

Abstract
All newly synthesized proteins are subject to quality control check-points, which prevent aberrant polypeptides from harming the cell. For proteins that ultimately reside in the cytoplasm, components that also reside in the cytoplasm were known for many years to mediate quality control. Early biochemical and genetic data indicated that misfolded proteins were selected by molecular chaperones and then targeted to the proteasome (in eukaryotes) or to proteasome-like particles (in bacteria) for degradation. What was less clear was how secreted and integral membrane proteins, which in eukaryotes enter the endoplasmic reticulum (ER), were subject to quality control decisions. In this review, we highlight early studies that ultimately led to the discovery that secreted and integral membrane proteins also utilize several components that constitute the cytoplasmic quality machinery. This component of the cellular quality control pathway is known as ER associated degradation, or ERAD.
1. Introduction
Every newly synthesized protein must transition from a nascent polypeptide chain, which lacks significant secondary structure, into its folded and stable conformation. During this process even relatively simple proteins are susceptible to inappropriate interactions with the complex mixture of macromolecules inside the cell. In turn, larger and more complex proteins usually possess multiple domains that need to fold independently before the final tertiary structure is attained, and proteins in multi-subunit complexes must identify their partners. To assist in this process the cell has evolved a complex and diverse set of molecular chaperones. Molecular chaperones most commonly interact with exposed, hydrophobic portions in soluble polypeptide chains, thus preventing protein aggregation [1–4]. Some molecular chaperones may even provide an energetically favorable folding environment. Other members of this protein family directly catalyze chemical reactions, such as disulfide bond formation or proline isomerization, which directly facilitates protein folding.
The endoplasmic reticulum (ER) is a major folding organelle in the cell. All proteins destined to reside in membranes or to be secreted pass through this organelle, leading to a high concentration of different folding intermediates in various stages of completion [1, 2]. The folding of integral membrane proteins is challenging, as hydrophobic domains must be inserted into a lipid environment in their proper topological orientation, while soluble portions of the protein must fold in solution. Initial steps during the folding pathway take place co-translationally, but in at least some cases the final conformation can be attained only once translation is complete [5]. As might be anticipated, for most multi- spanning membrane proteins this process is quite inefficient and even wild type proteins under ideal conditions undergo many folding problems. Indeed, a growing body of evidence indicates that only a fraction of the total population of many membrane proteins reach maturation and are stable [6–9].
Because a significant population of proteins misfold or become damaged after synthesis—and because these aberrant species might exhibit toxic effects on cellular health—all cells possess a quality control system that identifies and degrades polypeptides that fail to mature [10, 11]. The two major pathways for protein disposal in eukaryotes are the lysosome and the cytoplasmic proteasome, and the existence and importance of these pathways have long been appreciated. It is also clear that other organelles, such as the mitochondria, the ER, and the Golgi apparatus, harbor specific proteases. Because the ER and Golgi represent compartments through which proteins travel en route to the plasma membrane or to the extracellular space, the proteases in these organelles are primarily devoted to maturation events that generate active forms of proteins that pass through the secretory pathway, but these proteases may not be suitable to mediate quality control.
As secreted and integral membrane proteins are synthesized, an embedded hydrophobic signal sequence delivers the nascent chain-ribosome complex to the ER membrane, an event that requires the function of the signal recognition particle (SRP) and an ER associated SRP receptor [12, 13]. At the ER, ongoing translation is coupled to import, or translocation, of the nascent protein into the ER membrane (if the protein is an integral membrane protein) or into the ER lumen (if the protein is soluble). Approximately one-third of all eukaryotic proteins follow this path [14], which places a significant burden on the ER to ensure that these proteins fold efficiently. But, as noted above, protein folding is inefficient, and this event is magnified when one considers that secreted proteins may possess genetic mutations and that cellular stresses, which are quite commonly encountered, may overwhelm the ability of molecular chaperones to function efficiently. Consequently, for many years, it was assumed that a protein quality control system existed within the ER that would identify and degrade misfolded proteins. An efficient ER quality control machine is vital as the cargo that passes through this compartment includes essential plasma membrane receptors, secreted signaling molecules and regulatory particles that control trafficking and delivery to organelles within the cell. In fact, the quality control machinery is so robust that slowly folding, wild type proteins can be degraded [2, 5, 15]. From an evolutionary perspective, it is better for the cell to remove potentially toxic protein than to risk the threat posed by these species in order to save energy and increase the efficiency of protein production.
For many years, significant efforts were directed toward identifying the resident machinery and protease(s) that mediate protein quality control in the ER. In this review, we highlight the conclusions obtained from a select number of these important papers. In the end, however, it was clear that proteins that failed to pass ER quality control were not exclusively selected by ER resident factors, and that degradation occurred— surprisingly—in the cytoplasm and was mediated by the proteasome. This pathway was then referred to as ER Associated Degradation (ERAD) [16]. Before we survey early work in this field, we will first discuss general features underlying the selection and degradation of ERAD substrates.
2. Basic Features Underlying ER Associated Degradation
The elimination of misfolded proteins from the ER is a complex multi-step process that involves the coordination of many proteins in both the ER and the cytoplasm [17–21]. To enter the ERAD pathway a protein must first be recognized. It must then be targeted for transport across or from the ER membrane. As it is dislocated from the ER membrane, the ERAD substrate interacts with a ubiquitin ligase and becomes polyubiquitinated. The substrate must then be delivered to the cytoplasmic proteasome, de-ubiquitinated, and finally fed into the proteasome for destruction.
The first step during ER quality control is substrate recognition. Recognition is most commonly mediated by molecular chaperones, which interact with a hydrophobic region that should normally be buried within the protein, is involved in an interaction with another subunit of a multi-protein complex, or is embedded within the lipid bilayer. Members of the Hsp70 chaperone family and Hsp40 co-chaperones are most commonly employed for ERAD substrate selection, although Hsp90 and the lumenal protein disulfide isomerase (PDI) also engage some ERAD substrates and seem to be involved in quality control decisions (see for example [22–25]). It is still unclear if Hsp90 binding can mediate degradation or if futile Hsp90 cycling ultimately leads to Hsp70 directed ERAD. Depending on the topology of the protein, i.e., whether it is a soluble substrate within the ER or is a membrane protein that exposes domains in both the ER lumen and in the cytoplasm (Fig. 1, A–B), cytoplasmic and/or ER lumenal Hsp70s and Hsp40s may be involved in substrate recognition. The central Hsp70 of the ER lumen, BiP, demonstrates the dual role of this class of chaperones. BiP is involved in the maturation and folding of proteins as well as in sensing chronically misfolded protein and targeting them for degradation [26]. Similarly, proper formation of disulfide bonds is integral to the correct folding of many secreted proteins and as such PDIs can be an important part of the quality control machinery [1, 27]. In addition, there is evidence that PDIs possess a chaperone function independent from their role in disulfide bond isomerization to facilitate ERAD [24, 28]
Figure 1.
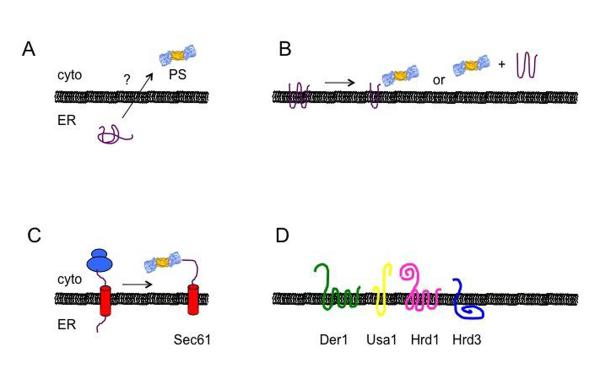
A. Soluble ERAD substrates must retrotranslocate across the ER bilayer in order to access the cytoplasmic proteasome (PS). B. Integral membrane proteins present domains that can directly access the proteasome, but membrane spanning segments must still be threaded from the lipid bilayer, regardless of whether degradation occurs at the membrane or in solution. C. The Sec61 translocation channel might be used for both protein translocation (left) and retrotranslocation (right). D. The components of the Hrd1 complex in yeast include Der1, Usa1 (which scaffolds and regulates Hrd1 oligomerization and function) and Hrd3, which serves as a dock for lectins and chaperones. The mammalian homolog of Hrd3 is SEL1, and Usa1 and Herp may function analogously. The Hrd1 complex also associates with Cdc48 (not shown), which extracts ERAD substrates. Also not shown is Ubx2, which associates with Hrd1 complex and helps anchor Cdc48 to the ER. In parts A–C, ERAD substrates are shown in purple. (See text for additional details and [39, 40, 180–182]).
Another form of substrate recognition occurs based on the state of the appended glycan chain on proteins within the ER [29, 30]. Most proteins that enter the secretory pathway (and that possess the Asn-X-Ser/Thr consensus sequence) are modified by the addition of a core oligosaccharide. The addition of sugar residues conveys information about the folding state of the protein to the ER quality control machinery [31, 32]. In the ER, three terminal glucose residues are then sequentially removed from the added oligosaccharide. If the protein fails to fold, an oligosaccharide transferase, UDP–glucose glycoprotein glucosyltransferase (UGGT), adds a glucose residue back onto the substrate by virtue of the fact that this enzyme also exhibits chaperone-like properties. Glucose re-addition by UGGT favors substrate binding to the lectin-like chaperones calnexin and calreticulin, which along with associated partners favor protein refolding [33, 34]. But if refolding fails to occur within a reasonable time frame, the substrate interacts with another lectin-like chaperone, the ER degradation enhancing α-mannosidase-like protein, EDEM1 [35].
EDEM1 functions with an ER Hsp40 homolog that also possesses disulfide reductase activity to target select, soluble ERAD substrates for retrotranslocation [36].
After the decision to degrade a protein has been made the protein must be transported out of the ER and into the cytoplasm (if it is soluble; Fig. 1A), or extracted from the ER membrane (if it is an integral membrane protein; Fig. 1B). This event has been referred to as retrotranslocation or dislocation. The retrotranslocation channel through which proteins move from the ER and into the cytoplasm is a focus of intense investigation, but there is still no clear consensus on which or even if a single protein constitutes this channel. Nevertheless, one retrotranslocation channel candidate is Sec61 [37], which is also used for protein translocation into the ER (Fig. 1C). Another candidate is the Hrd1 complex, which consists of several associated proteins in the ER membrane [38, 39](Fig. 1D). Notably, Hrd1 can be crosslinked to a soluble polypeptide as it is extracted from the ER, and also exhibits ubiquitin ligase activity [40], which is essential for the proteasome-mediated degradation of most ERAD substrates (also see below). What is clear is that the retrotranslocation of nearly all ERAD substrates requires the activity of the Cdc48/p97 complex [41–43]. Cdc48 is a hexameric AAA ATPase protein that together with its associated cofactors [44] is anchored to the ER membrane and interacts with ubiquitinated substrates. Sequential rounds of ATP binding and hydrolysis are then used to remove both soluble and lumenal proteins from the ER. Cdc48 associates with a series of cofactors that subsequently facilitate the transfer of ubiquitinated substrates to the proteasome [45], and also associates with the proteasome itself [46].
As noted above, protein ubiquitination is almost always required for proteasome degradation, and not surprisingly a key event during the delivery of an ERAD substrate for degradation is ubiquitination [47, 48]. Protein ubiquitination employs a series of enzymes; an E1 ubiquitin activating enzyme, E2 ubiquitin conjugating enzymes, and E3 ubiquitin ligases [49]. E3 ligases catalyze the covalent attachment of ubiquitin onto the substrate protein, either transferring ubiquitin directly itself or by bringing the substrate to an E2 conjugating enzyme. The E3 ubiquitin ligases are an abundant class of proteins and are thought provide diversity in substrate recognition, and over the years a subgroup of cellular E3s have been shown to play an important role during ERAD.
