

Abstract
ρ-Da1a is a three-finger fold toxin from green mamba venom that is highly selective for the α1A-adrenoceptor. This toxin has atypical pharmacological properties, including incomplete inhibition of 3H-prazosin or 125I-HEAT binding and insurmountable antagonist action. We aimed to clarify its mode of action at the α1A-adrenoceptor. The affinity (pKi 9.26) and selectivity of ρ-Da1a for the α1A-adrenoceptor were confirmed by comparing binding to human adrenoceptors expressed in eukaryotic cells. Equilibrium and kinetic binding experiments were used to demonstrate that ρ-Da1a, prazosin and HEAT compete at the α1A-adrenoceptor. ρ-Da1a did not affect the dissociation kinetics of 3H-prazosin or 125I-HEAT, and the IC50 of ρ-Da1a, determined by competition experiments, increased linearly with the concentration of radioligands used, while the residual binding by ρ-Da1a remained stable. The effect of ρ-Da1a on agonist-stimulated Ca2+ release was insurmountable in the presence of phenethylamine- or imidazoline-type agonists. Ten mutations in the orthosteric binding pocket of the α1A-adrenoceptor were evaluated for alterations in ρ-Da1a affinity. The D1063.32A and the S1885.42A/S1925.46A receptor mutations reduced toxin affinity moderately (6 and 7.6 times, respectively), while the F862.64A, F2886.51A and F3127.39A mutations diminished it dramatically by 18- to 93-fold. In addition, residue F862.64 was identified as a key interaction point for 125I-HEAT, as the variant F862.64A induced a 23-fold reduction in HEAT affinity. Unlike the M1 muscarinic acetylcholine receptor toxin MT7, ρ-Da1a interacts with the human α1A-adrenoceptor orthosteric pocket and shares receptor interaction points with antagonist (F862.64, F2886.51 and F3127.39) and agonist (F2886.51 and F3127.39) ligands. Its selectivity for the α1A-adrenoceptor may result, at least partly, from its interaction with the residue F862.64, which appears to be important also for HEAT binding.
Introduction
Many toxins that interact with voltage- and ligand-gated ion channels display both high affinity and selectivity. For the last 50 years, these properties have been used to identify, purify and classify membrane targets and for structure/function studies. The particular properties of these toxins are now also being exploited pharmacologically, and some toxins are used as drugs and others are currently undergoing preclinical trials [1]–[4].
Although voltage- and ligand-gated ion channels are the main targets for neurotoxins, other targets, including G Protein-Coupled Receptors (GPCRs), have also been identified. The animal toxins active on GPCRs can be divided into two families [5]. Members of the first family, the sarafotoxins, conopressin or contulakin-G mimic the natural agonist of the targeted receptor: endothelin, vasopressin and neurotensin, respectively. The second family consists of highly reticulated toxins with folds that are unrelated to any natural ligands. Nine have been isolated from mamba venoms and are active against muscarinic acetylcholine receptors and adrenoceptors (ARs), [6]. Two other toxins: ρ-TIA, from Conus tulipa and β-cardiotoxin, from the snake Ophiophagus hannah, are active against α1-ARs [7] and β-ARs [8], respectively. We suspected that animal venoms are a potential source of novel GPCR binding agents, and developed a screening strategy, initially focused on the binding of green mamba venom to ARs. This screening led to the isolation of two novel snake toxins from Dendroaspis angusticeps: ρ-Da1a, previously called AdTx1, which is highly selective for the α1A-AR [9], and ρ-Da1b, selective for α2-ARs [10]. ρ-Da1a and ρ-Da1b are peptides of 65 and 66 residues, respectively, reticulated by four disulfide bridges, and are members of the three-finger-fold toxin family. The modes of action of these peptide ligands on ARs are not clear. In equilibrium binding experiments, neither ρ-Da1a nor ρ-Da1b fully inhibits radioligand binding [9],[10]. In addition, in isolated prostatic muscle, ρ-Da1a acts as an insurmountable antagonist [9], and cell-based assays indicate that ρ-Da1b is a non-competitive antagonist at the human α2A-AR [10].
Adrenergic and muscarinic toxins isolated from mamba snake venoms belong to the same three-finger-fold family and display substantial sequence identity (52–97%) [5]. The interactions between MT1, MT7 and M1 muscarinic receptors have been studied in detail [11]–[14]. Pharmacological studies indicate competition between MT1 and 3H-N- methylscopolamine [11], [14], [15]. In contrast, MT7 significantly affects the dissociation kinetics of 3H-N- methylscopolamine and 3H-acetylcholine [14], [16] and leaves residual binding in equilibrium binding experiments [11] suggesting an allosteric mode of action. As a negative allosteric modulator, MT7 reduces the efficacy and potency of carbamylcholine at M1 muscarinic receptors expressed in CHO cells [16] and interacts mainly with the extracellular loop 2 of this receptor [13], [17]. The smallest peptide ligand acting at ARs, ρ-TIA, is a 19-residue toxin from Conus tulipa, and has been classified as a non-competitive α1B-AR antagonist that accelerates 3H-prazosin dissociation kinetics and antagonizes α1B-AR activation by an insurmountable mechanism [7]. A recent experimentally-based model shows that ρ-TIA interacts primarily with extracellular loop 3 (ec3) of the α1B-AR, consistent with its allosteric properties [18]. Thus, both MT7 and ρ-TIA display a negative allosteric mode of action by interacting with extracellular loops of their receptor targets, namely ec2 of the M1 AChR, and ec3 of the α1B-AR. ρ-TIA, however, shows only 10 to 25-fold selectivity for the α1B-AR over the other α1-AR subtypes, and has been described as a competitive antagonist at the α1A-AR although it does not fully inhibit 125I-HEAT binding [19].
These observations have led to hypotheses regarding the mode of action of these peptide toxins at receptor targets. The aims of our study were to use equilibrium and kinetic binding experiments to establish the pharmacological behavior of ρ-Da1a at the α1A-AR, to define the effect of ρ-Da1a on agonist-stimulated Ca2+ release, and to use site-directed mutagenesis to analyze the α1A-AR binding site for this peptide toxin.
Experimental Procedures
125I-HEAT, 3H-prazosin, 3H-rauwolscine and 3H-CGP-12177 were purchased from PerkinElmer (Courtaboeuf, France). Non radioactive HEAT was obtained from Tocris (Ellisville, Missouri, USA), and 5-(N-ethyl-N-isopropyl-amiloride (EPA), prazosin, yohimbine, and propranonol were obtained from Sigma-Aldrich (St Quentin-Fallavier, France).
Protein quantification
Total protein and membrane protein concentrations were determined using the Bio-Rad protein assay, with bovine serum albumin as standard.
Site-directed mutagenesis
α1A-AR cDNA inserted in the prK5 vector was kindly provided by Michael Brownstein (Craig Venter Institute, Rockville, MD). Point mutations were introduced into the α1A-AR gene by sense and antisense primers (Sigma-Aldrich, St Quentin-Fallavier, France) containing the desired changes, using the QuikChange Site-Directed Mutagenesis kit. The incorporation of each mutation was verified by DNA sequencing. The variants F3087.35A and F3127.39A were generous gifts from Dr. Diane Perez (The Cleveland Clinic Foundation, Cleveland, Ohio, USA).
