | P3 | none | In house | Neutralization |
| CD4 | RPA-T4 | APC | BD Bioscience | Flow cytometry |
| CD8α | OKT8 | none | In house | Flow cytometry |
| CD8β | 2ST8.5H7 | none | Beckman Coulter | Flow cytometry |
| TRAV1-2 | 3C10 | PE | Biolegend | Flow cytometry |
Acknowledgments
Supported by the National Institute of Allergy and Infectious Diseases (AI04393, AR048632 to D.B.M. and K08 AI089858 to A.K.) the Burroughs Wellcome Fund for Translational Research, and Nederlands Wetenschappelijk Onderzoek (Meervoud 836.08.001 to I.V.R.) D.I.G. is supported by an NHMRC Senior Principal Research Fellowship, J.R. is supported by an NHMRC Australia Fellowship and S.G. is supported by an ARC Future Fellowship. CD1b protein was provided by the NIH Tetramer Facility. We thank M. Turner, C. Seshadri and the Shattuck Hospital for clinical collaboration and L. Tan for technical help and the beamline staff at the Australian synchrotron for assistance with data collection.
Footnotes
Author contributions
I.V.R. designed and performed experiments and prepared the manuscript; D.B.M. supervised the experiments and prepared the manuscript; A.K. developed tetramer methods and patient cohorts and performed PD1, CD161, and CD69 flowcytometric analyses; A.d.J. designed experiments and provided technical advice. M.B., S.G., D.G. and J.R. designed experiments contributed affinity and structural data for TCRs. D.G., J.R. and S.G. assisted in preparation of the manuscript. W.D.J performed Luminex experiments.
References
- 1.Beckman EM, et al. Recognition of a lipid antigen by CD1-restricted alpha beta+ T cells. Nature. 1994;372:691–694. doi: 10.1038/372691a0. [DOI] [PubMed] [Google Scholar]
- 2.Han M, Hannick LI, DiBrino M, Robinson MA. Polymorphism of human CD1 genes. Tissue Antigens. 1999;54:122–7. doi: 10.1034/j.1399-0039.1999.540202.x. [DOI] [PubMed] [Google Scholar]
- 3.Porcelli S, Yockey CE, Brenner MB, Balk SP. Analysis of T cell antigen receptor (TCR) expression by human peripheral blood CD4-8- alpha/beta T cells demonstrates preferential use of several V beta genes and an invariant TCR alpha chain. J Exp Med. 1993;178:1–16. doi: 10.1084/jem.178.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fowlkes BJ, et al. A novel population of T-cell receptor alpha beta-bearing thymocytes which predominantly expresses a single V beta gene family. Nature. 1987;329:251–4. doi: 10.1038/329251a0. [DOI] [PubMed] [Google Scholar]
- 5.Cardell S, et al. CD1-restricted CD4+ T cells in major histocompatibility complex class II-deficient mice. J Exp Med. 1995;182:993–1004. doi: 10.1084/jem.182.4.993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Van Rhijn I, et al. CD1d-restricted T cell activation by nonlipidic small molecules. Proc Natl Acad Sci U S A. 2004;101:13578–83. doi: 10.1073/pnas.0402838101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Grant EP, et al. Molecular recognition of lipid antigens by T cell receptors. J Exp Med. 1999;189:195–205. doi: 10.1084/jem.189.1.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.de Jong A, et al. CD1a-autoreactive T cells are a normal component of the human alphabeta T cell repertoire. Nat Immunol. 2010;11:1102–9. doi: 10.1038/ni.1956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.de Lalla C, et al. High-frequency and adaptive-like dynamics of human CD1 self-reactive T cells. Eur J Immunol. 2010;41:602–10. doi: 10.1002/eji.201041211. [DOI] [PubMed] [Google Scholar]
- 10.Vincent MS, Xiong X, Grant EP, Peng W, Brenner MB. CD1a-, b-, and c-restricted TCRs recognize both self and foreign antigens. J Immunol. 2005;175:6344–51. doi: 10.4049/jimmunol.175.10.6344. [DOI] [PubMed] [Google Scholar]
- 11.Godfrey DI, MacDonald HR, Kronenberg M, Smyth MJ, Van Kaer L. NKT cells: what’s in a name? Nat Rev Immunol. 2004;4:231–7. doi: 10.1038/nri1309. [DOI] [PubMed] [Google Scholar]
