

Abstract
Plant behaviors across levels of cellular organization, from biochemical components to tissues and organs, relate and reflect growth habitats. Quantification of the relationship between behaviors captured in various phenotypic characteristics and growth habitats can help reveal molecular mechanisms of plant adaptation. The aim of this article is to introduce the power of using statistics originally developed in the field of geographic variability analysis together with prominent network models in elucidating principles of biological organization. We provide a critical systematic review of the existing statistical and network-based approaches that can be employed to determine patterns of covariation from both uni- and multivariate phenotypic characteristics in plants. We demonstrate that parameter-independent network-based approaches result in robust insights about phenotypic covariation. These insights can be quantified and tested by applying well-established statistics combining the network structure with the phenotypic characteristics. We show that the reviewed network-based approaches are applicable from the level of genes to the study of individuals in a population of Arabidopsis thaliana. Finally, we demonstrate that the patterns of covariation can be generalized to quantifiable biological principles of organization. Therefore, these network-based approaches facilitate not only interpretation of large-scale data sets, but also prediction of biochemical and biological behaviors based on measurable characteristics.
INTRODUCTION
The behaviors of biochemical components, from DNA and proteins to metabolites, in biological entities across all levels of cellular organization, from single cells to whole organisms, partly depend on the space in which they operate and interact. Advances in the development of novel (semi-)automated technologies have facilitated high-throughput quantification not only of biochemical components, resulting in particular molecular phenotypes (e.g., gene expression, protein abundances, and metabolite levels), but also complex physiological traits (e.g., flowering time and yield) and morphological properties (e.g., cell and leaf shape and shoot size) in plants. The problem of identifying the extent to which spatial constraints shape plant behaviors, within and across individuals, resulting in particular genetic makeup and varying phenotypes captured in transcriptomics, proteomics, and metabolomics data sets, is particularly relevant in the study of plants as sessile organisms. Resolving this problem requires analyses of patterns of covariation in characteristics of interrelated biochemical/biological entities. For instance, one may investigate the relation between geographic distances and genetic differences, the relationship between covariation of expression levels of genes and their functional characterization, as well as the relation of chromosomal gene locations, chromatin remodeling, or DNA coiling and gene expression levels (Zupancic et al., 2001; Marshall, 2002; Blanco et al., 2008; Ha et al., 2011; Sobetzko et al., 2012).
To analyze the patterns of covariation in characteristics of studied entities (e.g., individuals of a population and molecular components), it is necessary to specify a measure quantifying the distance between two entities. The distance measure can be used to determine which entities are to be treated as related. The related entities can be represented by a network of nodes, denoting the entities, and edges, representing the relatedness. One can then investigate covariation for a given set of characteristics only over related entities. Moreover, due to the network abstraction, covariation can be examined not only in a geographic space, but also a more general setting.
It then becomes apparent that the networks and any patterns of covariation are expected to depend on the sampled entities and the distances between them. Since, for biological entities, and especially plants, spatial location usually determines the growth habitat to which they are exposed, it is important to examine if a pattern of covariation can be robustly found across diverse environmental conditions typical for the habitat. Moreover, characteristics of biochemical and biological entities depend on not only the spatial aspects but also the timing of physiological and developmental events (e.g., flowering time), which are often tightly regulated. Therefore, the determined patterns of covariation should be investigated for robustness with respect to the time-dependent behavior of the analyzed entities.
The identification of a biologically meaningful and robust pattern of covariation in characteristics of biochemical and biological entities renders it possible to predict the behavior of entities based on their measurable characteristics. Thus, such a pattern of covariation can be regarded as a principle of biological organization. For instance, a robust pattern of genetic diversity can be used to specify the likeliest geographic location of an individual specified only by its genetic makeup. Analogously, a robust pattern in a given molecular function of genes with similar expression levels can be employed to characterize genes of unknown function. Therefore, approaches for determining patterns of covariation can find wide application across different fields in biology, from molecular physiology and genetics to ecology.
Here, we aim to provide a critical systematic review of the existing statistical and network-based approaches for revealing and further investigating patterns of covariation. First, we show how one can use measured characteristics of entities to arrive at distance matrices. We then demonstrate that comparison of the resulting distance matrices with classical statistical approaches often does not result in robust patterns of covariation across various characteristics. We in turn systematically review the approaches that extract networks from the distance matrices and focus on the properties they must satisfy to ensure robustness of any pattern of covariation. We also present a detailed overview of the statistics, originating in geostatistics and geographic variability analysis, that can be employed to quantify network-based covariation of characteristics in uni- and multivariate settings. In addition, we illustrate how these statistics relate to other well-studied network properties. On the level of biological entities, we apply these methods to show how multivariate high-throughput data can be integrated across 92 diverse Arabidopsis thaliana accessions to reveal relations between molecular factors and geographic location, thus providing insights in local adaptation. On the molecular level, with the help of these methods, we show how similarities in gene expression reflect the function annotation in Arabidopsis, thus providing a quantifiable principle useful in predicting functional characterization.
FROM HIGH-THROUGHPUT DATA TO DISTANCE MATRICES
For the purpose of illustrating the main concepts of the reviewed methods, we assume that there are n entities. Each entity, i, 1 ≤ i ≤ n, is described by two properties with corresponding data profiles, denoted by Xi and Yi. The data profiles Xi and Yi are collections of p and q characteristics, represented as vectors. Gathering all Xi and Yi data profiles over all n entities results in two matrices, X and Y, respectively. For instance, if the entities are Arabidopsis accessions, the data matrix X may consist of the longitude and latitude of their geographic origin, while the data matrix Y may be given by the metabolic levels or single-nucleotide polymorphisms (SNPs) of each accession. On the other hand, if the entities are Arabidopsis genes, the data matrix X may consist of their expression levels across various experiments, while the data matrix Y may be given by the characterization of genes’ function annotation as terms of a chosen ontology (e.g., MapMan [Thimm et al., 2004] or GO [Harris et al., 2004]) for the considered genes; similarly, if the entities are metabolites, X and Y may include the levels under same experimental scenarios in Arabidopsis and tomato (Solanum lycopersicum), respectively. Therefore, the particular characteristics gathered in the data matrices X and Y would depend on the biological question.