Together, ER protein folding and quality control are essential processes that maintain cellular homeostasis. Not surprisingly the ERAD pathway has been linked to a growing number of human diseases [50]. While the specifics and dynamics of substrate recognition and degradation for many proteins are still being established at the molecular level, the basic machinery required for ERAD is now relatively well-defined, especially in yeast [19, 51, 52]. This is a notable achievement, given that only ~20 years ago the existence of the ERAD pathway was unknown. However, there were many early hints that ERAD existed, and in the following sections we trace the scientific developments that ultimately led to our current view of the ERAD pathway.
3. The Early History of the ERAD Pathway
Throughout most of the 1980s it was assumed that damaged or unneeded secretory proteins were disposed of exclusively by the lysosomal pathway. The lysosome also serves as a site for large-scale disposal of organelles and recycling of macromolecules in the process known as autophagy [53]. Lysosomes form from Golgi-derived vesicles and resident lysosomal proteases traffic through the secretory pathway and are ultimately delivered to the lysosome [54, 55]. It was thought that the disposal of secretory proteins proceeded by a similar route, and indeed the lysosome is the site of degradation for proteins that fail Golgi or plasma membrane quality control check-points [56].
A large number of cytoplasmic proteases also exist in the cell [57], and the most prominent one is the proteasome. The 26S proteasome is a large (~2.5 MDa) multicatalytic protease that is primarily cytoplasmic, although a significant proteasome population exists in the nucleus and—intriguingly—is associated with the ER membrane [58, 59]. Indiscriminant proteolysis is prevented by the preferential selection of polyubiquitinated substrates. The proteasome cap, or the 19S particle (also known as PA700) harbors ubiquitin receptors, deubiquitinating enzymes, and a series of AAA ATPases that unfold and deliver substrates into the 20S proteasome core [60]. In turn, the 20S core is a barrel-shaped chamber with two-fold symmetry that houses duplicate copies of 3 enzymes with unique proteolytic activities. Because the proteasome and cytoplasmic proteases were only considered to target cytoplasmic substrates, there was no reason to believe that these components would contribute to events underlying ER quality control, especially given the lysosome's central position as a component of the secretory pathway.
3.1 T cell antigen receptor
The first hints that a quality control mechanism was located in the early secretory pathway came from studies on the maturation of the T cell antigen receptor (TCR). TCR is located on the plasma membrane of T lymphocytes and is responsible for the recognition of antigens presented by the major histocompatibility complex (MHC). TCR is a heteromeric complex composed of several polypeptide chains [61]. The alpha and beta chains form a disulfide linked heterodimer that act as the antigen binding region. Three additional peptide chains (delta, gamma and epsilon) and a homodimer of two zeta chains come together to form the functional receptor complex. Maintenance of TCR at the surface of T cells is critical for immune system function.
Like all plasma membrane proteins, TCR subunits are directed to the ER cotranslationally and are inserted into the ER membrane. Only completely assembled complexes reach the cell surface [62] while partially assembled complexes are destroyed [63]. These observations hinted that the secretory pathway contains a protein quality control check-point that selectively recognizes and mediates the destruction of non-functional TCR complexes before they traffic to later steps of the secretory pathway.
In an important early study, the mechanism of how the cell degrades unassembled TCR subunits was investigated. Specifically, the fate of individual chains of the TCR complex was followed in both T cell hybridoma lines and fibroblasts [64]. The authors discovered that two distinct pathways ensured that only the properly formed complex reached the plasma membrane. In T cells, individual alpha chains were processed through the Golgi and subsequently degraded via the lysosome. However, when expressed in fibroblasts both the alpha chains and alpha-beta complexes were degraded. Degradation was unaffected by inhibitors of lysosomal proteolysis, indicating a separate disposal route in fibroblasts. The existence of this second pathway was confirmed when treatment of T cells with compounds that blocked ER to Golgi transport now resulted in the degradation of alpha, beta and delta chains through a lysosome-independent mechanism. These studies were consistent with other reports that hinted that the degradation of delta chain took place early in the secretory pathway [65] via a process then referred to as pre-Golgi or ER degradation. Nevertheless, direct evidence that degradation occurred exclusively in the ER or associated with the ER was lacking [66, 67].
Because vesicular traffic is difficult to control and measure in intact cells, permeabilized cell systems were next employed. Permeabilized cells still support the folding and assembly of proteins and can even carry out the redox dependent degradation of a secreted protein [68]. This system also allowed for the add-back of regulatory molecules and specific proteins. Importantly, permeabilization destroys vesicular traffic, so that ER-specific events can be monitored.
To definitively establish that ER export was dispensable for the degradation of unassembled members of the TCR, a chimeric tac-TCR alpha subunit was expressed and localized to the ER as assessed by immunofluorescence microscopy [69]. After treatment with cycloheximide to inhibit new protein synthesis, the fluorescent signal was rapidly lost without any appearance outside of the ER. After the plasma membrane was permeabilized with a pore forming toxin, SLO, a treatment previously shown to abolish export from the ER [70], Tac-TCR alpha subunit degradation was unaffected. These data were recapitulated with a second chimeric substrate, Tac-TCR beta. Therefore, vesicular transport from the ER was dispensable for the degradation of the unassembled TCR subunits.
In addition to TCR, many other cell surface receptors are multi-subunit complexes and the assembly of these complexes occurs in the ER [71]. Other early evidence indicated that the ER could expand if unfolded proteins accumulated, and that misfolded proteins associated with an Hsp70 in the ER, BiP [72–78]. Thus, the ER was now implicated as a potential site of general secretory protein quality control, and a factor that might mediate this event had been identified. These collective data provided some of the first and best previews of the ERAD pathway.
3.2 Human asialoglycoprotein receptor
Studies on another plasma membrane protein provided strong supporting evidence of ER quality control and explored how substrate degradation might proceed. The human asiaglycoprotein receptor (AGPR) is found in liver cells and binds and removes specific glycoproteins from circulation. AGPR activity is closely tied to hepatic function and decreased AGPR is a characteristic marker of a variety of liver disorders [79]. AGPR forms a hetero-oligomer and both subunits are glycosylated integral membrane proteins. The functional receptor is thought to be a trimer of two H1 and one H2 subunits [80, 81]. Similar to TCR, when expressed together the subunits were transported to cell surface but when expressed individually they were rapidly degraded [82, 83]. Degradation was insensitive to lysosomal protease inhibitors, and based on the glycosylated state of the orphaned AGPR subunits it appeared that they never reached the Golgi. Moreover, the subunits appeared to be cleaved. Proteolysis occurred in the ER and could be reconstituted in vitro. Interestingly, a 35 kDa cleaved fragment was stable but it was proteolyzed further in viable cells. The results from subsequent studies indicated that cleavage occurred in the ER and that complete degradation was robust in the absence of vesicular transport from the ER to the Golgi [84]. Thus, the fragment was degraded either in the ER or in an ER associated manner. Based on our current knowledge, one may speculate that the proteasome or another protease might have clipped the substrate, but that ongoing proteasomal degradation required high levels of ATP and/or proteasome integrity, which may have been deficient in vitro.
3.3 Ribophorin I
Concurrent with studies on TCR and AGPR, another integral membrane protein, the transmembrane glycoprotein Ribophorin I, was examined as a substrate for ER quality control. Unlike these other substrates, Ribophorin I is an ER resident protein that exhibits ribosome binding properties and facilitates delivery of substrates to the glycosylation machinery in the ER [85, 86]. The Kreibich lab discovered that C-terminally truncated versions of Ribophorin I were unstable, in contrast to the full-length protein, and degradation occurred in a non-lysosomal manner [87]. Interestingly, the two truncated Ribophorins examined in this study differed: One had a membrane anchor while the other was a soluble ER lumenal protein that lacked the membrane anchor. It appeared that the lumenal form of the protein had entered a pre-Golgi compartment and underwent a system of degradation that was biphasic. The authors suggested that degradation commenced in the ER but continued in this second compartment. Consistent with this model, treatment with the ionophores monensin and CCCP, which inhibit vesicular transport out of the ER [88–90], blocked the second degradation phase if performed at the beginning but not later after transport had occurred. These results suggested that transport between the compartments was required for the second degradation phase.
Another observation from this study was a demonstrated requirement for high calcium concentrations to support degradation. This result suggested that the calcium-rich environment within the ER is important for quality control. We now appreciate that important mediators of ERAD, such as BiP, calnexin, and calreticulin are calcium binding proteins. Depletion of calcium also induces the unfolded protein response [91] [92], which alters the expression of ER chaperones and other components and might lead to complex effects on protein degradation.
3.4 MHC I and Human Cytomegalovirus
An important source of insight into the ERAD pathway was obtained from a series of studies of another cell membrane receptor, this time the major histocompatability complex class I (MHC I). MHC I is found on the surface of all nucleated cells and is an important part of the immune system. MHC I binds to short peptide sequences generated inside the cell via the proteasome and presents them at the cell surface. These peptide antigens can be derived from breakdown of endogenous proteins, or in the case of an infected cell, from foreign proteins expressed by an invading organism [93, 94]. These MHC I antigen complexes are monitored by cytotoxic T cells of the host organism and recognition of foreign peptides trigger an immune response to eliminate the infected cell [95].
Over the course of evolution a number of viruses have developed ways to evade detection and the MHC I complex is one target as the virus attempts to dampen the immune response [96]. One virus known to target MHC I is the human cytomegalovirus (HCMV). Previous studies had shown that cells infected with HCMV had downregulated levels of MHC I protein [97]. A follow up study demonstrated that the US11 viral gene product could similarly downregulate MHC I when transfected into human cells [98]. In a seminal study the investigation into US11 revealed the mechanism by which it targeted MHC I [99]. Specifically, in the presence of US11, the heavy chain of the MHC I complex was rapidly degraded while in control cells the heavy chain was stable. The MHC I heavy chains failed to reach the Golgi and appeared to be degraded before leaving the ER, as chemically blocking ER to Golgi export did not stabilize the heavy chains. In contrast, the MHC I light chains were stable and in fact were secreted from the cell in increased amounts without its heavy chain partner, indicating that US11 specifically acted on the fate of the heavy chain. Even more surprising was that upon addition of inhibitors of the proteasome, degradation intermediates became detectable indicating the possibility that heavy chain breakdown occurred in the cytoplasm, even though US11 resided within the ER. Moreover, glycans were removed from heavy chain intermediates in US11 transfected cells, demonstrating that US11 triggered MHC I heavy chain deglycosylation, and dislocation out of the ER. Fractionation of cellular components confirmed this observation as US11 remained associated with ER microsomes, and heavy chain degradation intermediates were found in cytosolic fractions only in US11 transfected cells. Thus, US11 had downregulated surface expression of MHC I by turning the heavy chains into ERAD substrates. The authors of this study made the very insightful commentary that the pathway to destroy MHC I heavy chains may have been simply co-opted by the virus and that it existed as a more general pathway to dispose of unwanted membrane proteins. Follow up studies characterized another HCMV gene that also targeted MHC I heavy chains [100] and we now know that co-opting of the ER quality control machinery is a common mechanism of viral attempts to evade host cell immune response [101].
3.5 HMG-CoA reductase
Lipid synthesis takes place primarily in the ER. The synthesis of one lipid class, the sterols, is also primarily ER-localized and the rate-determining step in this anabolic pathway is catalyzed by HMG-CoA reductase (HMGR) [102]. The product of HMGR is mevalonate. HMGR is an ER integral membrane protein with 8 membrane spanning domains, and its activity was known to be down regulated by degradation in response to metabolic signals from sterol biosynthetic intermediates, such as mevalonate [103, 104].