Cell culture and membrane preparation
CHO cells stably expressing α1-ARs were kindly provided by Dr. Hervé Paris (INSERM U858, Toulouse, France) and were grown in a 50∶50 Dulbecco's Modified Eagle's Medium (DMEM)/Ham's F12 medium supplemented with 10% (v/v) foetal bovine serum (FBS), glutamine (2 mM), penicillin (100 units/ml) and streptomycin (100 µg/ml) at 37°C with 5% CO2. COS-7 cells were grown at 37°C under 5% CO2 in Dulbecco's modified Eagle's medium containing 10% fetal calf serum, 1% penicillin and 1% glutamine (Sigma-Aldrich, St Quentin-Fallavier, France). At 80% confluence, the cells were transfected using a calcium phosphate precipitation method for transient expression of the genetic construct. After 48 h incubation at 37°C, cells were harvested and the membranes were prepared as follow. Cells were washed with ice-cold phosphate buffer and centrifuged at 1700 g for 10 min (4°C). The pellet was suspended in ice-cold buffer (1 mM EDTA, 25 mM sodium phosphate, and 5 mM MgCl2, pH 7.4) and homogenized using an Potter-Elvehjem homogenizer (Fisher Scientific Labosi, Elancourt, France). The homogenate was centrifuged at 1700 g for 15 min (4°C). The sediment was resuspended in buffer, homogenized, and centrifuged at 1700 g for 15 min (4°C). The combined supernatants were centrifuged at 35,000 g for 30 min (4°C), and the pellet was suspended in the same buffer (0.1 ml/dish). The CHO cells used for Ca2+ release experiments also stably express the human α1A-AR (Bmax 531±94 fmol/mg protein, pKD for 125I-HEAT 9.2±0.09 [20]. Cells were grown in a 50∶50 Dulbecco's Modified Eagle's Medium (DMEM)/Ham's F12 medium supplemented with 10% (v/v) foetal bovine serum (FBS), glutamine (2 mM), penicillin (100 units/ml) and streptomycin (100 µg/ml) at 37°C with 5% CO2. Media was changed every 2–3 days and cells were passaged when confluent with 0.05% trypsin and 0.02% EDTA.
Binding assays
We used 3H-prazosin and 125I-HEAT (all incubations were done in the dark) as selective ligands for α1-ARs, 3H-rauwolscine for α2-ARs and 3H-CCGP-12177 for β-ARs. Non-specific binding to α1, α2 and β-ARs was measured in presence of prazosin (10 µM), yohimbine (10 µM) and propanolol (10 µM), respectively. Binding experiments were performed in a 100 µL reaction mix at room temperature in buffer composed of 50 mM Tris-HCl, pH 7.4, 10 mM MgCl2, 1 g/L BSA. Reactions were stopped by filtration through 96 GF/C filter plates pre-incubated with 0.5% polyethylenimine. An aliquot of 25 µL of Microscint 0 was added onto each dry filter and the radioactivity was quantified on a TopCount beta counter with a 33% yield (PerkinElmer, Courtaboeuf, France). Saturation binding assays were performed using a fixed amount of receptors and a series of concentrations of 125I-HEAT with an incubation time of 1 h. Competition binding assays were performed by mixing the radioligand (2 nM of 3H-prazosin or 3H-rauwolscine, 0.2–1.3 nM of 125I-HEAT, 6 nM of 3H-CGP-12177) with a range of competitor concentrations before adding membranes (α1A-AR: 1 µg for 3H-prazosin and 0.1 µg for 125I-HEAT, α1B: 3 µg, α1D: 29 µg, α2A: 140 µg; α2B: 100 µg, α2C: 3 µg, β1: 3 µg, β2: 1.5 µg, or α1A-mutants: 0.1–1 µg), for 16 h of incubation. Dissociation kinetics experiments were performed by pre-equilibrating 125I-HEAT (400 pM) or 3H-prazosin (2 nM) for 3 hours with α1A-AR COS-7 cell membranes (0.2 or 1 µg, respectively). Radiotracer dissociation was then measured following addition of HEAT (5 µM) or prazosin (10 µM) alone or with ρ-Da1a (2.5 µM), 5-(N-ethyl-N-isopropyl)-amiloride (EPA, 150 µM) or adrenaline (2 mM).
Measurement of intracellular Ca2+ concentration
CHO-K1 cells expressing the α1A-AR were seeded at 2×104 cells per well in 96-well plates overnight. The following morning, the media was removed and cells washed three times in a modified Hanks' buffered saline solution (HBSS; composition in mM: NaCl 150, KCl 2.6, MgCl2.2H2O 1.18, D-glucose 10, Hepes 10, CaCl2.2H2O 2.2, probenecid 2, pH 7.4) containing BSA 0.5% (w/v). In light-diminished conditions cells were treated with fluoro-4 (0.1% v/v in modified HBSS, 1 h, 37°C). Excess fluoro-4 not taken up by the cells was removed by washing twice in modified HBSS and then cells incubated for a further 30 min in the absence or presence of differing concentrations of ρ-Da1a before the assay plate was transferred to a FlexStation (Molecular Devices, Palo Alto CA, USA). Real-time fluorescence measurements were recorded every 1.7 seconds over 200 seconds, with agonist (noradrenaline, phenylephrine, A61603 or oxymetazoline) additions occurring after 17 seconds, using an excitation wavelength of 485 nm and reading emission wavelength of 520 nm. All experiments were performed in duplicate. Agonist responses represent the difference between basal fluorescence and peak [Ca2+]i measurements expressed as a percentage of the response to A23187 (1 µM) in each experiment.
Data analysis
Binding data were analyzed by nonlinear regression using the KaleidaGraph 4.0 software (Synergy software, Reading, PA). pKD values and Bmax (number of binding sites) were determined by applying a nonlinear regression to data obtained with saturation binding assays. The nonlinear regression used was BS = (Bmax*A)/KD+A, where BS is the specific binding, Bmax the number of binding sites, A the concentration of radioligand, and KD the dissociation constant of the radioligand. Data resulting from competition binding assays were analyzed using the Hill equation for IC50 and curve slope estimations. The binding affinity (pKi) of ρ-Da1a was determined from the IC50 value of inhibition curves using the Cheng and Prussof equation [21]. The linear curves were analyzed with IC50 = Ki+(L/KD)*Ki. Dissociation kinetics were analyzed using a simple equation of exponential decay BS * exp (-Koff*t), where BS is the specific binding at time zero and Koff is the dissociation rate constant. Results are expressed as mean ± s.e. mean from n independent experiments. One-way Anova test was used to compare values. A p<0.05 was accepted for statistical significance.
Values for intracellular Ca2+ release are expressed as mean ± s.e. mean from n independent experiments. Data were analysed using non-linear curve fitting (Graph Pad PRISM v5.02) to obtain pEC15 values for the [Ca2+]i assays. Antagonists such as ρ-Da1a that have slow dissociation kinetics are prone to display hemi-equilibrium artifacts in functional transient responses such as measurement of intracellular Ca2+ levels. As such, when competing with an agonist, the maximal response achieved by the agonist reduces in the presence of higher antagonist concentrations due to the inaccessibility of a large pool of the receptors in the time taken for the transient response to occur [22]. This affects the ability of a Schild analysis to estimate the pKB of ρ-Da1a. In order to account for this, the pKB value for ρ-Da1a was calculated by the modified Lew-Angus method [23] using pEC15 values, based on the extent of reduction in agonist maximal responses in the presence of ρ-Da1a. The pEC15 values were plotted against the concentration of antagonist and non-linear regression applied [23], [24] to estimate pKB values for ρ-Da1a against each of the four different agonists.
Homology modeling
A model of the α1A-AR was generated with MODELLER [25]. The receptor with the most similar sequence to the α1A-AR is the β2-AR subtype, with an overall amino acid sequence identity of 21% [26], [27], identity within 7TM domain from helix 1 to helix 8 (excluding intracellular ic3 loop) 38%, sequence similarity 61% [BLASTP]. Nine β2-AR structures are available (2RH1, 3D4S, 3KJ6, 3NY8, 3NY9, 3NYA, 3PDS, 3P0G, 3SN6) and are very similar (Cα RMSDs<1.5 Å for 253 residues). We used the X-ray structure with the highest resolution (2RH1) as a template [28].
Results
ρ-Da1a, (previously AdTx1) [9], was renamed according to a rational nomenclature [29]. A recombinant expression system producing the toxin with an extra glycine residue at its N-terminus was developed (Figure S1 in File S1). Recombinant ρ-Da1a displays the same affinity as the chemically synthesized toxin, indicating that the N-terminal glycine has no consequences for function. The pharmacological experiments reported in this study were performed with the recombinant form of the toxin.
