

Ribosome profiling provides a quantitative readout of mRNA segments bound by ribosomes in vivo. This work describes a cost-effective variant of the method and use it to address unresolved questions in maize plastid gene expression, including the translational coupling of overlapping reading frames, the ability of unspliced chloroplast RNAs to engage in translation, and mechanisms of ribosome pausing.
Abstract
The profiling of ribosome footprints by deep sequencing has revolutionized the analysis of translation by mapping ribosomes with high resolution on a genome-wide scale. We present a variation on this approach that offers a rapid and cost-effective alternative for the genome-wide profiling of chloroplast ribosomes. Ribosome footprints from leaf tissue are hybridized to oligonucleotide tiling microarrays of the plastid ORFeome and report the abundance and translational status of every chloroplast mRNA. Each assay replaces several time-consuming traditional methods while also providing information that was previously inaccessible. To illustrate the utility of the approach, we show that it detects known defects in chloroplast gene expression in several nuclear mutants of maize (Zea mays) and that it reveals previously unsuspected defects. Furthermore, it provided firm answers to several lingering questions in chloroplast gene expression: (1) the overlapping atpB/atpE open reading frames, whose translation had been proposed to be coupled, are translated independently in vivo; (2) splicing is not a prerequisite for translation initiation on an intron-containing chloroplast RNA; and (3) a feedback control mechanism that links the synthesis of ATP synthase subunits in Chlamydomonas reinhardtii does not exist in maize. An analogous approach is likely to be useful for studies of mitochondrial gene expression.
INTRODUCTION
The genome-wide analysis of translation received relatively little attention until Ingolia et al. (2009) combined deep sequencing with classical methods for mapping the positions of bound ribosomes, in a powerful new approach called ribosome profiling. Ribosome profiling provides a quantitative readout of mRNA regions that are protected from nucleases by ribosomes (ribosome footprints) at single-nucleotide resolution. The method has revealed fundamental aspects of ribosome behavior as well as the widespread impact of translational regulation in yeast, metazoans, and bacteria (Ingolia et al., 2009, 2011; Brar et al., 2012; Lee et al., 2012; Li et al., 2012).
The mapping of ribosome footprints by deep sequencing provides an excellent tool for the detailed analysis of complex translatomes. However, it is unnecessarily labor intensive and costly if the focus is more limited. For instance, mitochondrial and chloroplast genomes encode fewer than 100 proteins, and the translation of organellar mRNAs is commonly assayed by RNA gel blotting of polysome fractions or by pulse labeling (Barkan, 1998). Even so, it is impractical to monitor every organellar gene in this manner, and the resolution of these assays is limited. In particular, polysome analyses cannot discriminate translation activity in different regions of a reading frame or among different open reading frames on the same polycistronic mRNA.
Here, we present a modification of the ribosome profiling method that maps and quantifies chloroplast ribosome footprints by hybridization to high-resolution tiling microarrays. The resolution and dynamic range of this method are inferior to those of the deep sequencing approach, but these disadvantages are offset by substantial savings in cost and time. We used characterized chloroplast gene expression mutants to show that this method allows an accurate genome-wide examination of the translation and abundance of chloroplast mRNAs. The method easily discriminates different protein-coding regions on polycistronic mRNAs and different regions within single reading frames. We demonstrate the power of the approach by using it to elucidate several characteristics of chloroplast translation and to reveal additional functions for chloroplast translational activators that had already been studied in depth. The results resolve questions concerning the translational coupling of overlapping reading frames in the bicistronic atpB/E mRNA and the ability of unspliced chloroplast RNAs to engage in translation, and they suggest a mechanistic basis for the ribosome pausing that has been reported on chloroplast mRNAs.
RESULTS
Translational regulation is a major feature of chloroplast gene expression and involves numerous nucleus-encoded mRNA-specific translational activators (reviewed in Barkan, 2011). These translational activators have been discovered via genetic screens that begin with the recovery of nonphotosynthetic mutants, followed by molecular analyses that pinpoint the basis for the photosynthetic defects. Pulse labeling, reporter gene fusions, and polysome analyses have revealed chloroplast translation defects in various nuclear mutants (Barkan et al., 1994; Stampacchia et al., 1997; Drapier et al., 2007), but these methods are not suited to analysis of the complete chloroplast translatome, and they each suffer from substantial limitations. Application of the Ingolia ribosome profiling method to chloroplast translation could bypass the limitations of traditional methods while also allowing previously intractable questions to be addressed. However, the conversion of ribosome footprints into a sequencing library and the sequencing itself are time-consuming and costly. Furthermore, the resolution and depth of analysis offered by this method go well beyond what is necessary to address most questions in organelle translation. Chloroplast genomes account for only ∼0.1% of the gene complexity in land plants, with roughly 20% of the ribosome content deriving from chloroplasts in leaf tissue. Thus, the majority of reads obtained when profiling plant cell ribosomes by deep sequencing would be irrelevant to chloroplast translation. In addition, the single-nucleotide resolution offered by the sequencing approach is warranted in only specialized cases, and a resolution of ∼30 nucleotides would suffice to answer many questions.
With these issues in mind, we devised a rapid and cost-effective approach that combines the purification of ribosome footprints with microarray hybridization (Figure 1). Total leaf lysates are incubated with micrococcal nuclease to degrade nuclease-accessible mRNA and cleave polysomes into monosomes. Monosomes are purified by pelleting through a Suc cushion, and the ribosome footprints (RNA fragments of ∼30 nucleotides) are purified from this material. The RNA is labeled with a fluorescent dye in a simple procedure that does not involve conversion to cDNA, and the labeled ribosome footprints are hybridized to a custom microarray containing overlapping 50-mer oligonucleotides complementary to all known chloroplast open reading frames (ORFs). Consecutive array probes overlap by 20 nucleotides in order to detect ribosome footprints at every position. This design provides a resolution of ∼30 nucleotides, with each probe capturing the footprints of ribosomes translating ∼10 consecutive codons. The entire procedure from the time of tissue harvest through microarray scanning takes <5 d, and up to six experimental samples and matched controls are conveniently prepared and analyzed in parallel.
Figure 1.

Overview of the Microarray-Based Ribosome Profiling Approach and Its Application for Studying Mutants with Defects in Chloroplast Gene Expression.