HMGR degradation can take place in permeabilized cells [105], and to investigate this process further a chimeric protein was created with the HMGR membrane domains fused to beta-galactoside; the resulting protein resided in the ER [106]. Pulse chase studies in transfected cells demonstrated that the fusion protein remained mevalonate-responsive. The cells were then permeabilized with digitonin, a reagent that permeabilizes the plasma membrane by virtue of its interaction with cholesterol but leaves the ER membrane intact [107, 108]. The enzyme in the permeabilized system remained mevalonate responsive, indicating that degradation occurred in the absence of vesicular traffic from the ER. This result was consistent with an earlier study showing that HMGR degradation was unaffected by Brefeldin A treatment, which blocks export from the ER [109]. While these results indicated that HMGR degradation was ER-associated, there was an important hint that other components might be involved in triggering degradation. For example, the authors observed that metabolic induction of HMGR degradation was only seen when mevalonate treatment was performed before cell permeabilization, indicating that the degradation signal had to be set in motion before the loss of cytosolic components. The cytosolic factors remain unknown but the requirement for mysterious, soluble factors in these systems became a common theme in related, subsequent studies. In retrospect, these factors might have been proteins that support maximal substrate ubiquitination, or proteasome associated components or chaperones that aid in efficient substrate-targeting to the proteasome.
Another observation in this study [106] was that the degradation of HMGR seemed to be ATP-independent, which was inconsistent with previous studies [105]. In this case the lack of an ATP requirement could be connected to the observation that mevalonate treatment only led to degradation if added before membrane permeabilization. Thus, it is possible that the signal to degrade HMGR had already been implemented, the substrate had already been ubiquitinated, and the proteasome had already been recruited to HMGR at the ER membrane. Moreover, as observed for Ribophorin I (see above), the melvonate-triggered degradation of HMGR was highly dependent the concentration of intercellular calcium [110]. HMGR degradation was also reported to be inhibited by a “cysteine protease inhibitor”, ALLN (N-acetyl-leucyl-leucyl-norleucinal) [111], which also inhibits the proteasome. Overall, these pioneering studies on HMGR indicated that the ERAD pathway does not simply degrade misfolded or damaged proteins, but can be employed to regulate essential cellular processes. Ultimately, however, the first, definitive evidence that HMGR degradation occurs via the ERAD pathway emerged from pioneering genetic studies performed in yeast by Hampton and colleagues [112] and later in mammalian cells by a variety of investigators [113]
3.6 Cytochrome P450
The selectivity underlying ER quality control decisions became quite evident when the fates of different isoforms of the rat liver cytochrome P450 were examined. The ethanol inducible CYP2E1 is degraded in hepatocyctes with a half-life of ~9 hours whereas the CYP2B1 isozyme is longer lived with a half-life of ~21 hours. The two proteins were subsequently found to be degraded by completely different mechanisms. The longer lived 2B1 isoform was degraded by the lysosome [114, 115]. However, the disappearance of the more rapidly degraded 2E1 isoform was unaltered by treatment with a variety of lysosomal protease inhibitors [116]. Moreover, the inclusion of imidazole, a 2E1 substrate, stabilized the protein while the addition of glucagon led to phosphorylation of 2E1 and increased degradation. In contrast, the lysosome-targeted 2B1 protein was not subject to regulation by imidazole or glucagon. These results indicated that the 2E1 form of the cytochrome underwent non-lysosomal degradation that was highly regulated. The data also supported the notion that the conformational state of the protein might confer recognition by the degradation machinery, as substrate binding decreased and hormone-induced phosphorylation increased its degradation.
3.7 Expression of yeast prepro-alpha factor in mammalian cells
Many secreted proteins are zymogens or pro-hormones that must be processed. To develop a new model in which the link between hormone processing and quality control might be examined, yeast mating hormone precursor, prepro-alpha factor, was expressed in rat pituitary cells [117]. In yeast, the prepro-alpha factor signal sequence is cleaved in the ER, generating pro-alpha factor, which is then triply glycosylated and exported to the Golgi where the pro domain is removed and the core oligosaccharide is elaborated. In pituitary cells the protein entered the ER and underwent initial processing and core glycosylation, as in yeast. However, glycosylated pro-alpha factor was rapidly degraded and no mature product was detected. The authors found that chemical inhibition of ER mannosidases led to almost complete stabilization of the core glycosylated pro-alpha factor whereas inhibition of ER glucosidases accelerated the already rapid degradation of this protein; the use of these chemical reagents—which proved essential for this study—was pioneered by the Helenius laboratory, who had been concurrently been examining how viral proteins are subject to ER quality control [118, 119]. Based on these results, the authors speculated that glycosylated pro-alpha factor was subject to quality control because glycan processing differs between yeast and man. Instead, the data are more consistent with the current view of how ERAD substrates are selected: As described above, inhibition of the glucosidases prevents interaction with calnexin and calreticulin, which only bind mono-glucosylated substrates and facilitate folding, whereas inhibition of mannosidase activity blocks delivery via EDEM1 for retrotranslocation. Nevertheless, this study provided evidence that post-translational modifications may serve as a signal to regulate protein degradation in the early secretory pathway.
3.8 Peptide substrates
For many years, protein transport across the ER membrane was thought to be a one-way street; i.e., proteins were translocated into the ER but could only leave once they were packaged into vesicle carriers. The first hint that proteins—or formally, a peptide—could retrotranslocate was provided by examining the fate of a synthetic peptide containing a three residue amino acid sequence (an Asn-Tyr-Thr tripeptide) that acts as an acceptor site for N-linked glycosylation. This tripeptide had been used as a tool to monitor transport through the secretory pathway in mammalian cells [120], and at about the same time an in vitro system had been established to reconstitute ER to Golgi protein transport using yeast components [121]. When the transport of the peptide and pro-alpha factor in a modified version of this in vitro system was examined, an important discovery was made: The protein and tri-peptide both exited the ER, but they did so by different mechanisms [122]. Based on sedimentation analysis pro-alpha factor was membrane associated whereas the peptide was freed from the membrane pool. Also, antibodies that compromise ER vesicle release blocked pro-alpha factor release, but did not prevent peptide export, and the peptide possessed core oligosaccaharides conjugated in the ER but lacked Golgi-specific modifications. Finally, peptide export required cytosol, ATP, and physiological temperatures. The authors recognized that the peptide might be employed as a tool to characterize this novel mechanism of peptide export from the ER, and in fact had serendipitously identified one of the key steps during ERAD, the retrotranslocation of substrates from the ER. Subsequent studies would establish that this process is mediated by proteinaceous factors on the cytoplasmic face of the ER [123], and that the peptide exporting activity is conserved [124].
3.9 Alpha-1 antitrypsin
One of the first hints that ER protein degradation might be directly connected to the pathology underlying a human disease was obtained from studies on alpha-1 antitrypsin (AAT). AAT is a protein produced and secreted by hepatocytes and is the second most abundant serum protein. In the serum AAT acts an inhibitor of neutrophil elastase [125]. AAT also provides a vital function in the lung, where it keeps the levels of elastase in check. In fact, AAT deficiency leads to a chronic breakdown of lung tissue and emphysema.
A common disease-causing AAT mutation is known as the Z variant. The AAT-Z mutant protein is inefficiently secreted and can accumulate in the ER [126–128]. The misfolded, ER-retained AAT-Z species can form insoluble aggregates, which in some cases results in liver disease, but it was also noted that a significant portion of the protein was degraded in a pre-Golgi compartment [129, 130]. Therefore, individuals who exclusively express AAT-Z have compromised lung function. In later years, AAT-Z was analyzed as one of the first soluble ER proteins found to be retrotranslocated and degraded by the proteasome, an event that could be recapitulated in both a yeast AAT-Z expression system and in transfected mammalian cells [16, 131]. Since then, AAT-Z and another mutant form of AAT, known as null Hong Kong (NHK), have been heavily utilized as model ERAD substrates, and much of what is now understood about the quality control of glycosylated proteins in the ER has been derived from the use of the NHK variant. It should also be noted that AAT-Z is a substrate that can be disposed of by multiple degradation pathways [132]. While AAT-Z was one of the model substrates to first demonstrate the existence of ERAD, it has also clearly been shown that AAT-Z enters the autophagy pathway and is degraded in the lysosome [133, 134]. The pathway chosen—ERAD versus autophagy—seems to be related to the extent of misfolding, with more soluble forms being disposed of by ERAD and more aggregated and insoluble AAT-Z being routed to autophagy [135].
3.10 Immunoglobulin light chain
Another soluble secreted protein that can be targeted for degradation in the ER is the unassembled immunoglobulin light chain. During normal B lymphocyte differentiation there is an asynchronous expression of heavy and light immunoglobulin subunits. Excess light chains relative to the partner heavy chains are usually secreted without assembly [136]. However the sequence of light chain varies naturally to accommodate a vast immune repertoire. Some of these sequence changes result in folding conformations that can destabilize the light chain structure and even lead to aggregation [137, 138]. A number of malignant cell lines were isolated that had lost expression of heavy chain but light chain expression persisted. One such cell line is the murine lymphoma CH12kappa. In this line, the kappa light chains are produced but fail to be secreted [139]. To determine the mechanism underlying this phenomenon, Argon and colleagues [140] examined the fate of kappa light chains under a variety of conditions. First, they discovered that the failure to be processed was an intrinsic property of the unassembled kappa light chain. After exclusive expression of kappa in another myeloma line or in fibroblasts, the protein remained secretion-incompetent and was degraded. Pulse chase studies showed the free kappa chains to be degraded with no lag time and at a significantly greater rate than other light chains expressed in the same system. However the kappa light chains were not defective; when the corresponding heavy chain partner was co-expressed the light chains were assembled and functional immunoglobulin was secreted. Treatment with monensin, m-chlorophenylhydrazone, or brefeldin A, which disrupt ER to Golgi or inter-Golgi transport, had no effect on kappa degradation, suggesting that transit beyond the ER was dispensable for degradation. As anticipated, then, inhibitors of endosomal and lysosomal proteases had no effect on kappa degradation, and the kappa light chains co-localized with the ER resident chaperone, BiP, as determined by immunofluorescence microscopy. Further, the light chains associated with BiP, consistent with other studies that had implicated BiP as a component of light chain quality control [141, 142]. However, the light chains also associated with GRP94 after chemical crosslinking and immunoprecipitation [140]. GRP94 is another abundant ER resident chaperone, and recently this Hsp90 homolog was found to reside in a complex with a network of other factors that play a role in ERAD substrate selection [143].
3.11 ER-60, the long-sought ER protease?
Based on the studies discussed in the preceding sections, there was clearly significant interest in identifying an ER resident, quality control protease. At this time, the concept of the proteasome or another cytosolic protease degrading ER membrane proteins—let alone ER lumenal proteins—was not entertained. Several of the papers described above had implicated serine and/or cysteine proteases, suggesting the involvement of at least one ER protease. To identify these proteases, the quality controls of misfolded, mutant versions of human lysozyme were examined. Unlike wild type lysozyme, the mutant forms are retained in the ER and degraded [144]. Degradation of mutant lysozyme was then shown to be sensitive to cysteine protease inhibitors and chemical crosslinking identified a protein known as ER-60 as well as protein disulfide isomerase (PDI) as lysozyme partners [145]. ER-60 had previously been isolated from rat liver and appeared to exhibit cysteine protease activity [146]. In vitro assays suggested that ER-60 could degrade chemically denatured mutant lysozyme but not the wild type substrate. Based on these promising results and the fact that PDI aids in the maturation of disulfide-bonded secreted proteins [1], it was thought that ER-60 was the long sought ER quality control protease. However, evidence for a broader role of ER60 in the degradation of other substrates never materialized, and it is likely that initial reports contained an unknown contaminating protease activity. In the end ER-60 was re-annotated as p57, which is a calnexin associated disulfide isomerase in the PDI family [147].
3.12 Degradation of a Sec61 mutant and a connection to the ubiquitin pathway
Each of the studies described in the previous sections employed biochemical techniques and utilized mammalian cells. A significant breakthrough in our understanding of the machinery that mediates ER protein degradation came about from genetic studies in yeast [148]. In this study, the fate of a mutant version of the Sec61 translocation channel was examined. The mutation in the SEC61 gene results in a destabilized protein at 37°C, and as a result sec61 yeast are inviable at this temperature. Thus, extragenic suppressors of the growth phenotype could be identified, which in turn might be responsible for the degradation of Sec61. One identified suppressor was Ubc6, which is a ubiquitin conjugating enzyme (E2) that associates with the ER membrane and whose active site faces the cytoplasm. The loss of Ubc6 also restored efficient ER translocation in the sec61 mutant, demonstrating that both the translocation channel's levels and function had been corrected. These results suggested that ER protein quality control might require cytoplasmic components and implicated the ubiquitin proteasome system in quality control. In a subsequent study, the Sommer group confirmed this supposition [149]. Conditionally mutant forms of both the Sec61 protein, as well as one of its partners, Sss1 [150], were subject to regulated degradation at non-permissive temperatures in a ubiquitin and proteasome-dependent manner. As anticipated, degradation took place at the ER membrane. Based on the results of these studies, it was suggested that the ubiquitin-proteasome system might be responsible for other examples of ER protein degradation, as observed in earlier studies.