For a pair of data profiles (vectors), Xi and Xj, from the entities i and j, 1 ≤ i, j ≤ n, a distance measure μ results in a number, denoted by μ(Xi, Xj), quantifying the distance between the two data profiles. A distance measure μ is symmetric if its value does not depend on the order of the data profiles, for example, μ(Xi, Xj) = μ(Xj, Xi). In the following, we assume that the distance measure μ is such that higher values denote larger distances. The Euclidean distance and modifications of Pearson correlation coefficient are commonly used distance measures.
COMPARISON OF DISTANCE MATRICES
Equipped with the concept of a distance measure, there are two possible approaches to investigate the relationship between the data matrices X and Y regarding the distances of the included data profiles. In the first approach, one relies on applying two (not necessarily different) distance measures, μX and μY, on the data profiles in the matrices X and Y, resulting in two distance matrices, DX and DY, respectively. One can test the congruence between the distance matrices using the Mantel test or RV coefficient, or determine an empirical variogram.
The Mantel test, often used in ecological studies (Reynolds, 2003; Cushman and Landguth, 2010), quantifies the correlation between two matrices over the same set of entities, as is the case here. Let 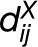 and
and 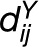 denote the distances between the data profiles of the entities i and j in X and Y, respectively. The Mantel correlation is given by the following expression (Mantel, 1967):
denote the distances between the data profiles of the entities i and j in X and Y, respectively. The Mantel correlation is given by the following expression (Mantel, 1967):
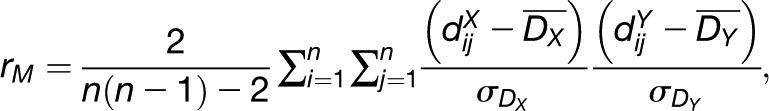 |
where 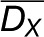 and
and 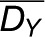 denote the means, while
denote the means, while 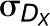 and
and 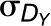 denote the standard deviations of DX and DY, respectively. Like Pearson correlation coefficient, rM takes values in the range [−1,1] whose statistical significance can be estimated empirically by permutation test (Smouse et al., 1986). However, it also shares the same disadvantages with Pearson correlation that presence of outliers may alter not only the value but also the sign of correlation (Gravetter and Wallnau, 2010).
denote the standard deviations of DX and DY, respectively. Like Pearson correlation coefficient, rM takes values in the range [−1,1] whose statistical significance can be estimated empirically by permutation test (Smouse et al., 1986). However, it also shares the same disadvantages with Pearson correlation that presence of outliers may alter not only the value but also the sign of correlation (Gravetter and Wallnau, 2010).
The RV coefficient characterizes the congruence between two matrices over the same set of entities n. It is given by the normalized scalar product of the two matrices, ranging in the interval [0,1], whose statistical significance can be determined analytically (Robert and Escoufier, 1976). With both statistics, the presence of a pattern of spatial covariation may be severely obscured by considering the covariation between pairs of entities that are not necessarily related (i.e., are at large distance), leading to nonrobust findings. To illustrate, let us consider as entities 20 Arabidopsis accessions with geographic origin in Germany, so that X contains their longitude and latitude and Y gathers the levels for 49 metabolites measured under near-optimal growth condition (see Supplemental Data Set 1 online). We next generate the distance matrices DX and DY from the geographic locations and the z-normalized metabolite profiles, respectively. The resulting Mantel correlation coefficient indicates an apparently positive but nonsignificant correlation. In addition, the RV coefficient for X and Y shows a small but nonsignificant congruence between the two matrices (Figure 1, inset).
Figure 1.

Statistics Based on Distance Matrices and Empirical Variogram.
The Mantel correlation coefficient and RV coefficient between the distance matrices DX and DY from the geographic locations and the z-normalized metabolite profiles, respectively, of 20 Arabidopsis accessions. Approximations of the Euclidean distances due to Earth curvature are performed by converting the longitude and latitude from radial units to miles by multiplying the values with 53 and 69.1 mi. The Mantel correlation is calculated between the two distance matrices via the ecodist R package (Goslee and Urban, 2007), whereas the RV coefficient for X and Y is determined via the FactoMineR package in R (Lê et al., 2008). The values are given in the inlay. The variogram is determined based on DX and DY with four bins and eight bins, each covering a range of 50 and 25 mi, respectively. The mean γ for each bin is represented by a point. The size of the point corresponds to the number of pairs in the bin. Furthermore, the sd of each bin is represented by error bars. The empirical variograms are determined by a modified function of the geoR R package (Ribeiro and Diggle, 2001).
FROM DISTANCE MATRICES TO VARIOGRAMS
Another technique of choice when analyzing covariation in space is based on the empirical variogram that quantifies how distances in a given property vary with spatial separation. Given the two distance matrices DX and DY, let N(k) denote the set of pairs of entities i and j, 1 ≤ i, j ≤ n, such that 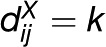 , and let |N(k)| be the number of such (i, j) pairs. The empirical variogram is then defined as follows (Clark, 1979):
, and let |N(k)| be the number of such (i, j) pairs. The empirical variogram is then defined as follows (Clark, 1979):
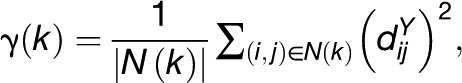 |
where k varies in the range of DX. In practice, instead of determining γ(k) for individual values of k, first the distances in DX are binned and the expression above is applied on pairs of entities whose differences lies in a corresponding bin. For instance, on the example of the 20 Arabidopsis accession with four bins, each covering a range of 50 miles (mi), as shown in Figure 1, we observe that from the first to the third bin, there is a slight increase in the mean γ, indicating that accessions further apart are more variable in their metabolic phenotype than those closer together. This behavior is lost for pairs of accessions having a large distance (fourth bin). By examining the results when eight bins, each covering a range of 25 mi in Figure 1, it appears that the mean in the first bin is larger compared with that of the second, suggesting that the relation between geographic distance and metabolic profiles is more diverse even for geographically close accessions. As with any procedure that depends on binning, as shown in the example, the results are sensitive with respect to the chosen size of the bin. Although there are optimal statistical designs for robust estimation of variograms (Cressie, 1993), this is of little value when dealing with a priori sampled and well-characterized Arabidopsis accessions.