Steps 1 through 3 are similar to the sequencing-based ribosome profiling method (Ingolia et al., 2012). Subsequently, the ribosome footprints are directly labeled (step 4) and hybridized to tiling microarrays that probe all protein-coding regions of the chloroplast genome (step 5). A screen capture of data from an analysis of a ppr10 mutant is shown; the red spot maps in the atpH RNA, whose translation and stability are known to rely on PPR10 (Pfalz et al., 2009). The microarray design is diagrammed below: 50-mer oligonucleotides cover all known protein-coding regions (ORF), with adjacent probes overlapping by 20 nucleotides (nt). WT, the wild type.
Microarray-Based Ribosome Profiling Detects Known and Unanticipated Targets of Chloroplast Translational Activators
We evaluated this approach by reanalyzing four nuclear mutants of maize (Zea mays) with known defects in the translation or stabilization of specific chloroplast mRNAs: atp1, atp4, ppr10, and crp1 (Barkan et al., 1994; McCormac and Barkan, 1999; Pfalz et al., 2009; Zoschke et al., 2012). Ribosome footprints were isolated from the leaves of mutant seedlings and from wild-type siblings grown in parallel. The RNAs were directly labeled with Cy5 (wild-type) or Cy3 (mutant); matched mutant and wild-type samples were combined and hybridized to custom microarrays as shown in Figure 1. Figure 2 shows the fluorescence signal as a function of position along a polycistronic transcription unit in a wild-type sample; the signals coincide with the boundaries of ORFs, providing evidence that they derive primarily from ribosome footprints. Figure 3 displays the results as the median ratio of signals in the wild type versus mutant, plotted according to position on the chloroplast genome (see also Supplemental Figure 1 online). Peaks in this plot identify regions that are expressed at reduced levels in the mutants due either to defects in translation or RNA accumulation. To distinguish defects in translation from defects in RNA accumulation, total RNA purified from the same tissue samples was profiled by hybridization to microarrays of the same design (Figure 3; see Supplemental Figure 2 online). The ratio of ribosome footprint signal to total RNA signal provides an estimate of translational efficiency.
Figure 2.
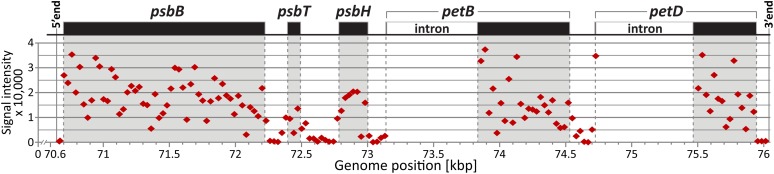
Distribution of Ribosome Footprint Signals in the Chloroplast psbB Gene Cluster.
A map of the psbB transcription unit in maize is shown at top. The predominant 5′ end upstream of psbB and 3′ end downstream of petD are marked (Zhelyazkova et al., 2012). Vertical dashed lines mark the positions of start codons, stop codons, and splice junctions. The normalized signals for the wild-type samples (F635) from the atp1 analysis are plotted according to genomic position. A full genome view is shown in Supplemental Figure 1D online. The signals within coding regions (gray shaded) are higher than those in transcribed but untranslated regions (unshaded). Intron sequences were not represented on the microarray.
[See online article for color version of this figure.]
Figure 3.

Microarray-Based Ribosome Profiling of Chloroplast mRNAs in atp1, atp4, ppr10, and crp1 Mutants.
(A) Map of the maize chloroplast genome illustrating protein-coding genes only. The map was created with OGDraw (Lohse et al., 2007). The map shows just one of the two large inverted repeat regions. Asterisks and crosses mark genes with previously described and newly identified gene expression defects, respectively. Dashed lines connect genes on the map with peaks in the plots below.
(B) and (C) Median ratios of ribosome footprint signals (Ribo footprints) in the wild type (WT) versus atp1 and atp4 mutants (F635/F532) are plotted as a function of genome position. Each plot shows normalized values obtained from two biological replicates. Peaks represent regions with fewer ribosome footprints in the mutants compared with the wild type.
(D) and (E) Top panels: Median ratios of ribosome footprint signals in the wild type versus ppr10 or crp1 mutants (F635/F532) are plotted as in (B) and (C). Whole-genome views of the ppr10 and crp1 ribosome footprint data with reduced y axis scales are given in Supplemental Figures 1B and 1C online, respectively, to better visualize minor peaks. Middle panels: Median ratios of total RNA signals in the wild type versus ppr10 or crp1 mutants (F635/F532) are plotted as a function of genome position. Each plot shows normalized values obtained from one experiment. Peaks represent regions with RNA accumulation defects in the mutants. Bottom panels: Relative translation efficiencies were calculated as the ratios of ribosome footprint ratios (shown in the top panels) to total RNA ratios (shown in the middle panels).
[See online article for color version of this figure.]
atp1 and atp4 mutants lack the chloroplast ATP synthase due to the failure to translate the chloroplast atpB mRNA (McCormac and Barkan, 1999; Zoschke et al., 2012). ATP4 is a pentatricopeptide repeat (PPR) protein that associates with the atpB 5′ untranslated region (UTR) (Zoschke et al., 2012), whereas the atp1 gene is still unidentified. The primary peaks in both ribosome profile data sets mapped to atpB, as anticipated (Figures 3B and 3C). In principle, these peaks could result from defects in atpB mRNA levels, atpB translation, or both. However, RNA gel blot hybridizations had shown that the atpB mRNA accumulates normally in these mutants (McCormac and Barkan, 1999; Zoschke et al., 2012), and we confirmed this to be true by slot-blot hybridization (Figures 4A and 4B) and microarray hybridization (see Supplemental Figures 2B and 2C online). The ribosome profiles of both mutants showed a minor loss of signal across atpA (Figures 3B and 3C); this is not due to a reduction in atpA mRNA (Figures 4A and 4B; see Supplemental Figures 2B and 2C online) and corroborates the mild atpA translation defects suggested previously (McCormac and Barkan, 1999; Zoschke et al., 2012). A minor peak at psaJ in the atp4 profile (Figure 3C) corresponds to a known RNA accumulation defect (Zoschke et al., 2012) and is visible in the microarray analysis of total RNA (see Supplemental Figure 2C online). Interestingly, the atp4 profile revealed a strong defect in the expression of rpl14 that had previously gone undetected (Figure 3C). RNA gel blot hybridizations showed this to result from the absence of dicistronic rpl16/rpl14 transcripts (Figure 5A). The absence of these processed RNAs is not accompanied by increased precursor accumulation, implying that ATP4 is required to stabilize dicistronic rpl16-rpl14 RNAs. A minor defect in psbA RNA accumulation was detected in the atp1 mutant (see Supplemental Figure 2B online); a similar reduction in psbA RNA was observed in several other mutants (see below), so we suspect this to be a secondary effect of compromised bioenergetics. Thus, this assay detected all known chloroplast gene expression defects in atp1 and atp4 mutants and alerted us to additional defects.