3.13 The cystic fibrosis transmembrane conductance regulator (CFTR)
Concurrent with these experiments, several laboratories began to investigate another substrate whose selection by the ER quality control machinery is linked to human disease. The cystic fibrosis transmembrane conductance regulator, CFTR, had been shown to be a highly unstable protein: Only 25% of the wild type protein matured in the ER and could traffic to the plasma membrane, thus escaping the ER degradation machinery [6]. In turn, a disease-associated deletion of Phe at position 508 in the protein (deltaF508-CFTR), resulted in the complete destruction of CFTR and the absence of this polytopic membrane protein at the plasma membrane in epithelial cells.
To define the pathway that leads to CFTR degradation, various protease inhibitors, including those that had recently shown activity against the cytoplasmic proteasome, were examined for their effects on CFTR [151]. The peptide aldehyde ALLN slowed but did not completely prevent the degradation of immature CFTR. However, a more specific proteasome inhibitor, MG132, completely blocked the degradation of immature CFTR. In no case did the prevention of degradation lead to the appearance of mature CFTR, suggesting that the restoration of functional protein required an earlier, corrective step in the quality control pathway; this supposition has been borne-out [152]. A direct link between CFTR degradation and the ubiquitination-proteasome pathway was also established in a companion study [153]. Here, the authors showed that immature CFTR was polyubiquitinated, and that concurrent with stabilization of CFTR by ALLN the ubiquitin signal significantly increased. In addition, co-expression of the K48R ubiquitin mutant, which is unable to form polyubiquitin chains, led to a massive increase in deltaF508 CFTR, strongly suggesting that polyubiquitination was required for CFTR degradation. Further, CFTR accumulated in a cell line that expressed a temperature sensitive allele of the E1 ubiquitin activating enzyme [154]. Taken together, these results provided the first direct evidence that misfolded ER membrane proteins access the cytoplasmic ubiquitin-proteasome system. Though the authors did not know the mechanism by which this membrane protein was selected for degradation, the large cytoplasmic domains of CFTR were known to interact with members of the Hsp70 chaperone family [155, 156]. It is now clear that a myriad of chaperones associate with immature forms of CFTR, particularly the delta508 variant, and select the proteins for ubiquitin-dependent degradation by the proteasome [157].
3.14 Yeast proalpha factor and the reconstitution of ER associated degradation
By the middle of the 1990s it was becoming apparent that a system to identify and degrade a variety of aberrant integral membrane proteins from the ER existed, and that the ubiquitin-proteasome might play a role in this process. It was unknown, however, how soluble secreted proteins within the ER were degraded since they seemed unable to access the cytoplasmic components that constituted the ubiquitin-proteasome system. Moreover, additional components required for ER protein degradation most probably existed but remained mysterious. Significant progress toward these ends was made with the development of an in vitro system using components derived from yeast [158].
As described above, the yeast prepheromone prepro-alpha factor translocates into the ER, and after the signal sequence is cleaved the resulting protein, pro-alpha factor, is glycosylated. However, if glycosylation is prevented, the protein is degraded in yeast [159]. Therefore, genetic and chemical tools were used to introduce an unglycosylated version of pro-alpha factor into isolated, ER-derived vesicles from yeast. When concentrated yeast cytosol and an ATP-regenerating system were added, the pro-alpha factor was rapidly and quantitatively degraded [158]. This system permitted the introduction of ER or cytosolic components prepared from mutant yeast strains, allowing for the testing of essentially any factor that might facilitate pro-alpha factor degradation. Calnexin as well as a number of other chaperones were subsequently identified as being critical for selecting pro-alpha factor for degradation [24, 25, 158, 160].
What remained curious was why degradation required the addition of cytoplasm, but based on data emerging from the literature it was possible that the substrate had somehow accessed the cytoplasmic space. Indeed, by introducing a centrifugation step during the degradation reaction, it was evident that pro-alpha factor had entered the cytoplasm in an ATP-dependent manner [158]. To test whether the substrate was subject to proteasomal degradation, cytosol from a proteasome mutant was examined in this in vitro system, as well as wild type cytosol incubated with a proteasome inhibitor. In both cases, pro-alpha factor degradation was significantly attenuated and the substrate accumulated in the cytosol. These data established that defective, soluble proteins in the ER could be selected, returned to the cytoplasm, and degraded by the proteasome [16]. Based on the fact that ER associated components were required for degradation, the process was called ERAD.
3.15 A mutant form of carboxypeptidase Y, CPY*
In the same year, another study in the yeast system demonstrated that a soluble protein within the ER could be selected, retrotranslocated, and degraded by the proteasome. The protein was a mutant form of the yeast vacuolar protease carboxypeptidase Y (CPY*) that was trapped in the ER and degraded [161]. Based on the ease with which CPY* degradation could be monitored in various mutant strain backgrounds, CPY* is now one of the most widely used ERAD substrates. In this first report, it was shown that CPY* degradation was dependant on an integral membrane ubiquitin conjugating enzyme in the ER, Ubc7, and on the 26S proteasome. Subsequent work established that CPY* degradation was BiP-dependent and might require the Sec61 translocation channel for delivery into the cytoplasm [162]. Genetic screens to isolate mutants that accumulated CPY* led to the isolation of an integral membrane protein that plays a critical role during the ERAD of many substrates [163]. Because CPY* is both glycosylated and disulfide bonded, this substrate has also been used to define more specifically how defects in folding result in ERAD (see for example [164, 165].
4. Concluding Remarks
In the 10 years from the mid-1980s, when the first reports on TCR subunit degradation appeared, to the mid-1990s, when the ubiquitin-proteasome system was implicated in the degradation of both soluble and integral membrane proteins in the early secretory pathway, the ER quality control field grew rapidly. Since then, a large and diverse group of ERAD substrates have been identified and the requirements for their disposal have been dissected. To date, there are almost 70 ERAD substrates linked to a variety of human diseases [50]. In the case of cystic fibrosis, an understanding of the quality control decisions in the ER has led to the identification of therapeutics that may be used to help alleviate disease [166].
Among the many components of the ERAD machinery that have been identified and characterized in the intervening years are the many subclasses of molecular chaperones which sense misfolded proteins and cooperate with and deliver these misfolded substrates to ER membrane associated E3 ubiquitin ligases. Although chaperones are generally thought to simply target exposed hydrophobic motifs on unfolded proteins, it remains mysterious why so many different chaperones may be needed to efficiently degrade a given ERAD substrate. It is still unclear exactly how non-glycosylated substrates are ultimately selected as substrates and this is a current area of active research [167]. Further, the number of proteins identified as E3 ligases has grown tremendously and indicate the large potential diversity in substrate recognition [48]. The E3 ligases themselves are part of large multi-protein complexes and have become appreciated as central players in the organization and regulation of protein quality control [17]. What is relatively mysterious, however, is the nature of the polyubiquitin chain that targets ERAD substrates from proteasomal degradation [168]. Lys-48 polyubiquitin linkages are generally thought to dictate proteasome targeting, but a mass spectrometry study also identified K-11 linkages as residing in ERAD substrates [169]. Whether mixed chains exist on ERAD substrates, and which E3s cooperate to append these various chains are unknown. We have also come further in our understanding of how glycosylation regulates the folding and degradation of secreted proteins. It is now known that not only the type of glycans present and the timing of addition are important, but the positioning of the sugar residues can have significant effects on the dynamics of degradation [170, 171]. Nevertheless, these studies have only been performed on a limited number of ERAD substrates.
While great progress has also been made in the understanding of the ERAD requirements for specific substrates this pursuit will become vital as new ERAD substrates are identified. In some cases substrate-specific factors, which are required for ERAD, exist [172]. It will be exciting to determine whether additional and as yet uncharacterized substrate-specific ERAD effectors exist. Another major “black box” in the field is the nature of the retrotranslocation channel. Several viable candidates for this channel are now known due to their interaction with membrane bound proteasomes or ERAD substrates targeted for proteasomal degradation. Proteasomes are localized throughout the cell and while some are free in the cytoplasm others associate with the ER membrane [173]. Several studies have provided evidence that the proteasome binds directly to the translocon channel Sec61 and at least for some substrates the same channel may be used for retrotranslation from the ER to the cytoplasm [37, 162, 174–176]. Other studies have suggested the involvement of other protein channels acting during retrotranslocation (e.g., Derlin and Hrd1 [40, 177, 178]). Therefore, it remains possible that multiple channels could be used for retrotranslocation or dislocation, depending on the specific substrate and with which quality control components are interacting.
The mechanism that leads to a soluble substrate first accessing the cytoplasmic space is another area that is still actively being investigated, though progress has been made on the pathway that at least some substrates follow from being selected for ERAD to being delivered to a retrotranslocon [179]. Similarly, the degree to which a protein must be unfolded before it is selected for ERAD is also unclear, as sensitive cellular read-outs for alterations in protein conformation are lacking. Finally, efforts to manipulate protein folding dynamics and to alter ERAD substrate selection in cell models and in animals are now of vital medical significance, and this represents a major aim in future research efforts.
