

Abstract
Background/Aims
The primary pathophysiologic abnormality in achalasia is known to be a loss of inhibitory myenteric ganglion cells, which may result from an immune-mediated response or neuronal degeneration. The aim of this study was to identify proteins suggestive of an immune-mediated response or neuronal degeneration in the serum of achalasia patients using a proteomic analysis.
Methods
Blood samples were collected from five symptomatic achalasia patients and five sex- and age-matched healthy controls. Serum proteomic analysis was conducted, and the protein spots were identified using matrix-assisted laser desorption ionization/time-of-flight and a proteomics analyzer. The serum level of C3 was measured by enzyme-linked immunosorbent assay in nine patients with achalasia and 18 sex- and age-matched healthy controls.
Results
Of the 658 matched protein spots, 28 spots were up-regulated over 2-fold in the serum from achalasia patients compared with that from controls. The up-regulated proteins included complement C4B5, complement C3, cyclin-dependent kinase 5, transthyretin, and alpha 2 macroglobulin. The serum levels of C3 in achalasia patients were significantly higher than those of controls.
Conclusions
The serum proteomic analysis of achalasia patients suggests an immune-mediated response or neuronal degeneration. Further validation studies in larger samples and the esophageal tissue of achalasia patients are required.
Keywords: Esophageal achalasia, Immune response, Neuronal degeneration, Proteomics
INTRODUCTION
Although the primary pathophysiologic abnormality of esophageal achalasia is a loss of the intrinsic inhibitory innervation of the lower esophageal sphincter (LES) and the smooth muscle segment of the esophageal body, its pathogenesis is not fully understood. Impairment in nitrergic inhibitory neurotransmission results in nonrelaxing LES and aperistalsis of the esophageal body, which are typical manometric findings of achalasia.1 Those findings are presumed to be caused by the neurodegenerative insult, which is believed to be of inflammatory origin.2 However, evidence for the etiology and pathogenesis of achalasia is lacking.
Proteomics is a powerful technology that can provide information about diagnosis, disease progression, and pathogenesis.3,4 The various biomarkers or key proteins for neuronal degeneration have been detected in patients with Alzheimer's disease using proteomic approaches.5 However, there is no report on the proteomic analysis in the serum of achalasia patients to explore the pathogenetic mechanism of achalasia.
Thus, in the present study, we aimed to compare protein profiles between achalasia patients and healthy controls using the serum proteomic analysis for the detection of key proteins suggestive of imune-mediated response or neuronal degeneration.
MATERIALS AND METHODS
1. Subjects
Peripheral blood samples of five symptomatic achalasia patients and five sex- and age-matched healthy controls were obtained. Median age of the patient group was 48 years (range, 30 to 68 years). Systemic infection, and acute and chronic inflammatory disorders were excluded by history, physical examinations and routine laboratory tests. Diagnosis of achalasia was made based on clinical, radiological, endoscopic, and manometric criteria. Manometric criteria included esophageal aperistalsis and poor LES relaxation. All the achalasia patients underwent pneumatic balloon dilation. Blood sampling was performed before the therapeutic procedures such as pneumatic balloon dilation. The blood samples obtained from the achalasia patients and healthy controls were immediately frozen in liquid nitrogen and stored at -80℃. The study protocol was approved by the Institutional Review Board of Ajou University Hospital.