Figure 4.

Slot-Blot Hybridization Assays Discriminate Translation and RNA Accumulation Defects in atp1, atp4, ppr10, and crs1 Mutants.
Three hundred nanograms of ribosome footprint RNA and 300 ng of total RNA from plants of the indicated genotype were applied to nylon membranes and hybridized to radiolabeled DNA probes covering large segments of the indicated ORFs (probes are described in Methods). The ratio of footprint signal in the wild type (WT) versus mutant (blue bars) indicates the overall ratio of expression. The ratio of total RNA in the wild type versus mutant (red bars) reveals differences in RNA level. Translational efficiency in the wild type versus mutant is indicated by the ratio of ratios [wild-type footprint/mutant footprint]/[wild type total RNA/mutant total RNA] (green bars).
Figure 5.

RNA Gel Blot Assays Confirm a Previously Unknown rpl14 Expression Defect Detected in Ribosome Profiles of atp4 Mutants.
Total leaf RNA (4 µg/lane) was analyzed by RNA gel blot hybridizations using probes corresponding to the indicated chloroplast genes. The positions of RNA size markers are shown (kilonucleotides [knt]). rRNAs were detected on the same filters by staining with methylene blue and are shown below the autoradiograms.
(A) Follow-up to atp4 ribosome profile data suggesting a defect in rpl14 expression (Figure 3C; see Supplemental Figure 2C online). Probes from the transcription unit harboring rpl14 (map at top) revealed the specific absence of spliced and unspliced dicistronic rpl16-rpl14 transcripts in atp4 mutants. The nonallelic atp1 mutant, which phenocopies the ATP synthase defect of atp4, does not show this RNA accumulation defect.
(B) Hybridization with an atpE-specific probe confirms that the atpE ORF is solely found on a dicistronic atpB/atpE transcript in maize. WT, the wild type.
PPR10 is a PPR protein that is also required for the accumulation of the chloroplast ATP synthase. PPR10 binds RNA in the atpI-atpH intergenic region and stabilizes the adjacent RNA by blocking exonucleases (Pfalz et al., 2009; Prikryl et al., 2011). An additional role for PPR10 in atpH translation was inferred based on polysome sedimentation data, but the presence of multiple ORFs on polycistronic atpH RNAs precluded firm conclusions about PPR10’s translational targets. The ribosome profile data revealed a dramatic defect in atpH expression in ppr10 mutants (Figure 3D). Comparison of the abundance of atpH ribosome footprints and total atpH RNA by microarray hybridization (Figure 3D) and by slot-blot hybridization (Figure 4C) showed that atpH mRNA in ppr10 mutants is translated very inefficiently. In fact, these results show that the loss of RNA makes only a small contribution to the atpH expression defect in ppr10 mutants, implying that PPR10 acts primarily at the level of translation to stimulate atpH expression. Comparison of ribosome footprint to total RNA levels (Figures 3D and 4C) show further that the translation of other ORFs on polycistronic atpH transcripts is no more than minimally affected in ppr10 mutants, clarifying the prior polysome data (Pfalz et al., 2009). In a second site of action, PPR10 binds the psaJ-rpl33 intergenic region and stabilizes upstream and downstream RNA (Pfalz et al., 2009). Although the magnitude of the defect in atpH expression dominates the ppr10 ribosome profile, the defect in psaJ expression is apparent in the microarray (Figure 3D) and slot-blot (Figure 4C) analyses of total RNA and by adjusting the scale on the y axis in the ribosome profile (see Supplemental Figure 1B online).
CRP1 is a PPR protein that stimulates the translation of the chloroplast petA and psaC RNAs and stabilizes processed RNAs with termini in the petB-petD intergenic region (Barkan et al., 1994; Fisk et al., 1999; Schmitz-Linneweber et al., 2005). The ribosome profile for crp1 mutants showed defects in petA, petD, and psaC expression (Figure 3E), as anticipated. The microarray analysis of total RNA confirmed that petA and psaC RNAs accumulate normally in crp1 mutants and that petD RNA levels are reduced (Figure 3E). These data show further that the loss of petD RNAs is minor in comparison with the defect in petD translation. Thus, CRP1 acts primarily at the level of translation to stimulate petD expression. Additionally, the data revealed a previously undetected defect in the expression of ndhE, which is adjacent to and cotranscribed with psaC (Figure 3E; see Supplemental Figure 1C online). Prior genome-wide RNA coimmunoprecipitation data had shown two peaks of CRP1 interaction in this region: one in the 5′UTR of psaC and one in the 5′UTR of ndhE (Schmitz-Linneweber et al., 2005). The ndhE signal had been assumed to result from its proximity to CRP1’s psaC binding site. However, in light of the decrease in ndhE translation detected in crp1’s ribosome profile, it now seems more likely that CRP1 does have an interaction site upstream of ndhE and that CRP1 modestly enhances ndhE translation. The ribosome profile also revealed an unanticipated defect in psbA RNA accumulation and translation in crp1 mutants (Figure 3E). However, we suspect that this is an indirect effect because the psbA RNA did not detectably coimmunoprecipitate with CRP1 in RNA coimmunoprecipitation assays (Schmitz-Linneweber et al., 2005), and a similar loss of psbA expression was observed in several other mutants.
Taken together, the reanalysis of the atp1, atp4, ppr10, and crp1 mutants demonstrates that microarray-based ribosome profiling provides a sensitive and accurate readout of chloroplast gene expression on a genome-wide scale, and that it is much faster and often more informative than assays used previously. Each assay replaces multiple time-consuming single-gene assays and resolves translation defects among different ORFs in the same polycistronic transcription unit. The atp4 and crp1 genes were already among the best characterized nuclear genes involved in chloroplast gene expression, in that their roles had been inferred based on genome-wide RNA coimmunoprecipitation data and on selected RNA gel blot and translation assays in the mutants (Barkan et al., 1994; Schmitz-Linneweber et al., 2005; Zoschke et al., 2012). The additional functions discovered here show the value of this genome-wide approach and suggest that many characterized nuclear genes involved in organelle gene expression have additional, as yet undiscovered functions.