Platt et al. (2010) used the concept of the variogram on X gathering the geographic location of 5707 plants and Y given by 135 SNPs spread across the genome, with μX given by the Euclidean distance and two versions of μY: the fraction of nonmatching alleles and the fraction of those belonging to the same haplogroup. To account for the drawbacks of using different binning sizes, Platt et al. examined the variograms with three sizes (i.e., 0.5, 10, and 150 km) for the North American and Eurasian accessions. By fitting linear and exponential models to the obtained empirical variograms without any smoothing, nonstandard in the field of geostatistics (Clark, 1979), the authors concluded that Arabidopsis exhibits a measure of isolation by distance. However, we emphasize that this claim and its robustness are not statistically and quantitatively supported with the applied approach.
FROM DISTANCE MATRICES TO PROXIMITY-BASED NETWORKS
In the second approach, one first applies a distance measure μX on the data matrix X and employs the resulting distance matrix DX to define which entities are to be considered as related. This procedure generates a network G, in which the set of nodes, V(G), represents the entities and edges (links), in the set, E(G), denote the pairs of entities which are related. The problem of detecting patterns of covariation then becomes one of investigating how the values in Y vary only over the neighbors (i.e., the entities whose corresponding nodes are connected by an edge in the network and not over all pairs of entities). In the following, we describe and illustrate some of the most prominently used classes of networks that capture different notions of relatedness.
The notion of relatedness will depend on the number of sampled entities as well as the type of data in X describing their positions. For instance, what may be considered related in geographic terms, where X gathers the geographic location of an entity, does not necessarily coincide with relatedness of X in terms of gene or metabolic regulation. Nevertheless, the relation of relatedness must satisfy a set of necessary properties in order to be applicable in a statistical setting: (1) no entity is related to itself, (2) if the entity i is related to the entity j, so is j to i, and (3) the relatedness between i and j is unique, in the sense that it does not depend on the choice of parameter values. These properties translate into the following network characteristics: (1) there are no loop edges on single nodes, (2) the edges are undirected, describing symmetric relationships, and (3) the network is unique. An additional property may also include the connectedness of the network, whereby any two nodes are connected by a path.
There are certainly several relations that satisfy these properties and operate on the distance matrix DX, resulting in different classes of networks. In the closest pairs (CP) network, a single edge is established between two entities i and j if their distance is the smallest one over all pairs of entities. Since this network includes a single edge, it is disconnected in all cases except when there exists an entity that is equidistant and closest to all other entities. While the problem of finding the CPs is computationally interesting, the CP network does not capture relatedness over all entities. For the 20 Arabidopsis accessions in Figure 1, the CP network is visualized in Figure 2A. In the nearest neighbors (NN) network, two entities i and j are connected by an edge if there exists no entity l for which the distance between i and l is smaller than the distance between i and j (i.e., 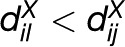 ). However, entity j being the nearest to entity i does not in general imply that i is the nearest to j, so this relation is not symmetric (Figure 2B). The NN network is in general disconnected and includes the CP network. Moreover, the generalization of the NN network to consider the k NNs does not suffer only from asymmetry, but also depends on the value of the parameter k, which violates the uniqueness property (see Figure 2B for k = 3). A heuristic resembling the kNN network, and suffering the same drawbacks, was recently employed by Anastasio et al. (2011) to determine Arabidopsis accessions with erroneous data on their origin based on their discrepancy from the isolation by distance observed by Platt et al. (2010).
). However, entity j being the nearest to entity i does not in general imply that i is the nearest to j, so this relation is not symmetric (Figure 2B). The NN network is in general disconnected and includes the CP network. Moreover, the generalization of the NN network to consider the k NNs does not suffer only from asymmetry, but also depends on the value of the parameter k, which violates the uniqueness property (see Figure 2B for k = 3). A heuristic resembling the kNN network, and suffering the same drawbacks, was recently employed by Anastasio et al. (2011) to determine Arabidopsis accessions with erroneous data on their origin based on their discrepancy from the isolation by distance observed by Platt et al. (2010).
Figure 2.

Visualization of Different Network Classes.
The nodes of each panel represent the geographic origin of the 20 Arabidopsis accessions, connected with an edge based on the different classes of networks.
(A) CP network (red edge) and SI network (blue edges).
(B) NN network illustrating asymmetric relations of connected edges, where, if x −> y, y is NN of x, but x is not NN of y. The k-nearest neighborhood network with k = 3 is shown with red edges.
(C) Gabriel neighborhood network (blue edges), MST network (red edges), and RN network (green edges).
(D) DT network.
The minimum spanning tree (MST) network spans all entities (i.e., the network is connected), so that the sum of the distances between the entities connected by an edge is the smallest over all connected networks (Figure 2C). The MST network is unique if all distances are distinct and contains the NN network as a subnetwork. Let di denote the smallest of the distances between entity i and any other entity. In the sphere of influence (SI) network, the entities i and j are connected by an edge if the spheres with radii di and dj centered in i and j, respectively, intersect in more than one point (Figure 2A). Like the MST network, the SI network also contains the NN network. While the SI relation is symmetric and defines a unique network, the resulting network is not necessarily connected (e.g., five components in Figure 2A).