Selectivity of ρ-Da1a
ρ-Da1a affinity was recently determined in tissue preparations, and using human and rat α1-ARs expressed in yeast [9]. To complete the ρ-Da1a selectivity profile, we expressed human ARs in eukaryotic cells and performed competition binding with additional receptor subtypes. The pKi values derived from these experiments [21] were: 9.19±0.09 for α1A-ARs, 7.28±0.09 for α1B-ARs, 6.85±0.08 nM for α2C-ARs, and 5.95±0.08 for α1D-ARs. No significant effect was observed with 10 µM of ρ-Da1a at α2A, α2B, β1, or β2-ARs. For α1A- and α1B-ARs, even the highest concentrations of ρ-Da1a did not completely inhibit 3H-prazosin binding, which remained at 18±3% and 17±1%, respectively (Fig. 1).
Figure 1. Pharmacological profile of ρ-Da1a binding to various human AR subtypes expressed in eukaryotic cells.

Binding inhibition curves for 3H-prazosin (2 nM), 3H-rauwolscine (2 nM) and 3H-CGP-12177 (6 nM) on hα1A- (1 µg, ○), hα1B- (3 µg, •), hα1D- (29 µg, □), hα2A- (140 µg, ◊), hα2B- (100 µg, Δ), hα2C- (3 µg, x), β1- (3 µg,▾) and β2-AR (1.5 µg, ▪) with recombinant ρ-Da1a. n = 4.
ρ-Da1a, prazosin and HEAT compete at α1A-ARs
3H-prazosin binding was fully inhibited by HEAT (pKi 9.66±0.08 nM, Hill slope 0.85, Fig. 2) and 125I-HEAT binding was fully displaced by prazosin (pKi 9.18±0.07 nM, Hill slope 0.95). However, as observed with 3H-prazosin, ρ-Da1a interacts very efficiently with the α1A-AR (pKi 9.26±0.07 nM, Hill slope 0.92, Fig. 2), but does not inhibit more than 80% of 125I-HEAT binding. This residual binding is stable with time, as we detected no variation with incubation times from 2 to 24 hours (data not shown).
Figure 2. Inhibition of 3H-prazosin (2 nM, 1 µg, open symbols) by HEAT (□), and ρ-Da1a (circle), and inhibition of 125I-HEAT (0.2 nM, 0.2 µg, full symbols) binding by prazosin (♦) and ρ-Da1a (circle) to α1A-AR.

n = 3.
Dissociation kinetic experiments are classically used to identify negative allosteric modulators [30], [31]. The influence of ρ-Da1a on the dissociation kinetics of 3H-prazosin and 125I-HEAT was studied in comparison with adrenaline and the negative allosteric modulator EPA (Fig. 3). 3H-prazosin dissociation from α1A-ARs was mono-exponential and the dissociation rate was 0.05±0.01 min−1. Consistent with previous studies [32], this value was increased 2.6 times in the presence of 150 µM EPA (Koff+ EPA = 0.15 min−1). In contrast, neither 2 mM adrenaline nor 2.5 µM ρ-Da1a affected the 3H-prazosin dissociation rate (Koff+adrenaline = 0.054 min−1; Koff+ρ-Da1a = 0.059 min−1, Fig. 3, n = 2) in the presence of excess prazosin. 125I-HEAT dissociation rates were measured in the absence (Koff = 0.062 min−1) and in the presence of ρ-Da1a (Koff HEAT+ρ-Da1a = 0.058 min−1), prazosin (Koff HEAT+prazosin = 0.06 min−1), and EPA (Koff HEAT+EPA = 0.37 min−1, Fig. 3, n = 2): neither prazosin nor ρ-Da1a affected the HEAT dissociation rate; whereas EPA increased the dissociation rate by six-fold.
Figure 3. Influence of various ligands on 3H-prazosin and 125I-HEAT dissociation.

Panel A: Dissociation of 3H-prazosin (2 nM) binding to α1A-AR (1 µg) in the presence of prazosin (10 µM, black), prazosin plus ρ-Da1a (2.5 µM, blue), prazosin plus adrenaline (2 mM, red) and prazosin plus EPA (150 µM, green). Panel B : dissociation of 125I-HEAT (0.4 nM) binding to α1A-AR (0.2 µg) in the presence of HEAT (5 µM, black), HEAT plus ρ-Da1a (2.5 µM, blue), HEAT plus prazosin (10 µM, red) and HEAT plus EPA (150 µM, green). n = 2.
The ρ-Da1a IC50 values were determined using various concentrations of radiotracers in competition binding experiments (Fig. 4). Eleven concentrations of 3H-prazosin (0.2, 0.5, 1.0, 1.86, 3.55, 4.53, 8.0, 9.14, 10, 13 and 16 nM) were dose-dependently inhibited by ρ-Da1a (IC50 of 2.4, 2.9, 2.6, 3.55, 10.2, 5.54, 18, 14, 20, 25 and 31 nM) with Hill slopes between 0.8 and 1.1. Residual binding in the presence of 3H-prazosin fluctuated between 15 to 25% of the total binding, but did not show any trend to concentration-dependence. The curve IC50ρ-Da1a as a function of 3H-prazosin concentration (L) fitted the linear regression IC50ρ-Da1a = 1.067+1.82*L, incompatible with a negative allosteric modulation (Fig. 4A). Using the equation IC50ρ-Da1a = Kiρ-Da1a+(Kiρ-Da1a * Lprazosin/Kdprazosin) [21], this experiment gave a Kiρ-Da1a of 1.067 nM (pKi 8.97) and a Kdprazosin of 0.586 nM (pKd 9.23). An analogous experiment was performed using 125I-HEAT. Ten concentrations (0.1, 0.13, 0.2, 0.3, 0.38, 0.4, 0.5, 0.7, 0.9 and 1.25 nM) were inhibited by ρ-Da1a (IC50 of 4.0, 2.75, 5.3, 3.23, 6.84, 8.0, 8.18, 11.5, 12.7 and 23.4 nM) with Hill slopes between 0.9 and 1.4. Residual binding fluctuated between 18 to 28% of the total binding (Fig. 4B). IC50ρ-Da1a as a function of 125I-HEAT concentrations fitted the equation IC50ρ-Da1a = 0.706+16.22*L which gave a Kiρ-Da1a of 0.706 nM (pKi 9.15) and a KdHEAT of 0.0435 nM (pKd 10.36).
Figure 4. Inhibition of the binding of a series of concentrations of 3H-prazosin and 125I-HEAT to α1A-AR by ρ-Da1a.

Panel A 3H-prazosin binding (from 0.2 to 16 nM) inhibited by ρ-Da1a. Panel B 125I-HEAT binding (from 0.1 to 1.25 nM) inhibited by ρ-Da1a. Panel C and D: Fitting, by the Cheng and Prusoff equation IC50 = Ki+Ki(L/Kd), of IC50 values as a function of the radiotracer concentrations.
Insurmountable antagonism of intracellular Ca2+ release by ρ-Da1a
We next tested the effect of ρ-Da1a on responses to noradrenaline and phenylephrine (phenethylamine agonists) and A61603 and oxymetazoline (imidazoline agonists). In CHO-K1 cells expressing the α1A-AR, all agonists stimulated Ca2+ release (Figure 5), with pEC50 and Emax values consistent with previous work [20] – noradrenaline pEC50 8.63±0.08, Emax (as a percentage of peak A23187 response) 77.8±1.5, phenylephrine pEC50 7.64±0.16, Emax 64.8±2.4, A61603 pEC50 10.17±0.07, Emax 75.5±1.3, and oxymetazoline pEC50 9.09±0.08, Emax 60.8±1.3. Increasing concentrations of ρ-Da1a reduced the maximal response to each of the four agonists while shifting the curves to the right. The pKB values for ρ-Da1a were calculated by a modified Lew-Angus method [23] and were similar irrespective of the agonist employed (7.71±0.05 vs noradrenaline; 7.60±0.04 vs phenylephrine; 7.66±0.10 vs A61603; 7.67±0.05 vs oxymetazoline).