Translation of Overlapping Chloroplast Reading Frames: atpE Translation Is Uncoupled from atpB in Vivo
Translation in chloroplasts shares many features with that in bacteria. For example, most chloroplast mRNAs are polycistronic, plastid ribosomes bind mRNA internally often via a Shine-Dalgarno interaction, and some cotranscribed ORFs overlap (Shinozaki et al., 1986; Hirose and Sugiura, 2004; Drechsel and Bock, 2011). The translation of overlapping ORFs in bacteria is typically coupled, in that translation of the second ORF is dependent on the translation of the first (reviewed in Jackson et al., 2007). However, there is conflicting data concerning the coupled translation of overlapping ORFs in chloroplasts: Analysis of the overlapping chloroplast atpB/atpE ORFs in heterologous expression systems pointed to coupled translation (Gatenby et al., 1989), whereas in vitro studies with a tobacco (Nicotiana tabacum) chloroplast translation extract suggested independent translation (Suzuki et al., 2011).
The ribosome profiles of the atp1 and atp4 mutants show that atpE translation is largely uncoupled from that of atpB in vivo: The ribosome footprints on the upstream atpB gene are severely reduced, whereas the abundance of footprints on the downstream atpE ORF is unaffected (Figure 6). The atpB and atpE ORFs are found solely on the same bicistronic RNA in maize (Figure 5B; Zoschke et al., 2012), so the polysome sedimentation assay used previously to study atpB/E translation in these mutants could not distinguish effects on atpB and atpE (McCormac and Barkan, 1999; Zoschke et al., 2012). The ability to resolve different ORFs on the same mRNA is a major advantage of ribosome profiling for studies of organellar and bacterial translation.
Figure 6.

Ribosome Profiling Data Showing Uncoupled Translation of the Overlapping atpB/E ORFs and Suggesting Ribosome Pausing at Shine-Dalgarno–Like Sequences.
A map of the atpB/E coding region in maize is shown at top, with the sequence at the overlap between the atpB stop codon (TGA, overlined) and atpE start codon (ATG, underlined) indicated. Vertical dashed lines mark the positions of start and stop codons. Peaks in the single-channel data that coincide with internal Shine-Dalgarno–like sequences are labeled by arrows (for details, see Results). The top diagrams show median ratios of ribosome footprint signals (Ribo footprints) in the wild type (WT) versus mutant (F635/F532) for the atpB/E coding region. The bottom diagrams show normalized ribosome footprint signals from wild-type or mutant samples in the atpB/E coding region. The full genome data are shown in Figures 3B and 3C and Supplemental Figures 1D and 1E online.
(A) Ribosome footprint data for the overlapping atpB and atpE ORFs in the atp1 mutant.
(B) Ribosome footprint data for the overlapping atpB and atpE ORFs in the atp4 mutant.
[See online article for color version of this figure.]
Evidence for Ribosome Pausing at Shine-Dalgarno–Like Sequences
To illustrate ribosome density as a function of position along the atpB/atpE mRNA, the normalized fluorescence intensities for the wild-type and mutant samples from the atp1 and atp4 experiments are plotted independently in Figure 6 (bottom panels). The shapes of the wild-type profiles are quite similar between the two independent experiments, as are the shapes of the mutant profiles, demonstrating that the fluctuations in signal across the atpB gene are quite reproducible (see Supplemental Figure 3 online). The single-channel plots reveal some interesting features. First, in wild-type chloroplasts, the median of the signals is considerably stronger in the atpB ORF than in the atpE ORF (∼2.5:1; Figure 6), implying that translation initiates more efficiently in the upstream atpB ORF. Second, despite the near absence of ribosome footprints across much of atpB in the mutants, there are several positions at which the mutant signals reach or exceed the wild-type signals, and these sometimes correspond to peaks in the wild-type plots (Figure 6). In a ribosome profiling-by-sequencing study in bacteria, ribosome footprint abundance varied by more than 10-fold within different regions of the same ORF (Li et al., 2012). The authors inferred that positions with high footprint signals are ribosome pause sites and that Shine-Dalgarno–like elements within ORFs account for much of this pausing. Our results suggest the same thing occurs in chloroplasts. The ideal Shine-Dalgarno element in maize chloroplasts is 5′-AGGAGG-3′ (Schwarz and Kossel, 1980). Interestingly, the probe centered at position 54,865 marks an isolated peak of ribosome footprints in all of the samples, and this is the only probe in the atpB ORF to contain this ideal Shine-Dalgarno sequence. This observation suggests that elongating ribosomes pause at this position or that ribosomes bind internally to this sequence. The prominent peaks at positions 55,593 and 55,514 in the mutant profiles are also noteworthy: Each of these peaks span two array elements whose overlap contains the next strongest matches to the ideal Shine-Dalgarno sequence in the atpB ORF (Figure 6; see Supplemental Table 1 online). A peak at 54,645 in both the wild-type and mutant data maps to the known Shine-Dalgarno element for atpE (Suzuki et al., 2011). In fact, every appearance of five contiguous nucleotides matching the ideal Shine-Dalgarno element in atpB corresponds with a peak in ribosome footprints (Figure 6; see Supplemental Table 1 online). A similar phenomenon can be observed for the petA RNA in crp1 mutants (see Supplemental Figure 4 and Supplemental Table 1 online).
These results suggest that chloroplast ribosomes pause at ORF-internal Shine-Dalgarno–like sequences. This phenomenon may contribute to ribosome pause events that have been proposed to correlate with prosthetic group attachment and membrane integration of certain chloroplast gene products (Kim et al., 1991; Stollar et al., 1994), although other factors are also likely to influence the rate of ribosome movement. A high-resolution examination of plastid ribosome positions by deep sequencing under various genetic and environmental conditions will be necessary to fully explore and clarify these issues.
Translation Initiates with Similar Efficiency on Spliced and Unspliced atpF mRNA
Chloroplast genomes in land plants contain ∼20 introns, many of which interrupt protein-coding genes (Shinozaki et al., 1986). Because RNA splicing and translation occur in the same compartment in chloroplasts, there is the possibility for translation to initiate on unspliced RNAs. Unspliced transcripts in chloroplasts accumulate to high levels (Barkan, 1989; Jenkins et al., 1997), and it has been unclear whether mechanisms have evolved to preclude their translation.