Highlights
Proteins in the secretory pathway are subject to quality control “decisions”
It was thought that only the lysosome mediated secretory protein quality control
These proteins are instead subject to degradation by the proteasome
This pathway is known as ER associated degradation, or ERAD
Acknowledgments
Work on ERAD in the Brodsky lab is funded by grants GM75061 and DK79307 (The Pittsburgh Center for Kidney Research) from the National Institutes of Health and by a grant from Cystic Fibrosis Foundation Therapeutics.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Braakman I, Bulleid NJ. Protein folding and modification in the mammalian endoplasmic reticulum. Annu Rev Biochem. 2011;80:71–99. doi: 10.1146/annurev-biochem-062209-093836. [DOI] [PubMed] [Google Scholar]
- [2].Brodsky JL, Skach R. Protein folding and quality control in the endoplasmic reticulum: Recent lessons from yeast and mammalian cell systems. Curr Opin Cell Biol. 2011;23:464–475. doi: 10.1016/j.ceb.2011.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Hartl FU, Bracher A, Hayer-Hartl M. Molecular chaperones in protein folding and proteostasis. Nature. 2011;475:324–332. doi: 10.1038/nature10317. [DOI] [PubMed] [Google Scholar]
- [4].Kampinga HH, Craig A. The HSP70 chaperone machinery: J proteins as drivers of functional specificity. Nat Rev Mol Cell Biol. 2010;11:579–592. doi: 10.1038/nrm2941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Okiyoneda T, Lukacs GL. Fixing cystic fibrosis by correcting CFTR domain assembly. J Cell Biol. 2012;199:199–204. doi: 10.1083/jcb.201208083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Cheng SH, Gregory RJ, Marshall J, Paul S, Souza DW, White GA, O'Riordan CR, Smith AE. Defective intracellular transport and processing of CFTR is the molecular basis of most cystic fibrosis. Cell. 1990;63:827–834. doi: 10.1016/0092-8674(90)90148-8. [DOI] [PubMed] [Google Scholar]
- [7].Staub O, Gautschi I, Ishikawa T, Breitschopf K, Ciechanover A, Schild L, Rotin D. Regulation of stability and function of the epithelial Na+ channel (ENaC) by ubiquitination. EMBO J. 1997;16:6325–6336. doi: 10.1093/emboj/16.21.6325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Petaja-Repo UE, Hogue M, Laperriere A, Walker P, Bouvier M. Export from the endoplasmic reticulum represents the limiting step in the maturation and cell surface expression of the human delta opioid receptor. J Biol Chem. 2000;275:13727–13736. doi: 10.1074/jbc.275.18.13727. [DOI] [PubMed] [Google Scholar]
- [9].Yan FF, Pratt EB, Chen PC, Wang F, Skach WR, David LL, Shyng SL. Role of Hsp90 in biogenesis of the beta-cell ATP-sensitive potassium channel complex. Mol Biol Cell. 2010;21:1945–1954. doi: 10.1091/mbc.E10-02-0116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Gottesman S, Wickner S, Maurizi MR. Protein quality control: triage by chaperones and proteases. Genes Dev. 1997;11:815–823. doi: 10.1101/gad.11.7.815. [DOI] [PubMed] [Google Scholar]
- [11].Ellgaard L, Molinari M, Helenius A. Setting the standards: quality control in the secretory pathway. Science. 1999;286:1882–1888. doi: 10.1126/science.286.5446.1882. [DOI] [PubMed] [Google Scholar]
- [12].Shao S, Hegde RS. Membrane protein insertion at the endoplasmic reticulum. Annu Rev Cell Dev Biol. 2011;27:25–56. doi: 10.1146/annurev-cellbio-092910-154125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Saraogi I, Shan SO. Molecular mechanism of co-translational protein targeting by the signal recognition particle. Traffic. 2011;12:535–542. doi: 10.1111/j.1600-0854.2011.01171.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Ghaemmaghami S, Huh WK, Bower K, Howson RW, Belle A, Dephoure N, O'Shea EK, Weissman JS. Global analysis of protein expression in yeast. Nature. 2003;425:737–741. doi: 10.1038/nature02046. [DOI] [PubMed] [Google Scholar]
- [15].Powers ET, Morimoto RI, Dillin A, Kelly JW, Balch WE. Biological and chemical approaches to diseases of proteostasis deficiency. Annu Rev Biochem. 2009;78:959–991. doi: 10.1146/annurev.biochem.052308.114844. [DOI] [PubMed] [Google Scholar]
- [16].Werner ED, Brodsky JL, McCracken AA. Proteasome-dependent endoplasmic reticulum-associated protein degradation: an unconventional route to a familiar fate. Proc Natl Acad Sci U S A. 1996;93:13797–13801. doi: 10.1073/pnas.93.24.13797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Smith MH, Ploegh HL, Weissman JS. Road to ruin: targeting proteins for degradation in the endoplasmic reticulum. Science. 2011;334:1086–1090. doi: 10.1126/science.1209235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Thibault G, Ng DT. The endoplasmic reticulum-associated degradation pathways of budding yeast. Cold Spring Harb Perspect Biol. 2012;4 doi: 10.1101/cshperspect.a013193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Vembar SS, Brodsky JL. One step at a time: endoplasmic reticulum-associated degradation. Nat Rev Mol Cell Biol. 2008;9:944–957. doi: 10.1038/nrm2546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Araki K, Nagata K. Protein folding and quality control in the ER. Cold Spring Harb Perspect Biol. 2012;4:a015438. doi: 10.1101/cshperspect.a015438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Meusser B, Hirsch C, Jarosch E, Sommer T. ERAD: the long road to destruction. Nat Cell Biol. 2005;7:766–772. doi: 10.1038/ncb0805-766. [DOI] [PubMed] [Google Scholar]
- [22].Loo MA, Jensen TJ, Cui L, Hou Y, Chang XB, Riordan JR. Perturbation of Hsp90 interaction with nascent CFTR prevents its maturation and accelerates its degradation by the proteasome. EMBO J. 1998;17:6879–6887. doi: 10.1093/emboj/17.23.6879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Youker RT, Walsh P, Beilharz T, Lithgow T, Brodsky JL. Distinct roles for the Hsp40 and Hsp90 molecular chaperones during cystic fibrosis transmembrane conductance regulator degradation in yeast. Mol Biol Cell. 2004;15:4787–4797. doi: 10.1091/mbc.E04-07-0584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Gillece P, Luz JM, Lennarz WJ, de La Cruz FJ, Romisch K. Export of a cysteine-free misfolded secretory protein from the endoplasmic reticulum for degradation requires interaction with protein disulfide isomerase. J Cell Biol. 1999;147:1443–1456. doi: 10.1083/jcb.147.7.1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Nishikawa S, Brodsky JL, Nakatsukasa K. Roles of molecular chaperones in endoplasmic reticulum (ER) quality control and ER-associated degradation (ERAD) J Biochem. 2005;137:551–555. doi: 10.1093/jb/mvi068. [DOI] [PubMed] [Google Scholar]
- [26].Otero JH, Lizak B, Hendershot LM. Life and death of a BiP substrate. Semin Cell Dev Biol. 2010;21:472–478. doi: 10.1016/j.semcdb.2009.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Feige MJ, Hendershot LM. Disulfide bonds in ER protein folding and homeostasis. Curr Opin Cell Biol. 2011;23:167–175. doi: 10.1016/j.ceb.2010.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Grubb S, Guo L, Fisher EA, Brodsky JL. Protein disulfide isomerases contribute differentially to the endoplasmic reticulum-associated degradation of apolipoprotein B and other substrates. Mol Biol Cell. 2012;23:520–532. doi: 10.1091/mbc.E11-08-0704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Aebi M, Bernasconi R, Clerc S, Molinari M. N-glycan structures: recognition and processing in the ER. Trends Biochem Sci. 2010;35:74–82. doi: 10.1016/j.tibs.2009.10.001. [DOI] [PubMed] [Google Scholar]
- [30].Pearse BR, Hebert DN. Lectin chaperones help direct the maturation of glycoproteins in the endoplasmic reticulum. Biochim Biophys Acta. 2010;1803:684–693. doi: 10.1016/j.bbamcr.2009.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Caramelo JJ, Parodi AJ. How sugars convey information on protein conformation in the endoplasmic reticulum. Semin Cell Dev Biol. 2007;18:732–742. doi: 10.1016/j.semcdb.2007.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Hebert DN, Molinari M. Flagging and docking: dual roles for N-glycans in protein quality control and cellular proteostasis. Trends Biochem Sci. 2012;37:404–410. doi: 10.1016/j.tibs.2012.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Parodi AJ. Role of N-oligosaccharide endoplasmic reticulum processing reactions in glycoprotein folding and degradation. Biochem J. 2000;348(Pt 1):1–13. [PMC free article] [PubMed] [Google Scholar]
- [34].Maattanen P, Gehring K, Bergeron JJ, Thomas DY. Protein quality control in the ER: the recognition of misfolded proteins. Semin Cell Dev Biol. 2010;21:500–511. doi: 10.1016/j.semcdb.2010.03.006. [DOI] [PubMed] [Google Scholar]
- [35].Sifers RN. Cell biology. Protein degradation unlocked. Science. 2003;299:1330–1331. doi: 10.1126/science.1082718. [DOI] [PubMed] [Google Scholar]
- [36].Hagiwara M, Maegawa K, Suzuki M, Ushioda R, Araki K, Matsumoto Y, Hoseki J, Nagata K, Inaba K. Structural basis of an ERAD pathway mediated by the ER-resident protein disulfide reductase ERdj5. Mol Cell. 2011;41:432–444. doi: 10.1016/j.molcel.2011.01.021. [DOI] [PubMed] [Google Scholar]
- [37].Schafer A, Wolf DH. Sec61p is part of the endoplasmic reticulum-associated degradation machinery. EMBO J. 2009;28:2874–2884. doi: 10.1038/emboj.2009.231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Carvalho P, Goder V, Rapoport TA. Distinct ubiquitin-ligase complexes define convergent pathways for the degradation of ER proteins. Cell. 2006;126:361–373. doi: 10.1016/j.cell.2006.05.043. [DOI] [PubMed] [Google Scholar]
- [39].Denic V, Quan EM, Weissman JS. A luminal surveillance complex that selects misfolded glycoproteins for ER-associated degradation. Cell. 2006;126:349–359. doi: 10.1016/j.cell.2006.05.045. [DOI] [PubMed] [Google Scholar]
- [40].Carvalho P, Stanley AM, Rapoport TA. Retrotranslocation of a misfolded luminal ER protein by the ubiquitin-ligase Hrd1p. Cell. 2010;143:579–591. doi: 10.1016/j.cell.2010.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Ye Y, Meyer HH, Rapoport TA. The AAA ATPase Cdc48/p97 and its partners transport proteins from the ER into the cytosol. Nature. 2001;414:652–656. doi: 10.1038/414652a. [DOI] [PubMed] [Google Scholar]
- [42].Bays NW, Wilhovsky SK, Goradia A, Hodgkiss-Harlow K, Hampton RY. HRD4/NPL4 is required for the proteasomal processing of ubiquitinated ER proteins. Mol Biol Cell. 2001;12:4114–4128. doi: 10.1091/mbc.12.12.4114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Rabinovich E, Kerem A, Frohlich KU, Diamant N, Bar-Nun S. AAA-ATPase p97/Cdc48p, a cytosolic chaperone required for endoplasmic reticulum-associated protein degradation. Mol Cell Biol. 2002;22:626–634. doi: 10.1128/MCB.22.2.626-634.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Stolz A, Hilt W, Buchberger A, Wolf DH. Cdc48: a power machine in protein degradation. Trends Biochem Sci. 2011;36:515–523. doi: 10.1016/j.tibs.2011.06.001. [DOI] [PubMed] [Google Scholar]
- [45].Raasi S, Wolf DH. Ubiquitin receptors and ERAD: a network of pathways to the proteasome. Semin Cell Dev Biol. 2007;18:780–791. doi: 10.1016/j.semcdb.2007.09.008. [DOI] [PubMed] [Google Scholar]