Figure 5. Concentration-response curves for stimulation of Ca2+ release by the α1A-AR.

Agonist responses represent the difference between basal fluorescence and the peak [Ca2+]i (reached within 20 sec of agonist addition), expressed as a percentage of the response to the Ca2+ ionophore A23187 (1 µM). Concentration-dependent Ca2+ release was stimulated by noradrenaline (panel A), phenylephrine (panel B), A61603 (panel C) or oxymetazoline (panel D). Concentration response curves were performed in the presence or absence of differing concentrations of ρ-Da1a (• control, ▪ 1 nM, ▴ 3 nM, ▾ 10 nM, ♦ 30 nM, ◊ 100 nM, ○ 300 nM). Values are means ± SEM of 3–4 independent experiments.
Molecular characterization of the ρ-Da1a/α1A-AR interaction
We investigated the involvement of residues within the orthosteric pocket of α1-ARs that are known to interact with agonists and/or antagonists: F862.64 [33], D1063.32 (D125 in α1B-AR) [34]–[36], F1875.41 [37], S1885.42 and S1925.46 [38], F2886.51 (F310 on α1B-AR) [18], [39], M2926.55 [40] and F3087.35 and F3127.39 [18], [41] (superscripts refer to the Ballesteros-Weinstein numbering system for residues in 7TM helices). In addition, we tested the positions F1935.47 and F2816.44, predicted to play a role in the stabilization of the active state of α1A-AR [42] (Table 1). In saturation binding experiments, 125I-HEAT affinity values for D1063.32A, F1935.47A, F2816.44A, F2886.51A, M2926.55A and F3087.35A were not significantly different from the wild type receptor. One mutated receptor, F1875.41A, had significantly higher affinity for 125I-HEAT with a pKD of 10.70±0.005 compared to wild type pKD 10.05±0.09 (Table 1, p<0.05). Only the F862.64A mutant showed a substantial 23-fold loss of 125I-HEAT affinity (pKD 8.68±0.09, p<0.05, Fig. 6, Table 1). The transiently transfected mutant receptors showed marked variability in expression level, with Bmax values ranging from 0.63 up to 29 pmol/mg protein compared to 11.3 pmol/mg protein for the wild type α1A-AR (Table 1), however there was no correlation between receptor abundance and the observed binding affinity of 125I-HEAT. For example, D1063.32A (0.63 pmol/mg protein) and F3087.35A (29 pmol/mg protein) both displayed a similar pKd to each other and to the wild type receptor.
Table 1. Effect of human α1A-AR mutations on receptor expression and affinity for HEAT and ρ-Da1a.
| Variant | Position | 125I- HEAT | HEAT | ρ-Da1a | ||||
| Bmax pmol/mg | pKd | Ratio | pKi | ratio | pKi | Ratio | ||
| WT | 11.3±2.3 | 10.05±0.14 | 1 | 9.57±0.08 | 1 | 9.26±0.07 | 1 | |
| F86A | 2.64 | 22.6±2.4 | 8.68±0.09 | 23 * | 8.21±0.09 | 23 * | 7.70±0.06 | 36 * |
| D106A | 3.32 | 0.63±0.05 | 9.82±0.11 | 1.7 | 9.74±0.12 | 0.67 | 8.48±0.11 | 6.0 * |
| F187A | 5.41 | 20.5±3.5 | 10.70±0.01 | 0.22 * | 9.12±0.07 | 1.4 | ||
| SS-AA | 5.42–5.46 | 13.3±3.3 | 10.15±0.01 | 0.78 | 8.38±0.09 | 7.6 * | ||
| F193A | 5.47 | 11.5±2.2 | 9,60±0.09 | 2.8 | 9.64±0.11 | 0.42 | ||
| F281A | 6.44 | 18.2±4.8 | 9.46±0.09 | 3.9 | 9.66±0.09 | 0.40 | ||
| F288A | 6.51 | 14.3±2.5 | 9.92±0.12 | 1.3 | 8.00±0.08 | 18 * | ||
| M292A | 6.55 | 15.8±3.5 | 9.69±0.09 | 2.2 | 9.41±0.11 | 0.71 | ||
| F308A | 7.35 | 29±4.2 | 9.66±0.11 | 2.4 | 9.15±0.07 | 1.3 | ||
| F312A | 7.39 | 3.2±0.12 | 10.40±0.10 | 0.44 | 7.28±0.10 | 93 * | ||
for p<0.05. Position refers to the Ballesteros-Weinstein numbering scheme for residues within TM domains of G protein-coupled receptors. n = 3–6.
Figure 6. Saturation experiments with 125I-HEAT on receptor variants.

Curves for competition of HEAT and ρ-Da1a with binding of 125I-HEAT are shown at wild type, D1063.32A and F862.64A receptors (Figure 7). As seen in the saturation binding experiment, HEAT had similar affinity for the D1063.32A variant (pKi 9.74±0.12) and the wild type receptor (pKi 9.57±0.08) but was strongly affected by the F862.64A mutation (pKi 8.21±0.09). ρ-Da1a affinity at the D1063.32A variant was reduced by 6-fold (pKi 8.48±0.11) compared to the wild type receptor while affinity at F862.64A was reduced by 36-fold (pKi 7.70±0.06). The mutation F862.64A affects both HEAT and ρ-Da1a affinities, suggesting that the structural organization of the receptor could have been perturbed by this modification. We used the radioligand 3H-prazosin to examine this point and found that prazosin affinity for the F862.64A mutant (pKd 9.21±0.07) was very close to the one measured for wild type receptor (9.26±0.05; data not shown). While this mutation may alter interactions between F862.64 and other aromatic residues within the orthosteric pocket [43], the retention of prazosin affinity indicates that there is no global effect on the native structural conformation of the receptor. As seen for the wild type α1A-AR, ρ-Da1a inhibited only 75–80% of 125I-HEAT binding in experiments with these mutant receptors.
Figure 7. Receptor affinities for ρ-Da1a (dash lines) and HEAT (solid lines) on mutated α1A-ARs.

Binding inhibition curves for 125I-HEAT binding to WT (200 pM, 0.2 µg, ○), D1063.32A (200 pM, 1 µg, □) and F862.64A (1.3 nM, 0.8 µg, •) receptor variants. n = 3–4.
ρ-Da1a affinities were tested on eight additional receptor variants (Fig. 8). At the F1875.41A, M2926.55A and F3087.35A variants, ρ-Da1a inhibited 125I-HEAT binding with affinities similar to the wild type (Table 1). However the F2886.51A and the F3127.39A variants decreased ρ-Da1a affinity by 18 and 93 times, with pKi of 8.00±0.08 and 7.28±0.10, respectively. Again, ρ-Da1a left residual binding between 20 and 30%, except for the F1875.41A variant (9±3%) receptor (Fig. 8).
Figure 8. Receptor affinities for ρ-Da1a on mutated α1A-ARs.

Binding inhibition curves for 125I-HEAT (200 pM) binding to WT (0.2 µg, black), F1875.41A (0.15 µg, light blue), the double S1885.42,S1925.46-AA (0.3 µg, dark blue), F1935.47A (0.25 µg, green), F2816.44A (0.15 µg, orange), F2886.51A (0.2 µg, red), M2926.55A (0.2 µg, purple), F3087.35A (0.1 µg, brown), F3127.39A (0.8 µg, grey), n = 3–4.
Discussion
ρ-Da1a is the first natural peptide shown to be selective for the α1A-AR. Due to its high selectivity and potent relaxing effect on isolated prostate smooth muscle [5], [9], [44], the peptide is in the process of therapeutic development. In recombinant expression systems, ρ-Da1a can be produced with a final yield of 5 mg per liter of culture. The recombinant toxin interacts with the α1A-AR in a similar manner to the chemically synthesized one.