We addressed this question by profiling chloroplast ribosomes in maize crs1 mutants, which fail to splice the intron in the chloroplast atpF pre-mRNA (Jenkins et al., 1997). The abundance of ribosome footprints across exon 1 is very similar in the wild-type and crs1 samples, whereas ribosome coverage of exon 2 is much higher in the wild type than in the mutant (Figure 7). The reduction in ribosomes on exon 2 of unspliced atpF RNA is to be expected because there is an in-frame stop codon mapping 25 nucleotides downstream from the 5′ splice junction. These results provide strong evidence that translation initiation and ribosome movement through exon 1 of spliced and unspliced atpF are very similar. The implications of the finding are discussed below.
Figure 7.

Translation of Unspliced atpF mRNA Detected by Ribosome Profiling of the crs1 Mutant.
A map of the atpF gene is shown at top. Vertical dashed lines mark the positions of the start codon, stop codon, and splice junctions. Intron sequences are marked by a gap in the map because they were not represented on the microarray. The top diagram shows the normalized ribosome footprint signals from wild-type (F635) or crs1 mutant (F532) samples in the atpF coding region. The bottom diagram shows median ratios of ribosome footprint signals in the wild type (WT) versus mutant (F635/F532) for the atpF coding region (the full genome data are shown in Supplemental Figures 1H and 2D online).
[See online article for color version of this figure.]
DISCUSSION
In this study, we describe a simple method that maps and quantifies chloroplast ribosome footprints with a resolution of ∼30 nucleotides. The accuracy and sensitivity of the method were demonstrated by using it to analyze chloroplast gene expression in several mutants that had already been thoroughly characterized with traditional methods. The method detected all known translation and RNA accumulation defects in these mutants and revealed others that had previously escaped detection. In addition, the technique provided firm answers to several fundamental questions in chloroplast gene expression that had been opaque to prior methods or for which existing data were inconclusive.
Utility of Microarray-Based Ribosome Profiling for Studying the Nuclear Control of Organelle Gene Expression
Genetic approaches have led to the discovery of numerous nucleus-encoded proteins that act at the posttranscriptional level to enhance the expression of subsets of chloroplast genes (reviewed in Barkan, 2011). The identification of the chloroplast gene expression defects in such mutants is often laborious, involving measures of RNA abundance, processing, and translation on a gene-by-gene basis with techniques such as RNA gel blotting, protein pulse labeling, and polysome analysis. Recent genome-wide methods have expanded this traditional toolkit (Schmitz-Linneweber et al., 2005; de Longevialle et al., 2008), but a method for the genome-wide analysis of chloroplast translation has been lacking. The approach described here is not only more comprehensive and rapid than previous assays, it also provides less ambiguous data in many instances. For example, protein pulse labeling is often used to assay rates of protein synthesis in chloroplasts, but many chloroplast gene products cannot be detected in this manner and the signal is inevitably a function of both translation rate and protein half-life. Thus, even where reduced protein labeling is observed, it is not certain that this does not result from rapid protein turnover. RNA gel blotting of size-fractionated polysomes has been used to detect defects in chloroplast translation, but it is impractical to assay a large number of ORFs in this manner, and the data are difficult to interpret when dealing with polycistronic mRNAs. For example, petA mRNA is found on polysomes of reduced size in crp1 mutants (Barkan et al., 1994), but because petA is found solely on polycistronic mRNAs it had been unclear which ORFs contributed to the change in polysome size. It is obvious from the ribosome profiling data presented here that the translation defect is specific to the petA ORF (Figure 3E). The ability to resolve different ORFs in a polycistronic mRNA is a major advantage of this method for studies of organellar and bacterial translation.
An additional advantage of this method is that it simultaneously monitors the abundance and ribosome association of each ORF. This is especially pertinent for the genetic analysis of chloroplast gene expression factors, many of which have dual activities in translation and RNA stabilization. For example, the PPR proteins ATP4 and CRP1 had previously been shown to affect the translation and stabilization of distinct chloroplast RNAs (Barkan et al., 1994; Schmitz-Linneweber et al., 2005; Zoschke et al., 2012). Despite the fact that these are among the most thoroughly characterized chloroplast gene expression factors, our reanalysis of atp4 and crp1 mutants discovered additional defects. The dramatic loss of dicistronic rpl16-rpl14 RNAs in atp4 mutants is a particularly noteworthy example (Figure 5A); expression of these genes had not been assayed previously because the atpB translation defect in atp4 mutants was sufficient to account for their specific loss of the ATP synthase complex (Zoschke et al., 2012). These results add to the emerging evidence that PPR proteins typically bind multiple RNAs in vivo and have multiple independent effects on organelle gene expression. Analogous comprehensive data for other PPR mutants will be crucial to arriving at a deep understanding of the functions, mechanisms, and evolution of proteins in this family. More generally, use of this method will be invaluable in the early stages of analyzing any nonphotosynthetic mutant by providing a global view of chloroplast gene expression that can focus subsequent experiments. It can be anticipated that the approach will be equally useful for studies of analogous phenomena in mitochondria, whose genome coding capacity and expression mechanisms share many features with those of chloroplasts.
Insights into Translation Mechanisms in Chloroplasts
The resolution of ribosomal profiling by microarray hybridization as described here, although lower than that obtained by deep sequencing, was more than sufficient to address several longstanding questions about chloroplast translation. For example, our results definitively show that translation of the maize atpB and atpE ORFs is not coupled in vivo, corroborating conclusions based on in vitro data (Suzuki et al., 2011) but differing from results obtained when these genes were expressed heterologously in bacteria (Gatenby et al., 1989). In addition, we were able to address for the first time whether splicing needs to precede translation initiation in chloroplasts. Unspliced RNAs accumulate to substantial levels in plastids, and they are found in the stromal compartment together with ribosomes. Translation of unspliced RNAs could, in principle, generate harmful proteins; in fact, it has been suggested that nuclear-cytoplasmic compartmentalization was driven by the pressure to separate splicing and translation (Martin and Koonin, 2006). By applying our ribosome profiling method to a mutant that fails to splice the chloroplast atpF mRNA, we showed that ribosome occupancy on the first exon of atpF RNA is virtually identical in the context of spliced and unspliced transcripts.