- [46].Verma R, Chen S, Feldman R, Schieltz D, Yates J, Dohmen J, Deshaies RJ. Proteasomal proteomics: identification of nucleotide-sensitive proteasome-interacting proteins by mass spectrometric analysis of affinity-purified proteasomes. Mol Biol Cell. 2000;11:3425–3439. doi: 10.1091/mbc.11.10.3425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Mehnert M, Sommer T, Jarosch E. ERAD ubiquitin ligases: multifunctional tools for protein quality control and waste disposal in the endoplasmic reticulum. Bioessays. 2010;32:905–913. doi: 10.1002/bies.201000046. [DOI] [PubMed] [Google Scholar]
- [48].Claessen JH, Kundrat L, Ploegh HL. Protein quality control in the ER: balancing the ubiquitin checkbook. Trends Cell Biol. 2012;22:22–32. doi: 10.1016/j.tcb.2011.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Varshavsky A. Discovery of cellular regulation by protein degradation. J Biol Chem. 2008;283:34469–34489. doi: 10.1074/jbc.X800009200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Guerriero CJ, Brodsky JL. The delicate balance between secreted protein folding and endoplasmic reticulum-associated degradation in human physiology. Physiol Rev. 2012;92:537–576. doi: 10.1152/physrev.00027.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Xie W, Ng DT. ERAD substrate recognition in budding yeast. Semin Cell Dev Biol. 2010;21:533–539. doi: 10.1016/j.semcdb.2010.02.007. [DOI] [PubMed] [Google Scholar]
- [52].Hampton RY, Sommer T. Finding the will and the way of ERAD substrate retrotranslocation. Curr Opin Cell Biol. 2012;24:460–466. doi: 10.1016/j.ceb.2012.05.010. [DOI] [PubMed] [Google Scholar]
- [53].Yang Z, Klionsky DJ. Eaten alive: a history of macroautophagy. Nat Cell Biol. 2010;12:814–822. doi: 10.1038/ncb0910-814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Marzella L, Glaumann H. Biogenesis, translocation, and function of lysosomal enzymes. Int Rev Exp Pathol. 1983;25:239–278. [PubMed] [Google Scholar]
- [55].Kornfeld S, Mellman I. The biogenesis of lysosomes. Annu Rev Cell Biol. 1989;5:483–525. doi: 10.1146/annurev.cb.05.110189.002411. [DOI] [PubMed] [Google Scholar]
- [56].Henne WM, Buchkovich NJ, Emr SD. The ESCRT pathway. Dev Cell. 2011;21:77–91. doi: 10.1016/j.devcel.2011.05.015. [DOI] [PubMed] [Google Scholar]
- [57].Pontremoli S, Melloni E. Extralysosomal protein degradation. Annu Rev Biochem. 1986;55:455–481. doi: 10.1146/annurev.bi.55.070186.002323. [DOI] [PubMed] [Google Scholar]
- [58].Enenkel C, Lehmann A, Kloetzel PM. Subcellular distribution of proteasomes implicates a major location of protein degradation in the nuclear envelope-ER network in yeast. EMBO J. 1998;17:6144–6154. doi: 10.1093/emboj/17.21.6144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Brooks P, Fuertes G, Murray RZ, Bose S, Knecht E, Rechsteiner MC, Hendil KB, Tanaka K, Dyson J, Rivett J. Subcellular localization of proteasomes and their regulatory complexes in mammalian cells. Biochem J. 2000;346(Pt 1):155–161. [PMC free article] [PubMed] [Google Scholar]
- [60].Finley D. Recognition and processing of ubiquitin-protein conjugates by the proteasome. Annu Rev Biochem. 2009;78:477–513. doi: 10.1146/annurev.biochem.78.081507.101607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Samelson LE, Harford JB, Klausner RD. Identification of the components of the murine T cell antigen receptor complex. Cell. 1985;43:223–231. doi: 10.1016/0092-8674(85)90027-3. [DOI] [PubMed] [Google Scholar]
- [62].Sussman JJ, Bonifacino JS, Lippincott-Schwartz J, Weissman AM, Saito T, Klausner RD, Ashwell JD. Failure to synthesize the T cell CD3-zeta chain: structure and function of a partial T cell receptor complex. Cell. 1988;52:85–95. doi: 10.1016/0092-8674(88)90533-8. [DOI] [PubMed] [Google Scholar]
- [63].Minami Y, Samelson LE, Klausner RD. Internalization and cycling of the T cell antigen receptor. Role of protein kinase C. J Biol Chem. 1987;262:13342–13347. [PubMed] [Google Scholar]
- [64].Lippincott-Schwartz J, Bonifacino JS, Yuan LC, Klausner RD. Degradation from the endoplasmic reticulum: disposing of newly synthesized proteins. Cell. 1988;54:209–220. doi: 10.1016/0092-8674(88)90553-3. [DOI] [PubMed] [Google Scholar]
- [65].Chen C, Bonifacino JS, Yuan LC, Klausner RD. Selective degradation of T cell antigen receptor chains retained in a pre-Golgi compartment. J Cell Biol. 1988;107:2149–2161. doi: 10.1083/jcb.107.6.2149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Klausner RD, Sitia R. Protein degradation in the endoplasmic reticulum. Cell. 1990;62:611–614. doi: 10.1016/0092-8674(90)90104-m. [DOI] [PubMed] [Google Scholar]
- [67].Bonifacino JS, Lippincott-Schwartz J. Degradation of proteins within the endoplasmic reticulum. Curr Opin Cell Biol. 1991;3:592–600. doi: 10.1016/0955-0674(91)90028-w. [DOI] [PubMed] [Google Scholar]
- [68].Wilson R, Allen AJ, Oliver J, Brookman JL, High S, Bulleid NJ. The translocation, folding, assembly and redox-dependent degradation of secretory and membrane proteins in semi-permeabilized mammalian cells. Biochem J. 1995;307(Pt 3):679–687. doi: 10.1042/bj3070679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Stafford FJ, Bonifacino JS. A permeabilized cell system identifies the endoplasmic reticulum as a site of protein degradation. J Cell Biol. 1991;115:1225–1236. doi: 10.1083/jcb.115.5.1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Beckers CJ, Keller DS, Balch WE. Semi-intact cells permeable to macromolecules: use in reconstitution of protein transport from the endoplasmic reticulum to the Golgi complex. Cell. 1987;50:523–534. doi: 10.1016/0092-8674(87)90025-0. [DOI] [PubMed] [Google Scholar]
- [71].Hurtley SM, Helenius A. Protein oligomerization in the endoplasmic reticulum. Annu Rev Cell Biol. 1989;5:277–307. doi: 10.1146/annurev.cb.05.110189.001425. [DOI] [PubMed] [Google Scholar]
- [72].Iozzo RV, Pacifici M. Ultrastructural localization of the major proteoglycan and type II procollagen in organelles and extracellular matrix of cultured chondroblasts. Histochemistry. 1986;86:113–122. doi: 10.1007/BF00493375. [DOI] [PubMed] [Google Scholar]
- [73].Pacifici M, Iozzo RV. Remodeling of the rough endoplasmic reticulum during stimulation of procollagen secretion by ascorbic acid in cultured chondrocytes. A biochemical and morphological study. J Biol Chem. 1988;263:2483–2492. [PubMed] [Google Scholar]
- [74].Sifers RN, Brashears-Macatee S, Kidd VJ, Muensch H, Woo SL. A frameshift mutation results in a truncated alpha 1-antitrypsin that is retained within the rough endoplasmic reticulum. J Biol Chem. 1988;263:7330–7335. [PubMed] [Google Scholar]
- [75].Machamer CE, Rose JK. Vesicular stomatitis virus G proteins with altered glycosylation sites display temperature-sensitive intracellular transport and are subject to aberrant intermolecular disulfide bonding. J Biol Chem. 1988;263:5955–5960. [PubMed] [Google Scholar]
- [76].Munro S, Pelham HR. An Hsp70-like protein in the ER: identity with the 78 kd glucose-regulated protein and immunoglobulin heavy chain binding protein. Cell. 1986;46:291–300. doi: 10.1016/0092-8674(86)90746-4. [DOI] [PubMed] [Google Scholar]
- [77].Kassenbrock CK, Garcia PD, Walter P, Kelly RB. Heavy-chain binding protein recognizes aberrant polypeptides translocated in vitro. Nature. 1988;333:90–93. doi: 10.1038/333090a0. [DOI] [PubMed] [Google Scholar]
- [78].Hurtley SM, Bole DG, Hoover-Litty H, Helenius A, Copeland CS. Interactions of misfolded influenza virus hemagglutinin with binding protein (BiP) J Cell Biol. 1989;108:2117–2126. doi: 10.1083/jcb.108.6.2117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Lee SM, Casey CA, McVicker BL. Impact of asialoglycoprotein receptor deficiency on the development of liver injury. World J Gastroenterol. 2009;15:1194–1200. doi: 10.3748/wjg.15.1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Bischoff J, Libresco S, Shia MA, Lodish HF. The H1 and H2 polypeptides associate to form the asialoglycoprotein receptor in human hepatoma cells. J Cell Biol. 1988;106:1067–1074. doi: 10.1083/jcb.106.4.1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Massarelli I, Chiellini F, Chiellini E, Bianucci AM. Three-Dimensional Models of the Oligomeric Human Asialoglycoprotein Receptor (ASGP-R) Int J Mol Sci. 2010;11:3867–3884. doi: 10.3390/ijms11103867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Amara JF, Lederkremer G, Lodish HF. Intracellular degradation of unassembled asialoglycoprotein receptor subunits: a pre-Golgi, nonlysosomal endoproteolytic cleavage. J Cell Biol. 1989;109:3315–3324. doi: 10.1083/jcb.109.6.3315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Wikstrom L, Lodish HF. Nonlysosomal, pre-Golgi degradation of unassembled asialoglycoprotein receptor subunits: a TLCK- and TPCK-sensitive cleavage within the ER. J Cell Biol. 1991;113:997–1007. doi: 10.1083/jcb.113.5.997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Wikstrom L, Lodish HF. Endoplasmic reticulum degradation of a subunit of the asialoglycoprotein receptor in vitro. Vesicular transport from endoplasmic reticulum is unnecessary. J Biol Chem. 1992;267:5–8. [PubMed] [Google Scholar]
- [85].Kelleher DJ, Gilmore R. An evolving view of the eukaryotic oligosaccharyltransferase. Glycobiology. 2006;16:47R–62R. doi: 10.1093/glycob/cwj066. [DOI] [PubMed] [Google Scholar]
- [86].Wilson CM, Roebuck Q, High S. Ribophorin I regulates substrate delivery to the oligosaccharyltransferase core. Proc Natl Acad Sci U S A. 2008;105:9534–9539. doi: 10.1073/pnas.0711846105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Tsao YS, Ivessa NE, Adesnik M, Sabatini DD, Kreibich G. Carboxy terminally truncated forms of ribophorin I are degraded in pre-Golgi compartments by a calcium-dependent process. J Cell Biol. 1992;116:57–67. doi: 10.1083/jcb.116.1.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Tartakoff AM, Vassalli P. Plasma cell immunoglobulin secretion: arrest is accompanied by alterations of the golgi complex. J Exp Med. 1977;146:1332–1345. doi: 10.1084/jem.146.5.1332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Tartakoff AM. Perturbation of vesicular traffic with the carboxylic ionophore monensin. Cell. 1983;32:1026–1028. doi: 10.1016/0092-8674(83)90286-6. [DOI] [PubMed] [Google Scholar]
- [90].Tartakoff AM. Perturbation of the structure and function of the Golgi complex by monovalent carboxylic ionophores. Methods Enzymol. 1983;98:47–59. doi: 10.1016/0076-6879(83)98138-7. [DOI] [PubMed] [Google Scholar]
- [91].Malhotra JD, Kaufman RJ. The endoplasmic reticulum and the unfolded protein response. Semin Cell Dev Biol. 2007;18:716–731. doi: 10.1016/j.semcdb.2007.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Prostko CR, Brostrom MA, Malara EM, Brostrom CO. Phosphorylation of eukaryotic initiation factor (eIF) 2 alpha and inhibition of eIF-2B in GH3 pituitary cells by perturbants of early protein processing that induce GRP78. J Biol Chem. 1992;267:16751–16754. [PubMed] [Google Scholar]
- [93].Rivett AJ. Proteasomes: multicatalytic proteinase complexes. Biochem J. 1993;291(Pt 1):1–10. doi: 10.1042/bj2910001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Heemels MT, Ploegh H. Generation, translocation, and presentation of MHC class I-restricted peptides. Annu Rev Biochem. 1995;64:463–491. doi: 10.1146/annurev.bi.64.070195.002335. [DOI] [PubMed] [Google Scholar]