The concept of relative neighborhood (RN) guarantees that the network is unique and connected. In an RN network, two entities i and j are connected by an edge if for any other entity l, l ≠ i, j, 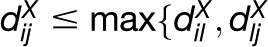 holds (i.e., l does not lie in the intersection of the two spheres centered at i and j of radius ) (Toussaint, 1980) (Figure 2C). Since the MST network is contained in the RN network, the latter is connected (Jaromczyk and Toussaint, 1992). Moreover, for a given distance matrix DX, the RN network is unique. The Gabriel neighborhood of two entities i and j is defined as the smallest sphere through i and j (Figure 2C). Since the sphere is of radius
holds (i.e., l does not lie in the intersection of the two spheres centered at i and j of radius ) (Toussaint, 1980) (Figure 2C). Since the MST network is contained in the RN network, the latter is connected (Jaromczyk and Toussaint, 1992). Moreover, for a given distance matrix DX, the RN network is unique. The Gabriel neighborhood of two entities i and j is defined as the smallest sphere through i and j (Figure 2C). Since the sphere is of radius 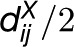 , the Gabriel neighborhood is contained in the RN. Like in the RN network, an edge is established between i and j in the Gabriel neighborhood network if the corresponding neighborhood is empty. Delaunay triangulation (DT) network is partitioning of the space into simplices in such a way that a simplex is a part of DT if the sphere through its nodes, representing the entities, contains no other nodes (Figure 2D). When the number of entities that lie on a sphere is larger than the dimension of the space, the DT network is not unique (de Berg et al., 2008). It has been shown that the RN network is contained in the DT network. For further extensions and discussion of the properties for these classes of networks, we direct the interested reader to Veltkamp (1992) and Aldous and Shun (2010).
, the Gabriel neighborhood is contained in the RN. Like in the RN network, an edge is established between i and j in the Gabriel neighborhood network if the corresponding neighborhood is empty. Delaunay triangulation (DT) network is partitioning of the space into simplices in such a way that a simplex is a part of DT if the sphere through its nodes, representing the entities, contains no other nodes (Figure 2D). When the number of entities that lie on a sphere is larger than the dimension of the space, the DT network is not unique (de Berg et al., 2008). It has been shown that the RN network is contained in the DT network. For further extensions and discussion of the properties for these classes of networks, we direct the interested reader to Veltkamp (1992) and Aldous and Shun (2010).
To introduce the next class of networks, we assume that each entity is associated with a subset of entities. The subset associated with entity i can be determined based on the distribution/ranking of distances in . In the class of proximity networks then two entities i and j are connected by an edge if the subset of i contains j and vice versa (Mutwil et al., 2011). Moreover, in the class of intersection networks, two entities i and j are adjacent if the intersection of the associated subsets is nonempty (Karoński et al., 1999). The proximity networks can be regarded as a symmetric variant of the NN network, while the intersection networks can be viewed as a complement of the empty neighborhood in the definitions above.
STATISTICS FOR CHARACTERISTICS OF NETWORK ENTITIES
The statistics capturing patterns of covariation of a property must explicitly consider the underlying relatedness of investigated entities. To discern such patterns, the statistics quantify how the property’s level for each entity covaries with those of its neighbors. They can be global, as in the case of the Moran’s I (Moran, 1950) and global G (Getis and Ord, 1992), or can take into account local effects, such as the Geary’s C (Geary, 1954) and local Moran’s I (also known as Anselin’s local indicators of spatial association; Anselin, 1995).
For now, let us assume that we are in a univariate setting, where each entity, i, 1 ≤ i ≤ n, is quantified by a single numeric value, yi, for a given characteristics. To test for spatial clustering, referring to a range of correlation values, one usually uses Moran’s I or global G statistics. Moran’s I is defined by the following expression:
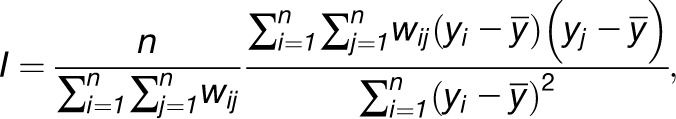 |
where wij denotes the weight of the edge (i, j) ∈ E(G) and takes a value of zero if the network does not include this edge. Usually, wij = 1 or wij = 1/k(i), where k(i) is the degree of the node representing the entity i in G. If wij = 1, then Moran’s I is equivalent to the Pearson correlation of a modified vector with the assumption of network homogeneity (see Supplemental Methods 1 online) and, interestingly, corresponds to the (weighted) degree-degree correlation (Newman, 2002). Therefore, its values are in the range [-1,1] and can be interpreted in the same way as correlations. Positive values indicate that the characteristic exhibits a clustered pattern, negative values suggest dispersal, and zero denotes homogenous distribution of values.
Here, we calculate Moran’s I for each metabolite (see Supplemental Table 1 online) in the 20 Arabidopsis accessions whose neighborhood structure is given by the RN network in Figure 2C. Glc, threhalose, and erythritol all show significant positive Moran’s I (0.67, 0.42, and 0.52, P value < 0.05), while Phe, Ile, and Glc demonstrate dispersal, random, and clustered behavior, respectively (Figure 3). Although useful in providing hints for some metabolic processes associated to local adaptation, these findings neglect the fact that metabolite levels arise from metabolic processes interconnected in a complex network.
Figure 3.

Metabolites Showing Clustered, Homogenous, and Dispersed Behavior.
Moran’s I and Geary’s C for Phe, Ile, and Glc in 20 Arabidopsis accessions demonstrating dispersal, random, and clustered behavior, respectively. We first obtained the underlying network, given by the RN network, based on their geographic origin with help of the spdep package in R (Bivand, 2013). The z-normalized metabolite profiles are used to calculate the statistics for the profiles with wij = 1/k(i). The color of the nodes in the network indicates the values of the profiles of the accessions for Phe (A), Ile (B), and Glc (C), respectively.
The global G statistics (also known as Getis-Ord General G) can be used to test the existence of concentrated high or low values in a given network and are defined as follows:
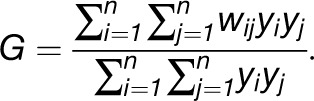 |
This expression includes the assumption that for every edge (i, j) ∈ E(G), 0 ≤ wij ≤ 1, the value of global G is in the range [0,1]. The equivalent zG-score, obtained by analytically determined mean and variance of the global G statistics, can be used to assess the presence of clustering: Statistically significant values whose zG-score is positive indicates that high values cluster together, while negative values support the concentration of small values. The global G statistics for the 20 accessions in Figure 2C indicate that in the case of trehalose and Asn, high values cluster together with wij = 1 if the two entities i and j are connected, and wij = 0, otherwise (see Supplemental Table 1 online).