A set of mechanisms known as Control by Epistasy of Synthesis coordinate the synthesis of subunits within the same photosynthetic enzyme complex in the chloroplasts of the alga Chlamydomonas reinhardtii (reviewed in Choquet and Wollman, 2009). However, the degree to which analogous mechanisms exist in land plants has been unclear. The ribosome profiles for the atp1 and atp4 mutants show that one example described in C. reinhardtii, the repression of atpA translation in the absence of atpB translation (Drapier et al., 2007), does not occur to a significant extent in maize. Our data confirmed the virtual absence of atpB translation in both mutants and showed that this was accompanied by only a minor decrease in ribosome coverage on atpA. The small effects that were observed at atpA may be a direct consequence of these mutations, as there is evidence that ATP4 is physically associated with the atpA 5′UTR in vivo (Zoschke et al., 2012). Two other mutants analyzed here, ppr10 and crs1, fail to synthesize ATP synthase subunits AtpH and AtpF, respectively, with no more than minimal effects on translation efficiencies for other chloroplast ATP synthase genes (Figure 3D; see Supplemental Figures 1B and 2D online). Taken together, our results show that defects in the synthesis of AtpB, AtpH, or AtpF have little if any effect on the translation of other chloroplast ORFs in maize.
Comparison of Ribosome Profiling-by-Microarray to Ribosome Profiling-by-Sequencing
The primary advantages of using microarrays rather than deep sequencing to profile ribosomes are savings in time and cost. Both strategies employ the same method for purifying ribosome footprints, but they diverge once the purified footprints are in hand. For the microarray method, the footprints are directly labeled with a simple protocol and hybridized to the array, with results returned the following day. Custom microarrays suitable for tiled probing of up to ∼100 ORFs can currently be obtained for about $50 each (see Methods). By contrast, the conversion of ribosome footprints into sequencing libraries involves complex protocols and/or costly kits (Ingolia et al., 2012). In addition the array-based method eliminates the need for depletion of rRNA fragments that copurify with the footprints (Ingolia et al., 2012). On the other hand, the resolution of our method, although significantly higher than that of polysome analyses, does not rival that of the deep sequencing approach, and sequencing also offers a greater dynamic range. However, in cases in which the ultra-high-resolution, depth of analysis, and large scale offered by deep sequencing are unnecessary, ribosome profiling-by-microarray offers a rapid and cost-effective alternative.
METHODS
Plant Material
Maize (Zea mays) plants were grown in soil in cycles of 16 h light (∼300 µmol m−2 s−1)/28°C and 8 h dark/26°C. One hour after the start of the light cycle on the eighth day after sowing, leaves 2 and 3 were harvested and snap-frozen in liquid nitrogen. Tissue was stored at −80°C until use. All mutant alleles were described previously (Barkan et al., 1994; Jenkins et al., 1997; McCormac and Barkan, 1999; Pfalz et al., 2009; Zoschke et al., 2012). The atp1, atp4, and ppr10 material was generated in allelism crosses involving the following alleles: atp1-1/-2, atp4-2/-3, and ppr10-1/-2, respectively. The crp1 and crs1 material came from self-pollination of heterozygous plants (crp1-1/+ and crs1-1/+, respectively). Phenotypically normal siblings segregating in the same plantings served as the control in each experiment.
Ribosome Footprint Preparation
The method for isolating ribosome footprints from leaf tissue combines our protocol for leaf polysome extraction (Barkan, 1998) with the Ingolia ribosome profiling protocol (Ingolia et al., 2012). Leaves from one or two maize seedlings (∼0.4 g fresh weight) were homogenized in liquid nitrogen with mortar and pestle and thawed in 5 mL of ribosome extraction buffer (0.2 M Suc, 0.2 M KCl, 40 mM Tris-acetate, pH 8.0, 10 mM MgCl2, 10 mM 2-mercaptoethanol, 2% polyoxyethylene [10] tridecyl ether, 1% Triton X-100, 100 µg/mL chloramphenicol, and 100 µg/mL cycloheximide). A 0.6-mL aliquot was removed and flash frozen for subsequent total RNA isolation and quantification by microarray or slot-blot hybridization. The remainder of the suspension was filtered through glass wool and centrifuged for 10 min at 15,000g and 4°C in a JA-20 rotor (Beckman) to remove cell debris. The supernatant was transferred to a new tube, supplemented with 25 μL 1 M CaCl2 and 750 units of Micrococcal nuclease (Roche), and incubated on a rotator at 23°C for 1 h to degrade unprotected RNA. The mixture was layered onto a 2-mL Suc cushion (1 M Suc, 0.1 M KCl, 40 mM Tris-acetate, pH 8.0, 15 mM MgCl2, 5 mM 2-mercaptoethanol, 100 µg/mL chloramphenicol, and 100 µg/mL cycloheximide) and centrifuged for 3 h at 55,000g at 4°C in a 80 Ti rotor (Beckman). The pellet (monosomes and larger particles) was dissolved in 0.6 mL of RNA extraction buffer I (0.1 M EGTA, pH 8.0, 0.1 M NaCl, 1% SDS, 10 mM Tris, pH 8.0, and 1 mM EDTA). RNA was isolated with Tri reagent (Molecular Research Center) following the manufacturer’s instructions. Ribosome footprints were separated from bulk rRNA by gel purification: 30 µg of RNA from the monosome fraction was electrophoresed through a 12% denaturing polyacrylamide gel, gel slices harboring RNAs between ∼23 and 45 nucleotides were excised, and RNA was eluted by overnight incubation in RNA extraction buffer II (10 mM Tris, pH 8.0, 1 mM EDTA, 0.1 M NaCl, and 0.2% SDS) at 4°C on a rotator. The recovered RNA was extracted with phenol/chloroform and precipitated with ethanol, suspended in 100 µL water, and stored at −80°C until use.
For array hybridization, total leaf RNA (8 µg) was fragmented by incubation in fragmentation buffer [40 mM Tris-OAc, pH 8.3, 100 mM KOAc, and 30 mM Mg(OAc)2] for 8 min at 95°C. The reaction was stopped by adding EDTA, pH 8.0, to a final concentration of 50 mM. The fragmented RNA was extracted with phenol/chloroform and precipitated with ethanol, suspended in 20 µL water, and stored at −80°C until use.