- [95].Townsend A, Bodmer H. Antigen recognition by class I-restricted T lymphocytes. Annu Rev Immunol. 1989;7:601–624. doi: 10.1146/annurev.iy.07.040189.003125. [DOI] [PubMed] [Google Scholar]
- [96].Maudsley DJ, Pound JD. Modulation of MHC antigen expression by viruses and oncogenes. Immunol Today. 1991;12:429–431. doi: 10.1016/0167-5699(91)90013-J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Beersma MF, Bijlmakers MJ, Ploegh HL. Human cytomegalovirus down-regulates HLA class I expression by reducing the stability of class I H chains. J Immunol. 1993;151:4455–4464. [PubMed] [Google Scholar]
- [98].Jones TR, Hanson LK, Sun L, Slater JS, Stenberg RM, Campbell AE. Multiple independent loci within the human cytomegalovirus unique short region down-regulate expression of major histocompatibility complex class I heavy chains. J Virol. 1995;69:4830–4841. doi: 10.1128/jvi.69.8.4830-4841.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Wiertz EJ, Jones TR, Sun L, Bogyo M, Geuze HJ, Ploegh HL. The human cytomegalovirus US11 gene product dislocates MHC class I heavy chains from the endoplasmic reticulum to the cytosol. Cell. 1996;84:769–779. doi: 10.1016/s0092-8674(00)81054-5. [DOI] [PubMed] [Google Scholar]
- [100].Machold RP, Wiertz EJ, Jones TR, Ploegh HL. The HCMV gene products US11 and US2 differ in their ability to attack allelic forms of murine major histocompatibility complex (MHC) class I heavy chains. J Exp Med. 1997;185:363–366. doi: 10.1084/jem.185.2.363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Chen M, Gerlier D. Viral hijacking of cellular ubiquitination pathways as an anti-innate immunity strategy. Viral Immunol. 2006;19:349–362. doi: 10.1089/vim.2006.19.349. [DOI] [PubMed] [Google Scholar]
- [102].Goldstein JL, Brown MS. Regulation of the mevalonate pathway. Nature. 1990;343:425–430. doi: 10.1038/343425a0. [DOI] [PubMed] [Google Scholar]
- [103].Nakanishi M, Goldstein JL, Brown MS. Multivalent control of 3-hydroxy-3-methylglutaryl coenzyme A reductase. Mevalonate-derived product inhibits translation of mRNA and accelerates degradation of enzyme. J Biol Chem. 1988;263:8929–8937. [PubMed] [Google Scholar]
- [104].Edwards PA, Lan SF, Fogelman AM. Alterations in the rates of synthesis and degradation of rat liver 3-hydroxy-3-methylglutaryl coenzyme A reductase produced by cholestyramine and mevinolin. J Biol Chem. 1983;258:10219–10222. [PubMed] [Google Scholar]
- [105].Leonard DA, Chen HW. ATP-dependent degradation of 3-hydroxy-3-methylglutaryl coenzyme A reductase in permeabilized cells. J Biol Chem. 1987;262:7914–7919. [PubMed] [Google Scholar]
- [106].Meigs TE, Simoni RD. Regulated degradation of 3-hydroxy-3-methylglutaryl-coenzyme A reductase in permeabilized cells. J Biol Chem. 1992;267:13547–13552. [PubMed] [Google Scholar]
- [107].Beaufay H, Amar-Costesec A, Thines-Sempoux D, Wibo M, Robbi M, Berthet J. Analytical study of microsomes and isolated subcellular membranes from rat liver. 3. Subfractionation of the microsomal fraction by isopycnic and differential centrifugation in density gradients. J Cell Biol. 1974;61:213–231. doi: 10.1083/jcb.61.1.213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Katz J, Wals PA. Studies with digitonin-treated rat hepatocytes (nude cells) J Cell Biochem. 1985;28:207–228. doi: 10.1002/jcb.240280304. [DOI] [PubMed] [Google Scholar]
- [109].Chun KT, Bar-Nun S, Simoni RD. The regulated degradation of 3-hydroxy-3-methylglutaryl-CoA reductase requires a short-lived protein and occurs in the endoplasmic reticulum. J Biol Chem. 1990;265:22004–22010. [PubMed] [Google Scholar]
- [110].Roitelman J, Bar-Nun S, Inoue S, Simoni RD. Involvement of calcium in the mevalonate-accelerated degradation of 3-hydroxy-3-methylglutaryl-CoA reductase. J Biol Chem. 1991;266:16085–16091. [PubMed] [Google Scholar]
- [111].Inoue S, Bar-Nun S, Roitelman J, Simoni RD. Inhibition of degradation of 3-hydroxy-3-methylglutaryl-coenzyme A reductase in vivo by cysteine protease inhibitors. J Biol Chem. 1991;266:13311–13317. [PubMed] [Google Scholar]
- [112].Hampton RY. Proteolysis and sterol regulation. Annu Rev Cell Dev Biol. 2002;18:345–378. doi: 10.1146/annurev.cellbio.18.032002.131219. [DOI] [PubMed] [Google Scholar]
- [113].Jo Y, Debose-Boyd RA. Control of cholesterol synthesis through regulated ER-associated degradation of HMG CoA reductase. Crit Rev Biochem Mol Biol. 2010;45:185–198. doi: 10.3109/10409238.2010.485605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Ronis MJ, Johansson I, Hultenby K, Lagercrantz J, Glaumann H, Ingelman-Sundberg M. Acetone-regulated synthesis and degradation of cytochrome P450E1 and cytochrome P4502B1 in rat liver [corrected] Eur J Biochem. 1991;198:383–389. doi: 10.1111/j.1432-1033.1991.tb16026.x. [DOI] [PubMed] [Google Scholar]
- [115].Masaki R, Yamamoto A, Tashiro Y. Cytochrome P-450 and NADPH-cytochrome P-450 reductase are degraded in the autolysosomes in rat liver. J Cell Biol. 1987;104:1207–1215. doi: 10.1083/jcb.104.5.1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Eliasson E, Mkrtchian S, Ingelman-Sundberg M. Hormone- and substrate-regulated intracellular degradation of cytochrome P450 (2E1) involving MgATP-activated rapid proteolysis in the endoplasmic reticulum membranes. J Biol Chem. 1992;267:15765–15769. [PubMed] [Google Scholar]
- [117].Su K, Stoller T, Rocco J, Zemsky J, Green R. Pre-Golgi degradation of yeast prepro-alpha-factor expressed in a mammalian cell. Influence of cell type-specific oligosaccharide processing on intracellular fate. J Biol Chem. 1993;268:14301–14309. [PubMed] [Google Scholar]
- [118].Hammond C, Braakman I, Helenius A. Role of N-linked oligosaccharide recognition, glucose trimming, and calnexin in glycoprotein folding and quality control. Proc Natl Acad Sci U S A. 1994;91:913–917. doi: 10.1073/pnas.91.3.913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Hebert DN, Foellmer B, Helenius A. Glucose trimming and reglucosylation determine glycoprotein association with calnexin in the endoplasmic reticulum. Cell. 1995;81:425–433. doi: 10.1016/0092-8674(95)90395-x. [DOI] [PubMed] [Google Scholar]
- [120].Wieland FT, Gleason ML, Serafini TA, Rothman JE. The rate of bulk flow from the endoplasmic reticulum to the cell surface. Cell. 1987;50:289–300. doi: 10.1016/0092-8674(87)90224-8. [DOI] [PubMed] [Google Scholar]
- [121].Baker D, Hicke L, Rexach M, Schleyer M, Schekman R. Reconstitution of SEC gene product-dependent intercompartmental protein transport. Cell. 1988;54:335–344. doi: 10.1016/0092-8674(88)90196-1. [DOI] [PubMed] [Google Scholar]
- [122].Romisch K, Schekman R. Distinct processes mediate glycoprotein and glycopeptide export from the endoplasmic reticulum in Saccharomyces cerevisiae. Proc Natl Acad Sci U S A. 1992;89:7227–7231. doi: 10.1073/pnas.89.15.7227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Ali BR, Tjernberg A, Chait BT, Field MC. A microsomal GTPase is required for glycopeptide export from the mammalian endoplasmic reticulum. J Biol Chem. 2000;275:33222–33230. doi: 10.1074/jbc.M003845200. [DOI] [PubMed] [Google Scholar]
- [124].Romisch K, Ali BR. Similar processes mediate glycopeptide export from the endoplasmic reticulum in mammalian cells and Saccharomyces cerevisiae. Proc Natl Acad Sci U S A. 1997;94:6730–6734. doi: 10.1073/pnas.94.13.6730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Perlmutter DH. Alpha-1-antitrypsin deficiency: importance of proteasomal and autophagic degradative pathways in disposal of liver disease-associated protein aggregates. Annu Rev Med. 2011;62:333–345. doi: 10.1146/annurev-med-042409-151920. [DOI] [PubMed] [Google Scholar]
- [126].Foreman RC, Judah JD, Colman A. Xenopus oocytes can synthesise but do not secrete the Z variant of human alpha 1-antitrypsin. FEBS Lett. 1984;168:84–88. doi: 10.1016/0014-5793(84)80211-2. [DOI] [PubMed] [Google Scholar]
- [127].Carlson JA, Rogers BB, Sifers RN, Finegold MJ, Clift SM, DeMayo FJ, Bullock DW, Woo SL. Accumulation of PiZ alpha 1-antitrypsin causes liver damage in transgenic mice. J Clin Invest. 1989;83:1183–1190. doi: 10.1172/JCI113999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].Lomas DA, Evans DL, Finch JT, Carrell RW. The mechanism of Z alpha 1-antitrypsin accumulation in the liver. Nature. 1992;357:605–607. doi: 10.1038/357605a0. [DOI] [PubMed] [Google Scholar]
- [129].Graham KS, Le A, Sifers RN. Accumulation of the insoluble PiZ variant of human alpha 1-antitrypsin within the hepatic endoplasmic reticulum does not elevate the steady-state level of grp78/BiP. J Biol Chem. 1990;265:20463–20468. [PubMed] [Google Scholar]
- [130].Le A, Graham KS, Sifers RN. Intracellular degradation of the transport-impaired human PiZ alpha 1-antitrypsin variant. Biochemical mapping of the degradative event among compartments of the secretory pathway. J Biol Chem. 1990;265:14001–14007. [PubMed] [Google Scholar]
- [131].Qu D, Teckman JH, Omura S, Perlmutter DH. Degradation of a mutant secretory protein, alpha1-antitrypsin Z, in the endoplasmic reticulum requires proteasome activity. J Biol Chem. 1996;271:22791–22795. doi: 10.1074/jbc.271.37.22791. [DOI] [PubMed] [Google Scholar]
- [132].Gelling CL, Brodsky JL. Mechanisms underlying the cellular clearance of antitrypsin Z: lessons from yeast expression systems. Proc Am Thorac Soc. 2010;7:363–367. doi: 10.1513/pats.201001-007AW. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].Teckman JH, Perlmutter DH. Retention of mutant alpha(1)-antitrypsin Z in endoplasmic reticulum is associated with an autophagic response. Am J Physiol Gastrointest Liver Physiol. 2000;279:G961–974. doi: 10.1152/ajpgi.2000.279.5.G961. [DOI] [PubMed] [Google Scholar]
- [134].Kamimoto T, Shoji S, Hidvegi T, Mizushima N, Umebayashi K, Perlmutter DH, Yoshimori T. Intracellular inclusions containing mutant alpha1-antitrypsin Z are propagated in the absence of autophagic activity. J Biol Chem. 2006;281:4467–4476. doi: 10.1074/jbc.M509409200. [DOI] [PubMed] [Google Scholar]
- [135].Kruse KB, Brodsky JL, McCracken AA. Characterization of an ERAD gene as VPS30/ATG6 reveals two alternative and functionally distinct protein quality control pathways: one for soluble Z variant of human alpha-1 proteinase inhibitor (A1PiZ) and another for aggregates of A1PiZ. Mol Biol Cell. 2006;17:203–212. doi: 10.1091/mbc.E04-09-0779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Stevens RH, Williamson AR. mRNA for H and L chains of immunoglobulin: specific control of H-chain production. Contemp Top Mol Immunol. 1974;3:85–110. doi: 10.1007/978-1-4684-2838-4_4. [DOI] [PubMed] [Google Scholar]
- [137].Dul JL, Burrone OR, Argon Y. A conditional secretory mutant in an Ig L chain is caused by replacement of tyrosine/phenylalanine 87 with histidine. J Immunol. 1992;149:1927–1933. [PubMed] [Google Scholar]
- [138].Davis DP, Gallo G, Vogen SM, Dul JL, Sciarretta KL, Kumar A, Raffen R, Stevens FJ, Argon Y. Both the environment and somatic mutations govern the aggregation pathway of pathogenic immunoglobulin light chain. J Mol Biol. 2001;313:1021–1034. doi: 10.1006/jmbi.2001.5092. [DOI] [PubMed] [Google Scholar]