A global statistic that is more sensitive to local spatial clustering is Geary’s C statistic, which is closely related to the inverse of Moran's I. It can be seen as the network equivalent of a variogram since
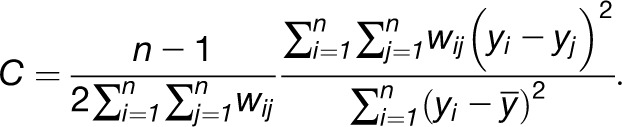 |
The values for this statistic are in the range [0,2], where 1 indicates lack of spatial clustering. Values smaller than 1 are interpreted as an indicator of clustering, while values larger than 1 suggest dispersal. An example of different values for Geary’s C is shown in Figure 3 based on analyzing the behavior of Phe, Ile, and Glc for the Arabidopsis accessions. The results for the other metabolites are similar to those obtained from Moran’s I.
Moran’s I and global G have their local counterparts [i.e., in a modified form, they can be used to assess clustering behavior with respect to a given property in a neighborhood N(i) of a given entity i]. This results in the following:
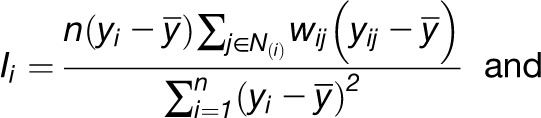 |
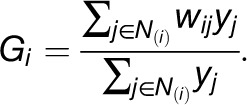 |
Like the global statistics, they can be transformed into z-scores to help interpret the local behavior of the characteristic.
FROM UNI- TO MULTIVARIATE SETTINGS
The statistics described above are suitable for application when a single characteristic of an entity is observed. With the advances in high-throughput technologies, biological entities are often described by vector profiles including different system level molecular phenotypes (e.g., transcriptomic, proteomic, and metabolomic). To render the described statistics useful in a multivariate setting, we employ the distance measure μY, which is used on the data matrix Y but only for the entities which are related (i.e., entities whose corresponding nodes are connected by an edge) (Kleessen et al., 2012). Given a network G, generated based on DY, in the simplest scenario, we determine the weight θij of each edge (i, j) ∈ E(G) as . Alternatively, one may consider combining the geographic distance with the weight θij. Irrespective of the scenario, each node is characterized by the mean of the weights of edges incident on it; in other words, each node i is assigned a weight, θi, such that 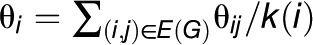 , where k(i) denotes the degree (number of neighbors) of the node i. The statistics reviewed above can then be used with θi instead of yi.
, where k(i) denotes the degree (number of neighbors) of the node i. The statistics reviewed above can then be used with θi instead of yi.
BRIDGING THE GAPS BETWEEN LEVELS OF BIOLOGICAL ORGANIZATION: TWO ILLUSTRATIONS
In the following, we apply the systematically reviewed network-based approaches together with the most widely used statistic in two scenarios. In the first, we investigate the geographic distribution of multivariate molecular phenotypes. In the second, we examine the relation between coexpressed genes, captured in a proximity network, and their functional characterizations.
Geographic Distribution of Multivariate Molecular Phenotypes
It was recently discovered that genetically similar Arabidopsis accessions are found closer to each other, suggesting a robust isolation by distance (Platt et al., 2010; Anastasio et al., 2011; Horton et al., 2012). However, these findings were obtained by relating the genetic and geographic distances either across all accession pairs or via parameter-dependent neighborhood structures (e.g., kNN-like networks) restricting the analysis to genotypic variation. The multivariate method allows for analyzing the relation of multivariate high-throughput data to the geographic origin of the accessions (Kleessen et al., 2012).
In the following, we use the RN network on 92 accessions with data sets including a collection of SNPs (Anastasio et al., 2011; Horton et al., 2012), flowering phenotypes (Atwell et al., 2010), and metabolic phenotypes consisting of levels for 49 metabolites. We chose the RN network model because it results in a unique network that does not depend on any parameters (unlike the kNN network model), it is sparser than other unique network models, and it results in a connected network. The metabolic phenotypes were obtained in three ex situ growth conditions: OpN (12 h light/12 h dark) in which nitrogen fertilization allows close to optimal growth; LiN (12 h light/12 h dark) in which growth is limited by nitrogen; and LiC (8 h light/12 h dark) with high nitrogen supply, with carbon-limited growth. Investigation of the results from the network-based approach shows a robust isolation by distance at the level of metabolic, flowering phenotypes, but not at the level of genetic variability for the analyzed accessions. Nevertheless, the isolation-by-distance model was confirmed also on SNP data using a larger set of 170 accessions (Horton et al., 2012), suggesting that the robustness of the isolation by distance based on SNPs depends on the number of sampled individuals.
Here, we expand these findings by focusing on chemical compound classes of metabolites and their role in biochemical processes including carbohydrates, organic acids, central amino acids, minor amino acids, miscellaneous, composition, as well as photorespiration, nitrogen assimilation, and amino acid metabolism (see Supplemental Methods 1 and Supplemental Table 2 online). Furthermore, we investigate the relation between geography and four phenotypes containing characteristics grouped according to their association with flowering (23 traits), defense (23), ion concentrations, named ionomics (18), and development (43). We focus on the later three categories, covering 39, 26, and 40 of the 92 accessions, respectively (see Supplemental Table 3 online).