- 12.Kasmar A, Van Rhijn I, Moody DB. The evolved functions of CD1 during infection. Curr Opin Immunol. 2009;21:397–403. doi: 10.1016/j.coi.2009.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Porcelli S, Morita CT, Brenner MB. CD1b restricts the response of human CD4-8- T lymphocytes to a microbial antigen. Nature. 1992;360:593–597. doi: 10.1038/360593a0. [DOI] [PubMed] [Google Scholar]
- 14.Gilleron M, et al. Diacylated Sulfoglycolipids Are Novel Mycobacterial Antigens Stimulating CD1-restricted T Cells during Infection with Mycobacterium tuberculosis. J Exp Med. 2004;199:649–659. doi: 10.1084/jem.20031097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Moody DB, et al. Structural requirements for glycolipid antigen recognition by CD1b-restricted T cells. Science. 1997;278:283–286. doi: 10.1126/science.278.5336.283. [DOI] [PubMed] [Google Scholar]
- 16.Layre E, et al. Mycolic acids constitute a scaffold for mycobacterial lipid antigens stimulating CD1-restricted T cells. Chem Biol. 2009;16:82–92. doi: 10.1016/j.chembiol.2008.11.008. [DOI] [PubMed] [Google Scholar]
- 17.Cohen NR, Garg S, Brenner MB. Antigen Presentation by CD1 Lipids, T Cells, and NKT Cells in Microbial Immunity. Adv Immunol. 2009;102:1–94. doi: 10.1016/S0065-2776(09)01201-2. [DOI] [PubMed] [Google Scholar]
- 18.Tilloy F, et al. An invariant T cell receptor alpha chain defines a novel TAP-independent major histocompatibility complex class Ib-restricted alpha/beta T cell subpopulation in mammals. J Exp Med. 1999;189:1907–21. doi: 10.1084/jem.189.12.1907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kasmar AG, et al. CD1b tetramers bind {alpha}{beta} T cell receptors to identify a mycobacterial glycolipid-reactive T cell repertoire in humans. J Exp Med. 2011;208:1741–7. doi: 10.1084/jem.20110665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Moody DB, et al. CD1b-mediated T cell recognition of a glycolipid antigen generated from mycobacterial lipid and host carbohydrate during infection. J Exp Med. 2000;192:965–976. doi: 10.1084/jem.192.7.965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ulrichs T, Moody DB, Grant E, Kaufmann SH, Porcelli SA. T-cell responses to CD1-presented lipid antigens in humans with Mycobacterium tuberculosis infection. Infect Immun. 2003;71:3076–3087. doi: 10.1128/IAI.71.6.3076-3087.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Van Rhijn I, et al. Low cross-reactivity of T-cell responses against lipids from Mycobacterium bovis and M. avium paratuberculosis during natural infection. Eur J Immunol. 2009;39:3031–41. doi: 10.1002/eji.200939619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Keane J, et al. Tuberculosis associated with infliximab, a tumor necrosis factor alpha-neutralizing agent. N Engl J Med. 2001;345:1098–104. doi: 10.1056/NEJMoa011110. [DOI] [PubMed] [Google Scholar]
- 24.Flynn JL, et al. An essential role for interferon gamma in resistance to Mycobacterium tuberculosis infection. J Exp Med. 1993;178:2249–2254. doi: 10.1084/jem.178.6.2249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stenger S, et al. An antimicrobial activity of cytolytic T cells mediated by granulysin. Science. 1998;282:121–125. doi: 10.1126/science.282.5386.121. [DOI] [PubMed] [Google Scholar]
- 26.Messi M, et al. Memory and flexibility of cytokine gene expression as separable properties of human T(H)1 and T(H)2 lymphocytes. Nat Immunol. 2003;4:78–86. doi: 10.1038/ni872. [DOI] [PubMed] [Google Scholar]
- 27.Mongkolsapaya J, et al. Antigen-specific expansion of cytotoxic T lymphocytes in acute measles virus infection. J Virol. 1999;73:67–71. doi: 10.1128/jvi.73.1.67-71.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Freeman JD, Warren RL, Webb JR, Nelson BH, Holt RA. Profiling the T-cell receptor beta-chain repertoire by massively parallel sequencing. Genome Res. 2009;19:1817–24. doi: 10.1101/gr.092924.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gras S, et al. The shaping of T cell receptor recognition by self-tolerance. Immunity. 2009;30:193–203. doi: 10.1016/j.immuni.2008.11.011. [DOI] [PubMed] [Google Scholar]