- [139].Arnold LW, Grdina TA, Whitmore AC, Haughton G. Ig isotype switching in B lymphocytes. Isolation and characterization of clonal variants of the murine Ly-1+ B cell lymphoma, CH12, expressing isotypes other than IgM. J Immunol. 1988;140:4355–4363. [PubMed] [Google Scholar]
- [140].Gardner AM, Aviel S, Argon Y. Rapid degradation of an unassembled immunoglobulin light chain is mediated by a serine protease and occurs in a pre-Golgi compartment. J Biol Chem. 1993;268:25940–25947. [PubMed] [Google Scholar]
- [141].Knittler MR, Haas IG. Interaction of BiP with newly synthesized immunoglobulin light chain molecules: cycles of sequential binding and release. EMBO J. 1992;11:1573–1581. doi: 10.1002/j.1460-2075.1992.tb05202.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [142].Skowronek MH, Hendershot LM, Haas IG. The variable domain of nonassembled Ig light chains determines both their half-life and binding to the chaperone BiP. Proc Natl Acad Sci U S A. 1998;95:1574–1578. doi: 10.1073/pnas.95.4.1574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [143].Christianson JC, Olzmann JA, Shaler TA, Sowa ME, Bennett EJ, Richter CM, Tyler RE, Greenblatt EJ, Harper JW, Kopito RR. Defining human ERAD networks through an integrative mapping strategy. Nat Cell Biol. 2012;14:93–105. doi: 10.1038/ncb2383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [144].Omura F, Otsu M, Yoshimori T, Tashiro Y, Kikuchi M. Non-lysosomal degradation of misfolded human lysozymes with and without an asparagine-linked glycosylation site. Eur J Biochem. 1992;210:591–599. doi: 10.1111/j.1432-1033.1992.tb17459.x. [DOI] [PubMed] [Google Scholar]
- [145].Otsu M, Urade R, Kito M, Omura F, Kikuchi M. A possible role of ER-60 protease in the degradation of misfolded proteins in the endoplasmic reticulum. J Biol Chem. 1995;270:14958–14961. doi: 10.1074/jbc.270.25.14958. [DOI] [PubMed] [Google Scholar]
- [146].Urade R, Nasu M, Moriyama T, Wada K, Kito M. Protein degradation by the phosphoinositide-specific phospholipase C-alpha family from rat liver endoplasmic reticulum. J Biol Chem. 1992;267:15152–15159. [PubMed] [Google Scholar]
- [147].Lindquist JA, Jensen ON, Mann M, Hammerling GJ. ER-60, a chaperone with thiol-dependent reductase activity involved in MHC class I assembly. EMBO J. 1998;17:2186–2195. doi: 10.1093/emboj/17.8.2186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [148].Sommer T, Jentsch S. A protein translocation defect linked to ubiquitin conjugation at the endoplasmic reticulum. Nature. 1993;365:176–179. doi: 10.1038/365176a0. [DOI] [PubMed] [Google Scholar]
- [149].Biederer T, Volkwein C, Sommer T. Degradation of subunits of the Sec61p complex, an integral component of the ER membrane, by the ubiquitin-proteasome pathway. EMBO J. 1996;15:2069–2076. [PMC free article] [PubMed] [Google Scholar]
- [150].Esnault Y, Feldheim D, Blondel MO, Schekman R, Kepes F. SSS1 encodes a stabilizing component of the Sec61 subcomplex of the yeast protein translocation apparatus. J Biol Chem. 1994;269:27478–27485. [PubMed] [Google Scholar]
- [151].Jensen TJ, Loo MA, Pind S, Williams DB, Goldberg AL, Riordan JR. Multiple proteolytic systems, including the proteasome, contribute to CFTR processing. Cell. 1995;83:129–135. doi: 10.1016/0092-8674(95)90241-4. [DOI] [PubMed] [Google Scholar]
- [152].Grove DE, Rosser MF, Ren HY, Naren AP, Cyr DM. Mechanisms for rescue of correctable folding defects in CFTRDelta F508. Mol Biol Cell. 2009;20:4059–4069. doi: 10.1091/mbc.E08-09-0929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [153].Ward CL, Omura S, Kopito RR. Degradation of CFTR by the ubiquitin-proteasome pathway. Cell. 1995;83:121–127. doi: 10.1016/0092-8674(95)90240-6. [DOI] [PubMed] [Google Scholar]
- [154].Finley D, Ciechanover A, Varshavsky A. Thermolability of ubiquitin-activating enzyme from the mammalian cell cycle mutant ts85. Cell. 1984;37:43–55. doi: 10.1016/0092-8674(84)90299-x. [DOI] [PubMed] [Google Scholar]
- [155].Yang Y, Janich S, Cohn JA, Wilson JM. The common variant of cystic fibrosis transmembrane conductance regulator is recognized by hsp70 and degraded in a pre-Golgi nonlysosomal compartment. Proc Natl Acad Sci U S A. 1993;90:9480–9484. doi: 10.1073/pnas.90.20.9480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [156].Strickland E, Qu BH, Millen L, Thomas PJ. The molecular chaperone Hsc70 assists the in vitro folding of the N-terminal nucleotide-binding domain of the cystic fibrosis transmembrane conductance regulator. J Biol Chem. 1997;272:25421–25424. doi: 10.1074/jbc.272.41.25421. [DOI] [PubMed] [Google Scholar]
- [157].Wang X, Venable J, LaPointe P, Hutt DM, Koulov AV, Coppinger J, Gurkan C, Kellner W, Matteson J, Plutner H, Riordan JR, Kelly JW, Yates JR, 3rd, Balch WE. Hsp90 cochaperone Aha1 downregulation rescues misfolding of CFTR in cystic fibrosis. Cell. 2006;127:803–815. doi: 10.1016/j.cell.2006.09.043. [DOI] [PubMed] [Google Scholar]
- [158].McCracken AA, Brodsky JL. Assembly of ER-associated protein degradation in vitro: dependence on cytosol, calnexin, and ATP. J Cell Biol. 1996;132:291–298. doi: 10.1083/jcb.132.3.291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [159].Caplan S, Green R, Rocco J, Kurjan J. Glycosylation and structure of the yeast MF alpha 1 alpha-factor precursor is important for efficient transport through the secretory pathway. J Bacteriol. 1991;173:627–635. doi: 10.1128/jb.173.2.627-635.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [160].Brodsky JL, Werner ED, Dubas ME, Goeckeler JL, Kruse KB, McCracken AA. The requirement for molecular chaperones during endoplasmic reticulum-associated protein degradation demonstrates that protein export and import are mechanistically distinct. J Biol Chem. 1999;274:3453–3460. doi: 10.1074/jbc.274.6.3453. [DOI] [PubMed] [Google Scholar]
- [161].Hiller MM, Finger A, Schweiger M, Wolf DH. ER degradation of a misfolded luminal protein by the cytosolic ubiquitin-proteasome pathway. Science. 1996;273:1725–1728. doi: 10.1126/science.273.5282.1725. [DOI] [PubMed] [Google Scholar]
- [162].Plemper RK, Bohmler S, Bordallo J, Sommer T, Wolf DH. Mutant analysis links the translocon and BiP to retrograde protein transport for ER degradation. Nature. 1997;388:891–895. doi: 10.1038/42276. [DOI] [PubMed] [Google Scholar]
- [163].Knop M, Finger A, Braun T, Hellmuth K, Wolf DH. Der1, a novel protein specifically required for endoplasmic reticulum degradation in yeast. EMBO J. 1996;15:753–763. [PMC free article] [PubMed] [Google Scholar]
- [164].Spear ED, Ng DT. Stress tolerance of misfolded carboxypeptidase Y requires maintenance of protein trafficking and degradative pathways. Mol Biol Cell. 2003;14:2756–2767. doi: 10.1091/mbc.E02-11-0717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [165].Stolz A, Wolf DH. Use of CPY and its derivatives to study protein quality control in various cell compartments. Methods Mol Biol. 2012;832:489–504. doi: 10.1007/978-1-61779-474-2_35. [DOI] [PubMed] [Google Scholar]
- [166].Van Goor F, Hadida S, Grootenhuis PD, Burton B, Stack JH, Straley KS, Decker CJ, Miller M, McCartney J, Olson ER, Wine JJ, Frizzell RA, Ashlock M, Negulescu PA. Correction of the F508del-CFTR protein processing defect in vitro by the investigational drug VX-809. Proc Natl Acad Sci U S A. 2011;108:18843–18848. doi: 10.1073/pnas.1105787108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [167].Okuda-Shimizu Y, Hendershot LM. Characterization of an ERAD pathway for nonglycosylated BiP substrates, which require Herp. Mol Cell. 2007;28:544–554. doi: 10.1016/j.molcel.2007.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [168].Kravtsova-Ivantsiv Y, Ciechanover A. Non-canonical ubiquitin-based signals for proteasomal degradation. J Cell Sci. 2012;125:539–548. doi: 10.1242/jcs.093567. [DOI] [PubMed] [Google Scholar]
- [169].Xu P, Duong DM, Seyfried NT, Cheng D, Xie Y, Robert J, Rush J, Hochstrasser M, Finley D, Peng J. Quantitative proteomics reveals the function of unconventional ubiquitin chains in proteasomal degradation. Cell. 2009;137:133–145. doi: 10.1016/j.cell.2009.01.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [170].Kostova Z, Wolf DH. Importance of carbohydrate positioning in the recognition of mutated CPY for ER-associated degradation. J Cell Sci. 2005;118:1485–1492. doi: 10.1242/jcs.01740. [DOI] [PubMed] [Google Scholar]
- [171].Spear ED, Ng DT. Single, context-specific glycans can target misfolded glycoproteins for ER-associated degradation. J Cell Biol. 2005;169:73–82. doi: 10.1083/jcb.200411136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [172].Alzayady KJ, Wojcikiewicz RJ. The role of Ca2+ in triggering inositol 1,4,5-trisphosphate receptor ubiquitination. Biochem J. 2005;392:601–606. doi: 10.1042/BJ20050949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [173].Rivett AJ. Intracellular distribution of proteasomes. Curr Opin Immunol. 1998;10:110–114. doi: 10.1016/s0952-7915(98)80040-x. [DOI] [PubMed] [Google Scholar]
- [174].Kalies KU, Allan S, Sergeyenko T, Kroger H, Romisch K. The protein translocation channel binds proteasomes to the endoplasmic reticulum membrane. EMBO J. 2005;24:2284–2293. doi: 10.1038/sj.emboj.7600731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [175].Ng W, Sergeyenko T, Zeng N, Brown JD, Romisch K. Characterization of the proteasome interaction with the Sec61 channel in the endoplasmic reticulum. J Cell Sci. 2007;120:682–691. doi: 10.1242/jcs.03351. [DOI] [PubMed] [Google Scholar]
- [176].Scott DC, Schekman R, et al. Role of Sec61p in the ER-associated degradation of short-lived transmembrane proteins. J Cell Biol. 2008;181:1095–1105. doi: 10.1083/jcb.200804053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [177].Ye Y, Shibata Y, Yun C, Ron D, Rapoport TA. A membrane protein complex mediates retrotranslocation from the ER lumen into the cytosol. Nature. 2004;429:841–847. doi: 10.1038/nature02656. [DOI] [PubMed] [Google Scholar]
- [178].Lilley BN, Ploegh HL. A membrane protein required for dislocation of misfolded proteins from the ER. Nature. 2004;429:834–840. doi: 10.1038/nature02592. [DOI] [PubMed] [Google Scholar]
- [179].Ushioda R, Hoseki J, Araki K, Jansen G, Thomas DY, Nagata K. ERdj5 is required as a disulfide reductase for degradation of misfolded proteins in the ER. Science. 2008;321:569–572. doi: 10.1126/science.1159293. [DOI] [PubMed] [Google Scholar]
- [180].Schuberth C, Buchberger A. Membrane-bound Ubx2 recruits Cdc48 to ubiquitin ligases and their substrates to ensure efficient ER-associated protein degradation. Nat Cell Biol. 2005;7:999–1006. doi: 10.1038/ncb1299. [DOI] [PubMed] [Google Scholar]
- [181].Neuber O, Jarosch E, Volkwein C, Walter J, Sommer T. Ubx2 links the Cdc48 complex to ER-associated protein degradation. Nat Cell Biol. 2005;7:993–998. doi: 10.1038/ncb1298. [DOI] [PubMed] [Google Scholar]
- [182].Horn SC, Hanna J, Hirsch C, Volkwein C, Schutz A, Heinemann U, Sommer T, Jarosch E. Usa1 functions as a scaffold of the HRD-ubiquitin ligase. Mol Cell. 2009;36:782–793. doi: 10.1016/j.molcel.2009.10.015. [DOI] [PubMed] [Google Scholar]