The relationship between the metabolic, defense-related, ionomics, and developmental phenotypes of the neighboring accessions is investigated using Moran’s I statistics on the RN network. We chose to focus on Moran’s I, first, because it is connected to (and abstracts) network topology, as described above, and secondly, the claims made based on this statistic are of global character. The values of Moran’s I indicate positive relations with geographic distance for the four phenotypes (Figure 4; see Supplemental Tables 4 and 5 online), suggesting a robust isolation by distance. The results for the metabolite classes corroborate the claim that not all metabolic processes response in the same way in the three growth conditions. For instance, the metabolites involved in nitrogen assimilation have a high Moran’s I in LiC and OpN with a high nitrogen supply, but the smallest in LiN where growth is limited by nitrogen (Figure 4). Moreover, metabolites participating in photorespiration show larger value for the Moran’s I in LiC compared with those in LiN and OpN. These findings suggest that the metabolic response to particular growth conditions is affected by geographic distance and reflect the adaptation to specific environments. The defense-related, ionomics, and developmental phenotypes demonstrate a similar behavior, with a larger Moran’s I value for the ionomics phenotypes.
Figure 4.

Moran’s I Statistics of Different Phenotypes for 92 Arabidopsis Accessions.
The results of the Moran’s I statistics between different classes of metabolites under three growth condition, LiN (blue), OpN (red), and LiC (green), and the underlying network are depicted. The metabolite classes are sorted in ascending order with respect to their values of Moran’s I in OpN; Moran’s I between defense-related (violet), ionomics (light blue), and developmental phenotypes (orange) covering 39, 26, and 40 of the 92 accessions, respectively. For details about the different phenotypes, we refer the reader to Supplemental Tables 3 to 5 in Atwell et al. (2010). All values of Moran’s I are significant with a P value < 0.05 (see Supplemental Tables 5 and 6 online).
Proximity Network-Based Statistics Validate the Guilt-by-Association Principle in Gene Networks
The guilt-by-association principle (GBA) postulates that two biological entities with similar quantitative behavior have similar functions (Eisen et al., 1998). This principle has been used for developing methods for automated prediction of gene functions based on gene expression profiles (Oliver, 2000; Lee et al., 2010; Zhou et al., 2002). While arguments have been provided both in favor of the general applicability of GBA in the case of networks obtained only using the Pearson correlation, referred to as relevance networks (Wolfe et al., 2005; Kinoshita and Obayashi, 2009), as well as questioning its validity (Gillis and Pavlidis, 2011, 2012). Interestingly, there exists no rigorous statistical assessment for the validity of this principle with respect to spatial clustering of biological functions in genome-wide coexpression networks. Such quantification will help determine which gene functions indeed follow the GBA principle, ultimately resulting in more accurate and more specific predictions.
Here, we illustrate how proximity networks extracted from gene expression data in combination with distance measures, quantifying the similarity of gene function annotation and the statistics for testing spatial clustering can be used to assess the GBA principle. The resulting quantities can be further interpreted in a network setting. To this end, we extracted the proximity network from a compendium of publically available microarray experiments in Arabidopsis, following the approach of Mutwil et al. (2011) (see Supplemental Methods 1 online).
In the extracted network, we focus on subsets of genes described by one of the six high-level categories from MapMan (Thimm et al., 2004), including regulation, hormones, cell wall, cellular response, primary metabolism, and secondary metabolism (Usadel et al., 2005). To assess the spatial clustering in a multivariate setting, the θi node weights are calculated from the semantic similarity of the gene neighbors in the proximity network (Figure 5). The semantic similarity was determined based on information-theoretic distance measures considering ancestral MapMan bins, as proposed by Klie and Nikoloski (2012) (see Supplemental Methods 1 online). The values for the Moran’s I presented in Table 1 indicate that the there is increasing clustering of genes with similar function starting from regulation to cellular response and secondary metabolism. These findings can be interpreted further with respect to the behavior of genes in each category: The genes involved in secondary metabolism, involving more linear pathways, show exceptionally high congruence with respect to both coexpression and gene function (Figure 5). Moreover, the genes involved in primary metabolism and cell wall–related functions, covering tightly transcriptionally regulated pathways, also show high values for Moran’s I. The broader categories of genes involved in regulation and crosstalk between pathways are, expectedly, showing the smallest Moran’s I, suggesting a weak relation between gene expression and characterization of their functions.
Figure 5.

Comparison of the Subnetworks Obtained for Genes Involved in Primary (Left) and Secondary (Right) Metabolism.
The color of a node/gene corresponds to the average semantic similarity of its neighboring nodes. Genes involved in both primary and secondary metabolism exhibit coexpression pattern that is in agreement with the function of the neighboring genes, resulting in high values of Moran’s I.
Table 1. Moran’s I Statistic and Typical (Sub)Network Properties of an Arabidopsis Coexpression Network.
| Moran's I |
Network Properties |
||||||
|---|---|---|---|---|---|---|---|
| Subnetworks | Observed | Expected | P Value | Rel. Density | Order (Genes) | Singletons | Components (Size >1) |
| Complete network | 0.456519 | -6.56E-05 | <1e-16 | 0.0013 | 15238 | 0 | 1 |
| Regulation | 0.399287 | −0.00017 | <1e-16 | 0.0015 | 5929 | 112 | 3 |
| Hormones | 0.442837 | −0.0061 | 0.000517 | 0.0054 | 165 | 112 | 14 |
| Cell wall | 0.478987 | −0.02326 | 0.007807 | 0.054 | 44 | 13 | 7 |
| Response | 0.705959 | −0.00055 | <1e-16 | 0.0022 | 1828 | 277 | 54 |
| Primary metabolism | 0.809655 | −0.00072 | <1e-16 | 0.008 | 1383 | 264 | 26 |
| Secondary metabolism | 0.967833 | −0.00238 | <1e-16 | 0.0087 | 421 | 172 | 23 |
The subnetworks correspond to high-level MapMan categories obtained from Usadel et al. (2005). The observed and expected values for the Moran’s I statistic along with its P values are given in the second, third, and fourth columns, respectively. The properties of the (sub)networks, listed in the first column, include the relative density (i.e., the number of edges divided by the total possible number of edges on the same number of nodes), order (i.e., number of genes), number of singleton nodes (i.e., isolated nodes without any neighbors), and the number of connected components (of size greater than one) and are given in the remaining columns.