The selectivity profile of ρ-Da1a for human ARs was established, and confirmed a sub-nanomolar affinity for the α1A-AR subtype: the order of selectivity is α1A>α1B>α2C>α1D>>α2A = α2B = β1 = β2. The affinities of ρ-Da1a for α1-ARs expressed in yeast or in mammalian cells were slightly different: 0.35 and 0.55 nM for α1A-AR, 420 nM and 1110 nM for α1D-AR, and 317 and 53 nM for α1B-AR respectively. These differences in toxin affinity may be related to differences in associated lipids or proteins in the membranes of these cells, as described for the μ-opioid [45] or the dopamine D2S receptors [46].
We have characterized the interaction between ρ-Da1a and the α1A-AR by a series of binding and functional experiments. Our findings from Ca2+ release assays, competition binding and radioligand dissociation curves in the presence of ρ-Da1a generally indicate competition between the toxin and small molecule ligands. On the other hand, ρ-Da1a was unable to completely inhibit orthosteric radioligand binding to α1A-ARs regardless of the time of incubation (2 to 24 h), the radioligand (3H-prazosin or 125I-HEAT), or the expression system (yeast, CHO, or COS-7 cells), a finding more consistent with non-competitive interaction. To examine functional antagonism by ρ-Da1a, we measured blockade of intracellular Ca2+ release following 30 min pre-incubation of CHO-α1A-AR cells with the toxin (Fig. 5). The observed insurmountable antagonism indicates that at higher concentrations of ρ-Da1a, a large proportion of receptors are inaccessible to agonist during the time taken for the transient Ca2+ response [22]. This effect is in part due to the slow dissociation kinetics of ρ-Da1a [9], which prevents the system from reaching equilibrium under the assay conditions [22]. The reduction in Emax is governed by the efficacy of each agonist – for example oxymetazoline is a high affinity, low efficacy agonist that displays a greater loss of maximal response in the presence of ρ-Da1a than high efficacy agonists such as noradrenaline and A61603. Essentially there is lower receptor reserve for responses to oxymetazoline than to noradrenaline or A61603. Despite these differences in reduction of Emax, the pKB values for ρ-Da1a blockade of Ca2+ release remained the same irrespective of the agonist used (between 7.6 and 7.71). These data conform to “non-permissive” antagonism, where receptor occupancy by the toxin prevents simultaneous orthosteric agonist interaction [47]. In the converse situation where a toxin (for example MT7) binds to a receptor at a site distinct from the orthosteric pocket, receptors are able to bind simultaneously both the toxin and an agonist – illustrating “permissive” antagonism characteristic of allosteric modulators [48]. As different agonists adopt distinct poses in the orthosteric binding site, they have the capacity to differentially affect the affinity of an allosteric modulator for the receptor, thus pKB values of the modulator are altered depending on the agonist used [48], [49]. Our finding that the pKB of ρ-Da1a is the same for four agonists belonging to two distinct structural classes, and known to display signaling bias at the α1A-AR [20], corroborates our other data showing that ρ-Da1a has no effect on the dissociation rate of either 3H-prazosin or 125I-HEAT, and that ρ-Da1a affinity for the α1A-AR is reduced by mutation of residues within the orthosteric pocket.
Allosteric modulators are generally characterized by effects on the dissociation rate of orthosteric radioligands in kinetic binding experiments. For example, MT7 significantly affects the dissociation kinetics of 3H-N-methylscopolamine and 3H-acetylcholine in membranes expressing the M1 AChR [14], [16], and the negative allosteric modulator EPA (5-(N-ethyl-N-isopropyl-amiloride) substantially increases the dissociation rate of both 3H-prazosin and 125I-HEAT from the α1A-AR (Fig. 3 in this study, [32]). In contrast, ρ-Da1a has no effect on the dissociation rate of 3H-prazosin or 125I-HEAT (Fig. 3). Reciprocally, prazosin has no effect on the 125I-ρDa1a dissociation rate [9], indicating competitive behavior. In equilibrium binding experiments, there was a linear relationship between the IC50 of ρ-Da1a and the concentration of 3H-prazosin or 125I-HEAT (Fig. 4, panel C and D), again consistent with competitive behavior [50].
The observed competition between ρ-Da1a and radioligands at the α1A-AR suggested that the toxin directly interacts with the orthosteric binding pocket. To provide further evidence for this proposal, ten positions belonging to the orthosteric pocket of the α1A-AR were tested for effects on ρ-Da1a affinity. Previous studies on the α1A-AR have shown that residues F1875.41 [37] and M2926.55 [40] are important for agonist and/or antagonist binding, and the two residues F1935.47 and F2816.44 [42] participate in stabilizing the active conformation of the receptor. Aside from an increase of 4.5 fold in HEAT affinity at the F1875.41A variant, none of these mutations affected either ρ-Da1a or HEAT affinity (Table 1).
The negative charge of D1063.32 is expected to interact with the positive charge of biogenic amines [51], and no binding of 3H-prazosin to the α1A-AR variants D1063.32A or D1063.32A/N167F is observed [36]. The homologous D1253.32A variant of the α1B-AR has been expressed but showed no change in affinity for HEAT [52], although another study has shown a total loss of affinity for HEAT [34]. In our study, HEAT affinity was not affected by alanine substitution of residue D1063.32, whereas toxin affinity was reduced six-fold. The TM5 residues S1885.42/S1925.46 are one helical turn apart, and have been implicated in agonist binding and receptor activation. While either single mutation, S1885.42A or S1925.46A does not alter agonist binding, the double mutation reduces agonist affinity [38]. In our hands, the double mutation moderately reduced ρ-Da1a affinity by 7.6-fold.
We found three key mutations that have major importance for ρ-Da1a binding: F862.64A, F2886.51A and F3127.39A. Residue F3127.39 in the α1A-AR has been described to interacts with prazosin and imidazoline-type agonists [41]. Phenylalanine F310 at position 6.51 in the α1B-AR (F288 in the α1A-AR) is a major determinant for the interaction with the aromatic ring of catecholamines and with α1-AR antagonists like prazosin and phentolamine [18], [39]. Thus, ρ-Da1a shares two major interaction points with prazosin, F2886.51 and F3127.39, and one with phenethylamine-type (F2886.51) and imidazoline-type (F3127.39) agonists. In addition, F862.64 is the only residue unique to the α1A-AR subtype that is important for toxin affinity. In α1B-, α2C- and α1D-ARs, this position is occupied by a leucine, an asparagine and a methionine, respectively. F862.64 was previously identified as a determinant for interaction of the α1A-AR with various antagonists [33]. While a F862.64M receptor mutant did not show any changes in HEAT affinity [33], the F862.64A one strongly decreased it while having no effect on prazosin affinity. This residue most likely contributes to the selectivity of ρ-Da1a for the α1A-AR subtype.
We constructed a homology model of the α1A-AR based on the β2-AR structure [28]. Green, orange and red denote residues with no, moderate or large influence on ρ-Da1a affinity (Fig. 9, panel A and B). Residue D1063.32 and the S1885.42/S1925.46 positions are about 16 Å from the surface of the receptor on one side of the orthosteric site, whereas positions F862.64, F2886.51 and F3127.39 that all interact strongly with ρ-Da1a are located on the opposite side and distributed from the surface down to a depth of 12 Å. As seen for toxin binding, mutation of F862.64 had a substantial effect on HEAT affinity, whereas mutation of F2886.51 had no effect, and mutation of F3127.39 to alanine caused a slight increase in the affinity of HEAT. Although we have yet to define additional residues that contribute to HEAT binding, these findings support the idea that ρ-Da1a and the radioligands have overlapping but distinct binding modes.
Figure 9. Homology modelling of the ρ-Da1a binding site in the α1A-AR and the MT7 toxin.