RNA Labeling and Microarray Analysis
Two micrograms of the gel-purified ribosome footprint RNA or the fragmented total RNA was directly labeled with Cy5 (wild-type) or Cy3 (mutant) fluorescent dye using the ULS aRNA labeling kit (Kreatech Diagnostics) following the manufacturer’s instructions. After purification, the volume of each labeled RNA was reduced to 3 to 5 μL in a vacuum centrifuge, and the wild-type and mutant samples of ribosome footprint RNA or total RNA, respectively, were combined in 40 μL hybridization buffer (2.25 M NaCl, 15 mM EDTA, pH 8.0, 89 mM NaH2PO4, 61 mM Na2HPO4, 10% deionized formamide, 0.01 mg/mL acetylated BSA, and 0.01% Tween 20), heated to 70°C for 10 min, and hybridized to tiling microarrays. The microarrays were produced by Mycroarray in 6 × 7k format and consist of overlapping 50-mer oligonucleotide probes (20-nucleotide overlap) in triplicate spots covering all protein-coding regions and some UTRs of the maize chloroplast genome (National Center for Biotechnology Information reference: NC_001666.2). The probe positions and sequences are given in Supplemental Data Set 1 online. The hybridization was performed in an Agilent microarray hybridization chamber (Agilent Technologies) overnight on a rotator at 40°C. The arrays were washed at room temperature twice for 1 min in 1× SSPE (15 mM NaCl, 5.9 mM NaH2HPO4, 4.1 mM Na2HPO4, and 1 mM EDTA, pH 8.0) and once for 30 s in 0.5× SSPE while shaking. The stringency of hybridization and washing conditions was checked in pre-experiments by hybridization with prelabeled control oligonucleotides provided with the microarrays (Mycroarray). Arrays were dried by centrifugation in a plate centrifuge at 500 rpm for 5 min and scanned with a GenePix 4000B microarray scanner (Axon Instruments). Data were analyzed with GenePix Pro 6.0 software (Axon Instruments) using the default local background subtraction method. Probe spots with background subtracted signals ≤0 were rejected from the analysis. For the ribosome footprint data, the median of ratios (F635/F532) were combined for two biological replicate experiments as described previously (Schmitz-Linneweber et al., 2005). The average of the combined median of ratios (F635/F532) was normalized to 1. Only probes with at least three (of six) spots (ribosome profiles) or two (of three) spots (total RNA controls) that passed the background filter are presented in the figures. The median of the normalized values is shown for the plots of the ratio of the wild type to mutant signal (F635/F532; Figures 3, 6, and 7; see Supplemental Figures 1 and 2 and Supplemental Data Set 1 online). For plots of single channel data (F635 or F532), the signals from the two channels were normalized to the average signal across all array elements for each channel on all arrays (Figures 2, 6, and 7; see Supplemental Figures 1, 3, and 4 and Supplemental Data Set 1 online). This is intended to minimize differences in signal intensities between the channels caused by different labeling or detection efficiencies. Plots of signal and GC content document that there is no general correlation between signal intensity and GC content (see Supplemental Figures 1, 3, and 4 online).
Blot Hybridization Analysis
For slot-blot hybridizations, 300 ng of purified ribosome footprint RNA and 300 ng of total RNA from the same leaf extraction were heated for 10 min to 85°C in 150 μL 1× SSC (1× SSC is 0.15 M NaCl and 0.015 M sodium citrate), cooled on ice, and spotted onto a nylon membrane as described previously (Schmitz-Linneweber et al., 2005). [α-32P]dCTP–labeled PCR probes were hybridized to slot-blot membranes overnight at 47°C as described (Schmitz-Linneweber et al., 2005). Filters were washed twice for 5 min in 5× SSC at 47°C and exposed to phosphor screens, which were scanned with a Storm 825 scanner (GE Healthcare). Signals were quantified with Quantity One software (Bio-Rad) using the local background subtraction method.
RNA gel blot hybridizations were performed with total leaf RNA as described previously (Barkan, 1998). DNA probes were PCR amplified from plasmids containing the following maize chloroplast genome regions (positions according to National Center for Biotechnology Information annotation NC_001666.2): atpH 34387 to 34616, atpF exon 1 35089 to 35229, atpF exon 2 36121 to 36454, atpF exon 1/2 35095 to 35241 joined to 36072 to 36479, atpA 36690 to 37679, atpE 54222 to 54600, atpB 54665 to 55265, rbcL 57036 to 57607, psaJ 66496 to 66631, rps8 78584 to 78877, rpl14 79182 to 79487, rpl16 exon 2 79519 to 79920, rpl16 intron 79921 to 80936, and rps3 81200 to 81714.
Supplemental Data
The following materials are available in the online version of this article.
Supplemental Figure 1. Alternative Representations of Ribosome Profile Data and Comparison of Signal Intensities to GC Content.
Supplemental Figure 2. Additional Plots of Microarray Data for Total RNA, Translation Efficiencies, and Ribosome Profiles.
Supplemental Figure 3. Alternative Representations of Ribosome Profile Data from Figure 6, Highlighting the Reproducibility of Results from Unrelated Wild-Type Samples, and the Similarity of Results from the Nonallelic atpB Translation Mutants atp1 and atp4.
Supplemental Figure 4. Ribosome Profiling Data Suggesting Ribosome Pausing at Shine-Dalgarno–Like Sequences in the petA ORF.
Supplemental Table 1. Examples of Potential Translational Pausing Sites Containing Internal Shine-Dalgarno–Like Sequences.
Supplemental Data Set 1. Data Sets of Ribosome Profiling and Total RNA Control Experiments.
Acknowledgments
We thank Tiffany Kroeger, Nicholas Stiffler, Rosalind Williams-Carrier, and Susan Belcher for excellent technical support. This work was supported by a postdoctoral fellowship to R.Z. from the German Research Foundation (Grant ZO 302/1-1) and Grant IOS-0922560 to A.B. from the National Science Foundation.
AUTHOR CONTRIBUTIONS
R.Z., K.P.W., and A.B. designed the research. R.Z. and K.P.W. performed the research. R.Z. and A.B. analyzed the data. R.Z. and A.B. wrote the article.