- 30.Wun KS, et al. A minimal binding footprint on CD1d-glycolipid is a basis for selection of the unique human NKT TCR. J Exp Med. 2008;205:939–49. doi: 10.1084/jem.20072141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Reantragoon R, et al. Structural insight into MR1-mediated recognition of the mucosal associated invariant T cell receptor. J Exp Med. 2012;209:761–74. doi: 10.1084/jem.20112095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kjer-Nielsen L, et al. MR1 presents microbial vitamin B metabolites to MAIT cells. Nature. 2012;491:717–23. doi: 10.1038/nature11605. [DOI] [PubMed] [Google Scholar]
- 33.Gold MC, et al. Human mucosal associated invariant T cells detect bacterially infected cells. PLoS Biol. 2010;8:e1000407. doi: 10.1371/journal.pbio.1000407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pellicci DG, et al. Differential recognition of CD1d-alpha-galactosyl ceramide by the V beta 8.2 and V beta 7 semi-invariant NKT T cell receptors. Immunity. 2009;31:47–59. doi: 10.1016/j.immuni.2009.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Martin E, et al. Stepwise development of MAIT cells in mouse and human. PLoS Biol. 2009;7:e54. doi: 10.1371/journal.pbio.1000054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cohen NR, et al. Shared and distinct transcriptional programs underlie the hybrid nature of iNKT cells. Nat Immunol. 2013;14:90–9. doi: 10.1038/ni.2490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Day CL, et al. PD-1 expression on HIV-specific T cells is associated with T-cell exhaustion and disease progression. Nature. 2006;443:350–4. doi: 10.1038/nature05115. [DOI] [PubMed] [Google Scholar]
- 38.Montamat-Sicotte DJ, et al. A mycolic acid-specific CD1-restricted T cell population contributes to acute and memory immune responses in human tuberculosis infection. J Clin Invest. 2011;121:2493–503. doi: 10.1172/JCI46216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Felio K, et al. CD1-restricted adaptive immune responses to Mycobacteria in human group 1 CD1 transgenic mice. J Exp Med. 2009;206:2497–509. doi: 10.1084/jem.20090898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dougan SK, Kaser A, Blumberg RS. CD1 expression on antigen-presenting cells. Curr Top Microbiol Immunol. 2007;314:113–41. doi: 10.1007/978-3-540-69511-0_5. [DOI] [PubMed] [Google Scholar]
- 41.Su LF, Kidd BA, Han A, Kotzin JJ, Davis MM. Virus-Specific CD4(+) Memory-Phenotype T Cells Are Abundant in Unexposed Adults. Immunity. 2013;38:373–83. doi: 10.1016/j.immuni.2012.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ly D, et al. CD1c tetramers detect ex vivo T cell responses to processed phosphomycoketide antigens. J Exp Med. 2013;210:729–41. doi: 10.1084/jem.20120624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Turner SJ, Doherty PC, McCluskey J, Rossjohn J. Structural determinants of T-cell receptor bias in immunity. Nat Rev Immunol. 2006;6:883–94. doi: 10.1038/nri1977. [DOI] [PubMed] [Google Scholar]
- 44.Venturi V, Price DA, Douek DC, Davenport MP. The molecular basis for public T-cell responses? Nat Rev Immunol. 2008;8:231–8. doi: 10.1038/nri2260. [DOI] [PubMed] [Google Scholar]
- 45.Klarenbeek PL, et al. Human T-cell memory consists mainly of unexpanded clones. Immunol Lett. 2010;133:42–8. doi: 10.1016/j.imlet.2010.06.011. [DOI] [PubMed] [Google Scholar]
- 46.Klarenbeek PL, et al. Inflamed target tissue provides a specific niche for highly expanded T-cell clones in early human autoimmune disease. Ann Rheum Dis. 2012;71:1088–93. doi: 10.1136/annrheumdis-2011-200612. [DOI] [PubMed] [Google Scholar]
- 47.Smart OS, et al. Exploiting structure similarity in refinement: automated NCS and target-structure restraints in BUSTER. Acta Crystallogr D Biol Crystallogr. 2012;68:368–80. doi: 10.1107/S0907444911056058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tynan FE, et al. A T cell receptor flattens a bulged antigenic peptide presented by a major histocompatibility complex class I molecule. Nat Immunol. 2007;8:268–76. doi: 10.1038/ni1432. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.