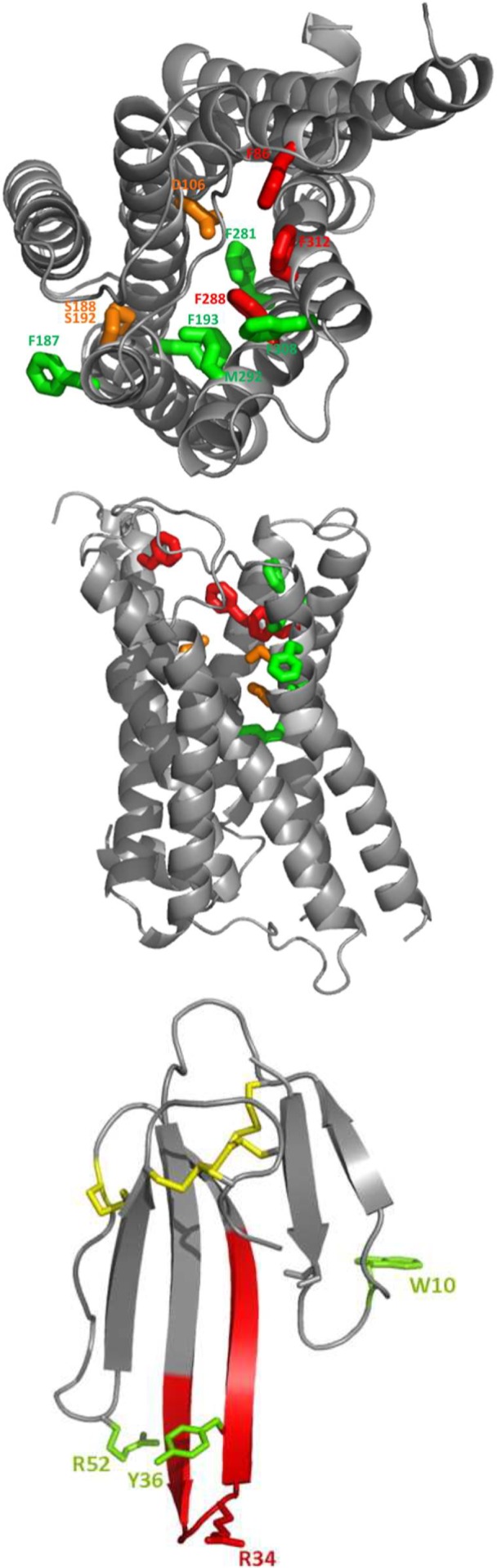
Views from the side of the TM bundle (Panel A), and from the top of the extracellular space (Panel B). F1875.41, F1935.47, F2816.44, M2926.55, F3087.35 in green. D1063.32 and the double S1885.42/S1925.46 in orange. F862.64, F2886.51 and F3127.39 in red. Panel C :3D structure of the three-finger fold MT7 toxin (2vlw) with the four conserved disulfide bridges in red.
A three-finger fold toxin can be represented by a 35 Å isosceles triangle of around 10 Å thickness (Fig. 9C). It is therefore much larger than classical small orthosteric ligands but nevertheless, ρ-Da1a is able to interact with positions within the orthosteric cavity of the α1A-AR. This is not the case for MT7, as the experimentally-based model of the MT7-M1 muscarinic receptor complex indicates an extracellular location of the toxin involving mainly the extracellular loop e2, in agreement with its allosteric properties [13]. The structural organization of the external part of GPCRs plays an important role in the access to the orthosteric site by agonists. Some receptors, like rhodopsin [53] or the S1P1 receptor [54] have their ligand-binding cavity substantially enclosed, compared to the chemokine CXCR4 receptor, for example, in which the extracellular loop conformation renders the binding cavity particularly open, facilitating the binding of large peptides [55]. The top view of the α1A-AR model (Fig. 9B) shows a relatively open receptor with external loops on the sides of the receptor, very similar to that proposed for the α1B-AR [18]. By comparison, M1 receptor modeling and mutational analysis indicate that extracellular loop 2 is important in binding of both orthosteric and allosteric ligands. The loop shows conformational flexibility but adopts closed conformations that affect access even of small molecule ligands [56]. Residues E170, R171, L174 and Y179 located in the e2 loop of the human M1 receptor collectively interact with MT7, with additional contributions from W91 in the e1 loop and W400 at the top of TM7 [13]. The more open conformation of the α1A-AR is certainly consistent with the capacity of ρ-Da1a to interact with residues inside the orthosteric pocket, however the large size and sub-nanomolar affinity of the toxin also suggest additional points of interaction with the receptor. A very recent publication describes how ρ-TIA, a conotoxin of 19 residues which acts as a negative allosteric modulator, interacts with the α1B-AR [18]. This small reticulated peptide binds primarily with the extracellular loop e3 of the α1B-AR and with the upper part of TM6 and TM7. ρ-TIA affinity is increased by the mutation F3106.51A in TM6, whereas our homologous mutation in the α1A-AR (F2886.51A) decreases ρ-Da1a affinity 18-fold. In TM7, ρ-TIA is sensitive to mutation at position F3307.35 of the α1B-AR (corresponding to F3087.35 in α1A-AR, not implicated in ρ-Da1a affinity) but not at position F3347.39 (corresponding to F3127.39 in α1A-AR). Mutation of the α1A-AR at residue F3127.39, which is one helical turn further from the extracellular face of the α1A-AR than F3087.35, produces a 93-fold reduction in ρ-Da1a affinity, highlighting the difference in binding mode of ρ-Da1a and the α1A-AR compared to ρ-TIA and the α1B-AR. Hence of the three animal toxins for which the mode of action has been described, the two negative allosteric modulators (MT7 and ρ-TIA) interact mostly with the external part of their receptor targets while ρ-Da1a interacts with the orthosteric binding site.
We found one discrepancy in our study, namely that ρ-Da1a shows incomplete competition with radioligands in equilibrium binding studies, a property normally characteristic of allosteric modulators. Our combined data, indicate that ρ-Da1a binds at least in part within the orthosteric pocket of the α1A-AR, however the large size of the toxin and/or its slow dissociation rate appear to cause altered pharmacology. In yeast membranes expressing the α1A-AR, both prazosin and ρ-Da1a cause complete displacement of 125I-ρ-Da1a, whereas like in CHO-K1 and COS-7 cells, ρ-Da1a displaces only 85% of 3H-prazosin binding [9]. Several three-finger snake toxins display similar incomplete competition for radioligand binding to GPCRs, however in the case of MT7 binding to the M1 muscarinic receptor, this residual binding is readily explained by an allosteric mode of interaction [11], [14], [16]. ρ-Da1b and MTα are also unable to fully inhibit 3H-rauwolscine binding to α2-ARs despite showing no effect on the 3H-rauwolscine dissociation rate, but their modes of action have still not been fully established [10], [57]. A third interesting case is that of ρ-TIA, which has an allosteric mode of action at α1B-ARs but has been described as a competitive antagonist of the α1A-AR in functional assays [19]. Despite this, ρ-TIA produces only 80% inhibition of 125I-HEAT binding in membranes from HEK-293 cells transfected with the α1A-AR. We initially thought that all α1A-ARs present in membrane preparations may be accessible to small molecule radioligands, but that a sub-population of the receptors might exist in conformations that are inaccessible to the larger toxin. This could reflect steric hindrance or a mixed allosteric/orthosteric mode of action of ρ-Da1a, however if this were the case, and the two populations of receptors were in equilibrium, the residual binding should change over time. We found that this was not the case, as the residual binding showed no time dependence over 2–24 hours. Thus the two receptor pools are not interchangeable, suggesting possible separation between distinct membrane compartments. This question remains to be resolved for ρ-Da1a but also for other toxins that display atypical pharmacological properties.
Key questions arising from our work are to determine which residues of ρ-Da1a bind within the α1A-AR orthosteric site, and whether additional regions bind to α1A-AR extracellular loops as seen for MT7 and ρ-TIA. Identification of additional receptor binding sites will be of interest because any such extra-orthosteric interaction may contribute to the α1A-AR selectivity of ρ-Da1a, as well as the insurmountable antagonism observed here in cell-based assays, on isolated rat [9] or human muscle and in in vivo experiments [44]. These questions will be addressed by characterization of mutated ρ-Da1a, by determining the crystal structure of the toxin, and by subsequent docking studies (for example [13]).