Glossary
- ORF
open reading frame
- PPR
pentatricopeptide repeat
- UTR
untranslated region
References
- Barkan A. (1989). Tissue-dependent plastid RNA splicing in maize: Transcripts from four plastid genes are predominantly unspliced in leaf meristems and roots. Plant Cell 1: 437–445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barkan A. (1998). Approaches to investigating nuclear genes that function in chloroplast biogenesis in land plants. Methods Enzymol. 297: 38–57 [Google Scholar]
- Barkan A. (2011). Expression of plastid genes: Organelle-specific elaborations on a prokaryotic scaffold. Plant Physiol. 155: 1520–1532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barkan A., Walker M., Nolasco M., Johnson D. (1994). A nuclear mutation in maize blocks the processing and translation of several chloroplast mRNAs and provides evidence for the differential translation of alternative mRNA forms. EMBO J. 13: 3170–3181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brar G.A., Yassour M., Friedman N., Regev A., Ingolia N.T., Weissman J.S. (2012). High-resolution view of the yeast meiotic program revealed by ribosome profiling. Science 335: 552–557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choquet, Y., and Wollman, F.A. (2009). The CES process. In Chlamydomonas Source Book, E. Harris, D.B. Stern, and G. Whitman, eds (New York: Academic Press/Elsevier), pp. 1027–1064. [Google Scholar]
- de Longevialle A.F., Hendrickson L., Taylor N.L., Delannoy E., Lurin C., Badger M., Millar A.H., Small I. (2008). The pentatricopeptide repeat gene OTP51 with two LAGLIDADG motifs is required for the cis-splicing of plastid ycf3 intron 2 in Arabidopsis thaliana. Plant J. 56: 157–168 [DOI] [PubMed] [Google Scholar]
- Drapier D., Rimbault B., Vallon O., Wollman F.A., Choquet Y. (2007). Intertwined translational regulations set uneven stoichiometry of chloroplast ATP synthase subunits. EMBO J. 26: 3581–3591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drechsel O., Bock R. (2011). Selection of Shine-Dalgarno sequences in plastids. Nucleic Acids Res. 39: 1427–1438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisk D.G., Walker M.B., Barkan A. (1999). Molecular cloning of the maize gene crp1 reveals similarity between regulators of mitochondrial and chloroplast gene expression. EMBO J. 18: 2621–2630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gatenby A.A., Rothstein S.J., Nomura M. (1989). Translational coupling of the maize chloroplast atpB and atpE genes. Proc. Natl. Acad. Sci. USA 86: 4066–4070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirose T., Sugiura M. (2004). Functional Shine-Dalgarno-like sequences for translational initiation of chloroplast mRNAs. Plant Cell Physiol. 45: 114–117 [DOI] [PubMed] [Google Scholar]
- Ingolia N.T., Brar G.A., Rouskin S., McGeachy A.M., Weissman J.S. (2012). The ribosome profiling strategy for monitoring translation in vivo by deep sequencing of ribosome-protected mRNA fragments. Nat. Protoc. 7: 1534–1550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingolia N.T., Ghaemmaghami S., Newman J.R., Weissman J.S. (2009). Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science 324: 218–223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingolia N.T., Lareau L.F., Weissman J.S. (2011). Ribosome profiling of mouse embryonic stem cells reveals the complexity and dynamics of mammalian proteomes. Cell 147: 789–802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson, R.J., Kaminski, A., and Pöyry, T.A.A. (2007). Coupled termination-reinitiation events in mRNA translation. In Translational Control in Biology and Medicine, M.B. Mathews, N. Sonenberg, and J.W.B. Hershey, eds (New York: Cold Spring Harbor Laboratory Press), pp. 197–223. [Google Scholar]
- Jenkins B.D., Kulhanek D.J., Barkan A. (1997). Nuclear mutations that block group II RNA splicing in maize chloroplasts reveal several intron classes with distinct requirements for splicing factors. Plant Cell 9: 283–296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J., Klein P.G., Mullet J.E. (1991). Ribosomes pause at specific sites during synthesis of membrane-bound chloroplast reaction center protein D1. J. Biol. Chem. 266: 14931–14938 [PubMed] [Google Scholar]
- Lee S., Liu B., Lee S., Huang S.X., Shen B., Qian S.B. (2012). Global mapping of translation initiation sites in mammalian cells at single-nucleotide resolution. Proc. Natl. Acad. Sci. USA 109: E2424–E2432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li G.W., Oh E., Weissman J.S. (2012). The anti-Shine-Dalgarno sequence drives translational pausing and codon choice in bacteria. Nature 484: 538–541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohse M., Drechsel O., Bock R. (2007). OrganellarGenomeDRAW (OGDRAW): A tool for the easy generation of high-quality custom graphical maps of plastid and mitochondrial genomes. Curr. Genet. 52: 267–274 [DOI] [PubMed] [Google Scholar]
- Martin W., Koonin E.V. (2006). Introns and the origin of nucleus-cytosol compartmentalization. Nature 440: 41–45 [DOI] [PubMed] [Google Scholar]
- McCormac D.J., Barkan A. (1999). A nuclear gene in maize required for the translation of the chloroplast atpB/E mRNA. Plant Cell 11: 1709–1716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfalz J., Bayraktar O.A., Prikryl J., Barkan A. (2009). Site-specific binding of a PPR protein defines and stabilizes 5′ and 3′ mRNA termini in chloroplasts. EMBO J. 28: 2042–2052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prikryl J., Rojas M., Schuster G., Barkan A. (2011). Mechanism of RNA stabilization and translational activation by a pentatricopeptide repeat protein. Proc. Natl. Acad. Sci. USA 108: 415–420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitz-Linneweber C., Williams-Carrier R., Barkan A. (2005). RNA immunoprecipitation and microarray analysis show a chloroplast pentatricopeptide repeat protein to be associated with the 5′ region of mRNAs whose translation it activates. Plant Cell 17: 2791–2804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz Z., Kossel H. (1980). The primary structure of 16S rDNA from Zea mays chloroplast is homologous to E. coli 16S rRNA. Nature 283: 739–742 [Google Scholar]
- Shinozaki K., et al. (1986). The complete nucleotide sequence of the tobacco chloroplast genome: Its gene organization and expression. EMBO J. 5: 2043–2049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stampacchia O., Girard-Bascou J., Zanasco J.L., Zerges W., Bennoun P., Rochaix J.D. (1997). A nuclear-encoded function essential for translation of the chloroplast psaB mRNA in Chlamydomonas. Plant Cell 9: 773–782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stollar N.E., Kim J.K., Hollingsworth M.J. (1994). Ribosomes pause during the expression of the large ATP synthase gene cluster in spinach chloroplasts. Plant Physiol. 105: 1167–1177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki H., Kuroda H., Yukawa Y., Sugiura M. (2011). The downstream atpE cistron is efficiently translated via its own cis-element in partially overlapping atpB-atpE dicistronic mRNAs in chloroplasts. Nucleic Acids Res. 39: 9405–9412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhelyazkova P., Hammani K., Rojas M., Voelker R., Vargas-Suárez M., Börner T., Barkan A. (2012). Protein-mediated protection as the predominant mechanism for defining processed mRNA termini in land plant chloroplasts. Nucleic Acids Res. 40: 3092–3105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoschke R., Kroeger T., Belcher S., Schöttler M.A., Barkan A., Schmitz-Linneweber C. (2012). The pentatricopeptide repeat-SMR protein ATP4 promotes translation of the chloroplast atpB/E mRNA. Plant J. 72: 547–558 [DOI] [PubMed] [Google Scholar]