FUTURE TECHNICAL AND MODELING CHALLENGES
The presented modeling approach aims at revealing patterns of covariation in uni- and multivariate characteristics of biochemical and biological entities embedded in space. This approach involves testing hypothesis based on global or local statistics that allow making claims about spatial clustering of the investigated multidimensional traits.
Here, we argued that to guarantee robustness of the claims, it is desirable that the spatial embedding of the entities is parameter-free and, thus, unique for the sampled entities. While there are network classes that could be used robustly to investigate molecular, physiological, and morphological characteristics, the challenge of determining objective criteria for selecting a network model remains. One possibility for overcoming this challenge would be simultaneously to investigate the enumerated statistics from the sparsest to the densest network model ensuring uniqueness. The statistics on networks of different density can in turn be used to determine the level of spatial proximity at which dramatic shift in the statistics is observed. One may then further examine the relevant properties of the neighbors enforced by a particular network class that may lead to relevant biological insights.
The reviewed network models applied in a geographic setting rely on the airline distance between two entities. However, geographic space is three dimensional and incorporates natural barriers, like mountains or bodies of water, with variable degree of relevance to different species. Moreover, geographic locations have other properties that vary in time due to climate effects. An interesting challenge is to extend the existing models to account for barriers given terrain descriptions as well as similarity of geographically proximal locations which share climate-related properties. In such a setting, the investigations of high-throughput data would shed light on the relation of geographic segregation and climate with the underlying molecular mechanisms of local adaptation.
Finally, the proposed approach for analysis of multidimensional data profiles simultaneously considers all traits and might not identify a pattern for the entire collection of traits. However, this does not imply that there are no patterns of covariation in a subset of traits. Automated selection of subsets of traits for which patterns are observed remains one of the key challenges. As with other statistical approaches, conclusions drawn from marrying off covariation measures and spatial embedding of entities is expected to depend on the sampling scheme for the entities. Therefore, the behavior of the network models and statistics in a bootstrap setting remains to be further investigated. Providing methods that could address these issues would further contribute to the resolving the grand challenge of identifying biologically meaningful principles that can be used to predict the behavior of biochemical and biological entities based on their high-throughput readouts.
Supplemental Data
The following materials are available in the online version of this article.
Supplemental Table 1. Moran’s I, Geary’s C, and Global G Statistics for 49 Metabolites of 20 Arabidopsis Accessions.
Supplemental Table 2. Assignment of Metabolites to Metabolite Classes.
Supplemental Table 3. List of Accessions Used in the Analyses of Metabolic, Defense-Related, Ionomics, and Developmental Phenotypes.
Supplemental Table 4. Moran’s I Statistic of 92 Arabidopsis Accessions for Different Metabolite Classes.
Supplemental Table 5. Moran’s I Statistic of 92 Arabidopsis Accessions for Defense-Related, Ionomics, and Developmental Phenotypes.
Supplemental Methods 1. Moran’s I and Pearson Correlation, Analysis of Metabolite Classes and Arabidopsis' Gene Coexpression Network.
Supplemental Data Set 1. Metabolic Profiles and Geographic Location of 20 Accessions with Geographic Origin in Germany in 12-h-Light/12-h-Dark Photoperiod under Nitrogen Supply Allowing Close to Optimal Growth (OpN).
Acknowledgments
We thank Alisdair R. Fernie and Roosa Laitinen from the Max Planck Institute of Molecular Plant Physiology for insightful discussions on the article.
AUTHOR CONTRIBUTIONS
Z.N. designed the study. S.Kle. and S.Kli. performed the analyses. All authors discussed the results and wrote the article.
References
- Aldous D.J., Shun J. (2010). Connected spatial networks over random points and a route-length statistic. Stat. Sci. 25: 275–288 [Google Scholar]
- Anastasio A.E., Platt A., Horton M., Grotewold E., Scholl R., Borevitz J.O., Nordborg M., Bergelson J. (2011). Source verification of mis-identified Arabidopsis thaliana accessions. Plant J. 67: 554–566 [DOI] [PubMed] [Google Scholar]
- Anselin L. (1995). Local indicators of spatial association-LISA. Geogr. Anal. 27: 93–115 [Google Scholar]
- Atwell S., et al. (2010). Genome-wide association study of 107 phenotypes in Arabidopsis thaliana inbred lines. Nature 465: 627–631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bivand R.S. (2002). Spatial econometric functions in R. J. Geogr. Syst. 4: 405–421 [Google Scholar]
- Blanco E., Pignatelli M., Beltran S., Punset A., Pérez-Lluch S., Serras F., Guigó R., Corominas M. (2008). Conserved chromosomal clustering of genes governed by chromatin regulators in Drosophila. Genome Biol. 9: R134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark, I. (1979). Practical Geostatistics. (London: Applied Science Publishers). [Google Scholar]
- Cressie, N. (1993). Statistics for Spatial Data. (New York: Wiley). [Google Scholar]
- Cushman S.A., Landguth E.L. (2010). Spurious correlations and inference in landscape genetics. Mol. Ecol. 19: 3592–3602 [DOI] [PubMed] [Google Scholar]
- de Berg, M., Cheong, O., van Kreveld, M., and Overmars, M. (2008). Delaunay triangulations. In Computational Geometry: Algorithms and Applications (Heidelberg: Springer), pp. 191–218. [Google Scholar]
- Eisen M.B., Spellman P.T., Brown P.O., Botstein D. (1998). Cluster analysis and display of genome-wide expression patterns. Proc. Natl. Acad. Sci. USA 95: 14863–14868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geary, R.C. (1954). The contiguity ratio and statistical mapping. The Incorporated Statistician 5: 115–127+129–146.