In conclusion, our findings demonstrate competitive behavior of the ρ-Da1a toxin at the α1A-AR and highlight the crucial role of residues located in the α1A-AR orthosteric site for the toxin interaction. Thus, despite the fact that ρ-Da1a and MT7 belong to the same three-finger fold structural family of toxins, and interact with homologous biogenic amine receptors, the mode of interaction with their respective targets is distinct. Evolution of snake toxins has thus not only generated a wide range of pharmacological activities from a unique peptide scaffold, but also various strategies to interact with similar molecular targets.
Supporting Information
Recombinant expression of ρ-Da1a.
(DOCX)
Acknowledgments
Dr. Michael Brownstein (JCVI), Dr. Diane Perez (The Cleveland clinic foundation, Ohio, USA) and Dr. Hervé Paris (INSERM, U858, Toulouse, France) for providing expression plasmids and cell lines.
Funding Statement
These authors have no support or funding to report.
References
- 1. Escoubas P, King GF (2009) Venomics as a drug discovery platform. Expert Rev Proteomics 6: 221–224. [DOI] [PubMed] [Google Scholar]
- 2. Halai R, Craik DJ (2009) Conotoxins: natural product drug leads. Nat Prod Rep 26: 526–536. [DOI] [PubMed] [Google Scholar]
- 3. Lewis RJ, Garcia ML (2003) Therapeutic potential of venom peptides. Nat Rev Drug Discov 2: 790–802. [DOI] [PubMed] [Google Scholar]
- 4. Shen GS, Layer RT, McCabe RT (2000) Conopeptides: From deadly venoms to novel therapeutics. Drug Discov Today 5: 98–106. [DOI] [PubMed] [Google Scholar]
- 5. Maïga A, Mourier G, Quinton L, Rouget C, Gales C, et al. (2012) G protein-coupled receptors, an unexploited animal toxin targets: Exploration of green mamba venom for novel drug candidates active against adrenoceptors. Toxicon 59: 487–496. [DOI] [PubMed] [Google Scholar]
- 6. Servent D, Fruchart-Gaillard C (2009) Muscarinic toxins: tools for the study of the pharmacological and functional properties of muscarinic receptors. J Neurochem 109: 1193–1202. [DOI] [PubMed] [Google Scholar]
- 7. Sharpe IA, Thomas L, Loughnan M, Motin L, Palant E, et al. (2003) Allosteric alpha 1-adrenoreceptor antagonism by the conopeptide rho-TIA. J Biol Chem 278: 34451–34457. [DOI] [PubMed] [Google Scholar]
- 8. Rajagopalan N, Pung YF, Zhu YZ, Wong PT, Kumar PP, et al. (2007) Beta-cardiotoxin: a new three-finger toxin from Ophiophagus hannah (king cobra) venom with beta-blocker activity. Faseb J 21: 3685–3695. [DOI] [PubMed] [Google Scholar]
- 9. Quinton L, Girard E, Maiga A, Rekik M, Lluel P, et al. (2010) Isolation and pharmacological characterization of AdTx1, a natural peptide displaying specific insurmountable antagonism of the alpha(1A)-adrenoceptor. Br J Pharmacol 159: 316–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Rouget C, Quinton L, Maiga A, Gales C, Masuyer G, et al. (2010) Identification of a novel snake peptide toxin displaying high affinity and antagonist behaviour for the alpha2-adrenoceptors. Br J Pharmacol 161: 1361–1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Fruchart-Gaillard C, Mourier G, Marquer C, Menez A, Servent D (2006) Identification of various allosteric interaction sites on M1 muscarinic receptor using 125I-Met35-oxidized muscarinic toxin 7. Mol Pharmacol 69: 1641–1651. [DOI] [PubMed] [Google Scholar]
- 12. Fruchart-Gaillard C, Mourier G, Marquer C, Stura E, Birdsall NJ, et al. (2008) Different interactions between MT7 toxin and the human muscarinic M1 receptor in its free and N-methylscopolamine-occupied states. Mol Pharmacol 74: 1554–1563. [DOI] [PubMed] [Google Scholar]
- 13. Marquer C, Fruchart-Gaillard C, Letellier G, Marcon E, Mourier G, et al. (2011) Structural Model of Ligand-G Protein-coupled Receptor (GPCR) Complex Based on Experimental Double Mutant Cycle Data: MT7 SNAKE TOXIN BOUND TO DIMERIC hM1 MUSCARINIC RECEPTOR. Journal of Biological Chemistry 286: 31661–31675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Mourier G, Dutertre S, Fruchart-Gaillard C, Menez A, Servent D (2003) Chemical synthesis of MT1 and MT7 muscarinic toxins: critical role of Arg-34 in their interaction with M1 muscarinic receptor. Mol Pharmacol 63: 26–35. [DOI] [PubMed] [Google Scholar]
- 15. Waelbroeck M, De Neef P, Domenach V, Vandermeers-Piret MC, Vandermeers A (1996) Binding of the labelled muscarinic toxin 125I-MT1 to rat brain muscarinic M1 receptors. Eur J Pharmacol 305: 187–192. [DOI] [PubMed] [Google Scholar]
- 16. Olianas MC, Adem A, Karlsson E, Onali P (2004) Action of the muscarinic toxin MT7 on agonist-bound muscarinic M1 receptors. European Journal of Pharmacology 487: 65–72. [DOI] [PubMed] [Google Scholar]
- 17. Kukkonen A (2004) Muscarinic Toxin 7 Selectivity Is Dictated by Extracellular Receptor Loops. Journal of Biological Chemistry 279: 50923–50929. [DOI] [PubMed] [Google Scholar]
- 18. Ragnarsson L, Wang CI, Andersson A, Fajarningsih D, Monks T, et al. (2013) Conopeptide rho-TIA Defines a New Allosteric Site on the Extracellular Surface of the alpha1B-Adrenoceptor. J Biol Chem 288: 1814–1827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lima V, Mueller A, Kamikihara SY, Raymundi V, Alewood D, et al. (2005) Differential antagonism by conotoxin rho-TIA of contractions mediated by distinct alpha1-adrenoceptor subtypes in rat vas deferens, spleen and aorta. Eur J Pharmacol 508: 183–192. [DOI] [PubMed] [Google Scholar]
- 20. Evans BA, Broxton N, Merlin J, Sato M, Hutchinson DS, et al. (2011) Quantification of functional selectivity at the human alpha(1A)-adrenoceptor. Mol Pharmacol 79: 298–307. [DOI] [PubMed] [Google Scholar]
- 21. Cheng Y, Prusoff WH (1973) Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem Pharmacol 22: 3099–3108. [DOI] [PubMed] [Google Scholar]
- 22. Christopoulos A, Parsons AM, Lew MJ, El-Fakahany EE (1999) The assessment of antagonist potency under conditions of transient response kinetics. Eur J Pharmacol 382: 217–227. [DOI] [PubMed] [Google Scholar]
- 23. Lew MJ, Angus JA (1995) Analysis of competitive agonist-antagonist interactions by nonlinear regression. Trends Pharmacol Sci 16: 328–337. [DOI] [PubMed] [Google Scholar]
- 24. Lew MJ, Angus JA (1997) An improved method for analysis of competitive agonist/antagonist interactions by non-linear regression. Ann N Y Acad Sci 812: 179–181. [DOI] [PubMed] [Google Scholar]
- 25. Sali A, Blundell TL (1993) Comparative protein modelling by satisfaction of spatial restraints. J Mol Biol 234: 779–815. [DOI] [PubMed] [Google Scholar]
- 26. Horn F, Bettler E, Oliveira L, Campagne F, Cohen FE, et al. (2003) GPCRDB information system for G protein-coupled receptors. Nucleic Acids Res 31: 294–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Vroling B, Sanders M, Baakman C, Borrmann A, Verhoeven S, et al. (2011) GPCRDB: information system for G protein-coupled receptors. Nucleic Acids Res 39: D309–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Cherezov V, Rosenbaum DM, Hanson MA, Rasmussen SG, Thian FS, et al. (2007) High-Resolution Crystal Structure of an Engineered Human {beta}2-Adrenergic G Protein Coupled Receptor. Science 318: 1258–1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. King GF, Gentz MC, Escoubas P, Nicholson GM (2008) A rational nomenclature for naming peptide toxins from spiders and other venomous animals. Toxicon 52: 264–276. [DOI] [PubMed] [Google Scholar]
- 30. Avlani V (2003) Application of a Kinetic Model to the Apparently Complex Behavior of Negative and Positive Allosteric Modulators of Muscarinic Acetylcholine Receptors. Journal of Pharmacology and Experimental Therapeutics 308: 1062–1072. [DOI] [PubMed] [Google Scholar]
- 31. Mohr K, Trankle C, Holzgrabe U (2003) Structure/activity relationships of M2 muscarinic allosteric modulators. Receptors Channels 9: 229–240. [PubMed] [Google Scholar]
- 32. Leppik RA, Mynett A, Lazareno S, Birdsall NJ (2000) Allosteric interactions between the antagonist prazosin and amiloride analogs at the human alpha(1A)-adrenergic receptor. Mol Pharmacol 57: 436–445. [DOI] [PubMed] [Google Scholar]
- 33. Hamaguchi N, True TA, Saussy DL Jr, Jeffs PW (1996) Phenylalanine in the second membrane-spanning domain of alpha 1A-adrenergic receptor determines subtype selectivity of dihydropyridine antagonists. Biochemistry 35: 14312–14317. [DOI] [PubMed] [Google Scholar]
- 34. Cavalli A, Fanelli F, Taddei C, De Benedetti PG, Cotecchia S (1996) Amino acids of the alpha1B-adrenergic receptor involved in agonist binding: differences in docking catecholamines to receptor subtypes. FEBS Lett 399: 9–13. [DOI] [PubMed] [Google Scholar]
- 35. Takahashi K, Hossain M, Ahmed M, Bhuiyan MA, Ohnuki T, et al. (2007) Asp125 and Thr130 in transmembrane domain 3 are major sites of alpha1b-adrenergic receptor antagonist binding. Biol Pharm Bull 30: 1891–1894. [DOI] [PubMed] [Google Scholar]
- 36. Ahmed M, Hossain M, Bhuiyan MA, Ishiguro M, Tanaka T, et al. (2008) Mutational analysis of the alpha 1a-adrenergic receptor binding pocket of antagonists by radioligand binding assay. Biol Pharm Bull 31: 598–601. [DOI] [PubMed] [Google Scholar]
- 37. Waugh DJ, Zhao MM, Zuscik MJ, Perez DM (2000) Novel aromatic residues in transmembrane domains IV and V involved in agonist binding at alpha(1a)-adrenergic receptors. J Biol Chem 275: 11698–11705. [DOI] [PubMed] [Google Scholar]
- 38. Hwa J, Perez DM (1996) The unique nature of the serine interactions for alpha 1-adrenergic receptor agonist binding and activation. J Biol Chem 271: 6322–6327. [DOI] [PubMed] [Google Scholar]
- 39. Chen S, Xu M, Lin F, Lee D, Riek P, et al. (1999) Phe310 in transmembrane VI of the alpha1B-adrenergic receptor is a key switch residue involved in activation and catecholamine ring aromatic bonding. J Biol Chem 274: 16320–16330. [DOI] [PubMed] [Google Scholar]
- 40. Hwa J, Graham RM, Perez DM (1995) Identification of critical determinants of alpha 1-adrenergic receptor subtype selective agonist binding. J Biol Chem 270: 23189–23195. [DOI] [PubMed] [Google Scholar]
- 41. Waugh DJ, Gaivin RJ, Zuscik MJ, Gonzalez-Cabrera P, Ross SA, et al. (2001) Phe-308 and Phe-312 in transmembrane domain 7 are major sites of alpha 1-adrenergic receptor antagonist binding. Imidazoline agonists bind like antagonists. J Biol Chem 276: 25366–25371. [DOI] [PubMed] [Google Scholar]
- 42. Wetzel JM, Salon JA, Tamm JA, Forray C, Craig D, et al. (1996) Modeling and mutagenesis of the human alpha 1a-adrenoceptor: orientation and function of transmembrane helix V sidechains. Receptors Channels 4: 165–177. [PubMed] [Google Scholar]
- 43. Shim JY, Bertalovitz AC, Kendall DA (2011) Identification of essential cannabinoid-binding domains: structural insights into early dynamic events in receptor activation. J Biol Chem 286: 33422–33435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Palea S, Maiga A, Guilloteau V, Rekik M, Guerard M, et al. (2012) Effects of rho-Da1a a peptidic alpha1a-adrenoceptor antagonist in human isolated prostatic adenoma and anesthetized rats. Br J Pharmacol in press [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Perret BG, Wagner R, Lecat S, Brillet K, Rabut G, et al. (2003) Expression of EGFP-amino-tagged human mu opioid receptor in Drosophila Schneider 2 cells: a potential expression system for large-scale production of G-protein coupled receptors. Protein Expression and Purification 31: 123–132. [DOI] [PubMed] [Google Scholar]
- 46. Grunewald S, Haase W, Molsberger E, Michel H, Reilender H (2004) Production of the Human D2S Receptor in the Methylotrophic Yeast P. pastoris? Receptors and Channels 10: 37–50. [DOI] [PubMed] [Google Scholar]
- 47. Kenakin T (2005) New concepts in drug discovery: collateral efficacy and permissive antagonism. Nat Rev Drug Discov 4: 919–927. [DOI] [PubMed] [Google Scholar]
- 48. Kenakin T, Miller LJ (2010) Seven transmembrane receptors as shapeshifting proteins: the impact of allosteric modulation and functional selectivity on new drug discovery. Pharmacol Rev 62: 265–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Compeer MG, Meens MJ, Hackeng TM, Neugebauer WA, Holtke C, et al. (2012) Agonist-dependent modulation of arterial endothelinA receptor function. Br J Pharmacol 166: 1833–1845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Lazareno S, Birdsall NJ (1995) Detection, quantitation, and verification of allosteric interactions of agents with labeled and unlabeled ligands at G protein-coupled receptors: interactions of strychnine and acetylcholine at muscarinic receptors. Mol Pharmacol 48: 362–378. [PubMed] [Google Scholar]
- 51. Strader CD, Sigal IS, Register RB, Candelore MR, Rands E, et al. (1987) Identification of residues required for ligand binding to the beta-adrenergic receptor. Proc Natl Acad Sci U S A 84: 4384–4388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Porter JE, Hwa J, Perez DM (1996) Activation of the alpha1b-adrenergic receptor is initiated by disruption of an interhelical salt bridge constraint. J Biol Chem 271: 28318–28323. [DOI] [PubMed] [Google Scholar]
- 53. Palczewski K, Kumasaka T, Hori T, Behnke CA, Motoshima H, et al. (2000) Crystal structure of rhodopsin: A G protein-coupled receptor. Science 289: 739–745. [DOI] [PubMed] [Google Scholar]
- 54. Hanson MA, Roth CB, Jo E, Griffith MT, Scott FL, et al. (2012) Crystal structure of a lipid G protein-coupled receptor. Science 335: 851–855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Wu B, Chien EY, Mol CD, Fenalti G, Liu W, et al. (2010) Structures of the CXCR4 chemokine GPCR with small-molecule and cyclic peptide antagonists. Science 330: 1066–1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Avlani VA, Gregory KJ, Morton CJ, Parker MW, Sexton PM, et al. (2007) Critical role for the second extracellular loop in the binding of both orthosteric and allosteric G protein-coupled receptor ligands. J Biol Chem 282: 25677–25686. [DOI] [PubMed] [Google Scholar]
- 57. Koivula K, Rondinelli S, Näsman J (2010) The three-finger toxin MTα is a selective α2B-adrenoceptor antagonist. Toxicon 56: 440–447. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Recombinant expression of ρ-Da1a.
(DOCX)