- Getis A., Ord J.K. (1992). The analysis of spatial association by use of distance statistics. Geogr. Anal. 24: 189–206 [Google Scholar]
- Gillis J., Pavlidis P. (2012). “Guilt by association” is the exception rather than the rule in gene networks. PLoS Comput. Biol. 8: e1002444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillis J., Pavlidis P. (2011). The impact of multifunctional genes on “guilt by association” analysis. PLoS ONE 6: e17258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goslee S.C., Urban D.L. (2007). The ecodist package for dissimilarity-based analysis of ecological data. J. Stat. Softw. 22: 1–19 [Google Scholar]
- Gravetter, F.J., and Wallnau, L.B. (2010). Essentials of Statistics for the Behavioral Science. (Belmont, CA: Cengage Learning). [Google Scholar]
- Ha M., Ng D.W.-K., Li W.-H., Chen Z.J. (2011). Coordinated histone modifications are associated with gene expression variation within and between species. Genome Res. 21: 590–598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris M.A., et al. Gene Ontology Consortium (2004). The Gene Ontology (GO) database and informatics resource. Nucleic Acids Res. 32 (Database issue): D258–D261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horton M.W., et al. (2012). Genome-wide patterns of genetic variation in worldwide Arabidopsis thaliana accessions from the RegMap panel. Nat. Genet. 44: 212–216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaromczyk J.W., Toussaint G.T. (1992). Relative neighborhood graphs and their relatives. Proc. IEEE 80: 1502–1517 [Google Scholar]
- Karoński M., Scheinerman E.R., Singer-Cohen K.B. (1999). On random intersection graphs: The subgraph problem. Combin. Probab. Comput. 8: 131–159 [Google Scholar]
- Kinoshita K., Obayashi T. (2009). Multi-dimensional correlations for gene coexpression and application to the large-scale data of Arabidopsis. Bioinformatics 25: 2677–2684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleessen S., Antonio C., Sulpice R., Laitinen R., Fernie A.R., Stitt M., Nikoloski Z. (2012). Structured patterns in geographic variability of metabolic phenotypes in Arabidopsis thaliana. Nat Commun 3: 1319. [DOI] [PubMed] [Google Scholar]
- Klie S., Nikoloski Z. (2012). The choice between MapMan and Gene Ontology for automated gene function prediction in plant science. Front. Genet. 3: 115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lê S., Josse J., Husson F. (2008). FactoMineR: An R package for multivariate analysis. J. Stat. Softw. 25: 1–18 [Google Scholar]
- Lee I., Ambaru B., Thakkar P., Marcotte E.M., Rhee S.Y. (2010). Rational association of genes with traits using a genome-scale gene network for Arabidopsis thaliana. Nat. Biotechnol. 28: 149–156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantel N. (1967). The detection of disease clustering and a generalized regression approach. Cancer Res. 27: 209–220 [PubMed] [Google Scholar]
- Marshall W.F. (2002). Order and disorder in the nucleus. Curr. Biol. 12: R185–R192 [DOI] [PubMed] [Google Scholar]
- Moran P.A.P. (1950). Notes on continuous stochastic phenomena. Biometrika 37: 17–23 [PubMed] [Google Scholar]
- Mutwil M., Klie S., Tohge T., Giorgi F.M., Wilkins O., Campbell M.M., Fernie A.R., Usadel B., Nikoloski Z., Persson S. (2011). PlaNet: Combined sequence and expression comparisons across plant networks derived from seven species. Plant Cell 23: 895–910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman M.E. (2002). Assortative mixing in networks. Phys. Rev. Lett. 89: 208701. [DOI] [PubMed] [Google Scholar]
- Oliver S. (2000). Guilt-by-association goes global. Nature 403: 601–603 [DOI] [PubMed] [Google Scholar]
- Platt A., et al. (2010). The scale of population structure in Arabidopsis thaliana. PLoS Genet. 6: e1000843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds C.E., Houle G. (2003). Mantel and partial Mantel tests suggest some factors that may control the local distribution of Aster laurentianus at Îles de la Madeleine, Québec. Plant Ecol. 164: 19–27 [Google Scholar]
- Ribeiro P.J., Jr, Diggle P.J. (2001). geoR: A package for geostatistical analysis. R-News 1: 15–18 [Google Scholar]
- Robert P., Escoufier Y. (1976). A unifying tool for linear multivariate statistical methods: The RV coefficient. J. R. Stat. Soc. Ser. C Appl. Stat. 25: 257–265 [Google Scholar]
- Smouse P.E., Long J.C., Sokal R.R. (1986). Multiple regression and correlation extensions of the mantel test of matrix correspondence. Syst. Zool. 35: 627–632 [Google Scholar]
- Sobetzko P., Travers A., Muskhelishvili G. (2012). Gene order and chromosome dynamics coordinate spatiotemporal gene expression during the bacterial growth cycle. Proc. Natl. Acad. Sci. USA 109: E42–E50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thimm O., Bläsing O., Gibon Y., Nagel A., Meyer S., Krüger P., Selbig J., Müller L.A., Rhee S.Y., Stitt M. (2004). MAPMAN: A user-driven tool to display genomics data sets onto diagrams of metabolic pathways and other biological processes. Plant J. 37: 914–939 [DOI] [PubMed] [Google Scholar]
- Toussaint G.T. (1980). The relative neighbourhood graph of a finite planar set. Pattern Recognit. 12: 261–268 [Google Scholar]
- Usadel B., et al. (2005). Extension of the visualization tool MapMan to allow statistical analysis of arrays, display of corresponding genes, and comparison with known responses. Plant Physiol. 138: 1195–1204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veltkamp R.C. (1992). The gamma-neighborhood graph. Comput. Geom. 1: 227–246 [Google Scholar]
- Wolfe C.J., Kohane I.S., Butte A.J. (2005). Systematic survey reveals general applicability of “guilt-by-association” within gene coexpression networks. BMC Bioinformatics 6: 227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X., Kao M.-C.J., Wong W.H. (2002). Transitive functional annotation by shortest-path analysis of gene expression data. Proc. Natl. Acad. Sci. USA 99: 12783–12788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zupancic M.L., Tran H., Hofmeister A.E.M. (2001). Chromosomal organization governs the timing of cell type-specific gene expression required for spore formation in Bacillus subtilis. Mol. Microbiol. 39: 1471–1481 [DOI] [PubMed] [Google Scholar]