2. Two-dimensional polyacrylamide gel electrophoresis and image analysis
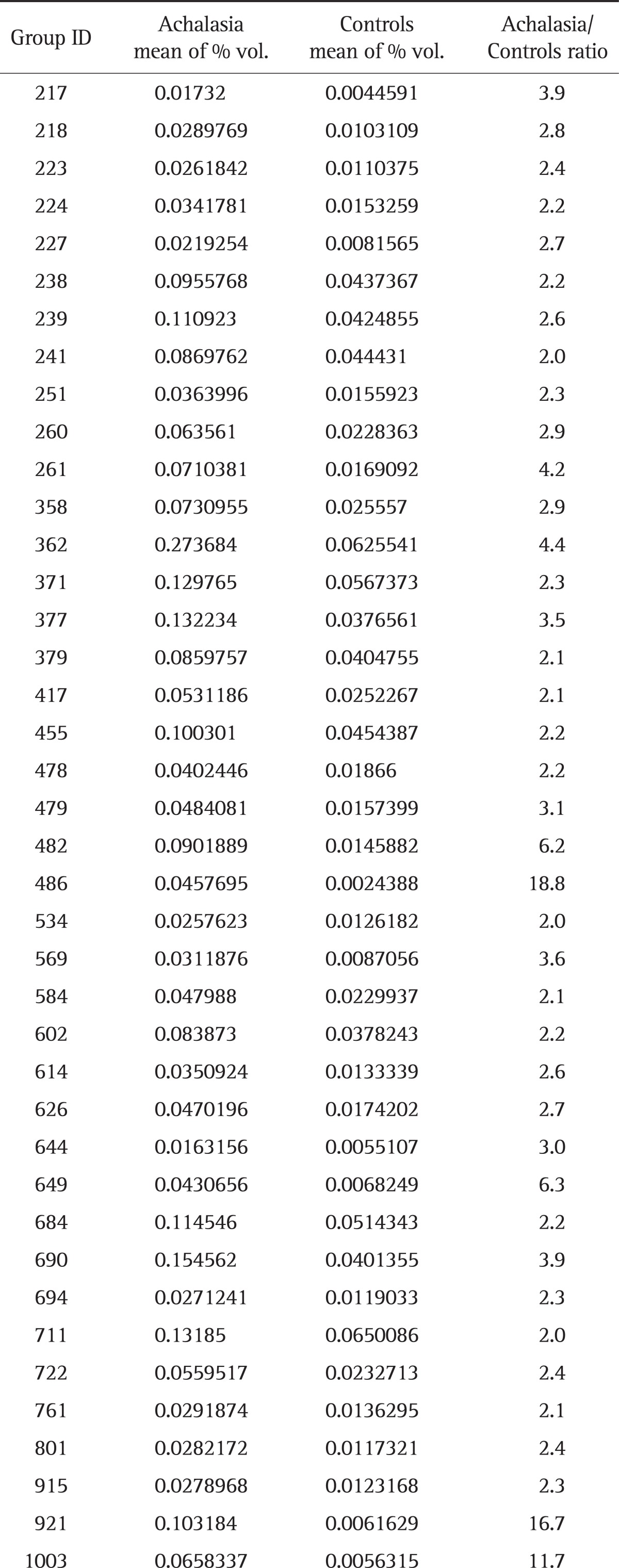
Aliquots of each sample solubilized in sample buffer (7 M urea, 2 M thiourea, 4.5% 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate, 100 mM 1,4-dithioerythritol, 40 mM Tris, and pH 8.8) were applied to immobilized pH 3 to 10 nonlinear gradient strips (Amersham Biosciences, Uppsala, Sweden). Isoelectric focusing was performed at 80,000 Vh. The second dimension electrophoresis was analyzed on 9% to 16% linear gradient polyacrylamide gels at a constant 40 mA per gel for approximately 5 hours. After protein fixation in 40% methanol and 5% phosphoric acid for 1 hour, gels were stained with Coomassie Brilliant Blue G250 for 12 hours, destained with H2O, and scanned in a Bio-Rad (Richmond, CA, USA) GS710 densitometer. Data were converted into electronic files and analyzed using an Image Master Platinum 5.0 image analysis program (Amersham Biosciences). The spot intensity was calculated by integrating the optical density (OD) over the spot area (the spot "volume"). We selected the protein spots that showed a greater than 2-fold increase of the spot volume in achalasia samples, compared with control samples. The criteria of 2-fold increase was arbitrarily determined. Table 1 shows the mean percentage volume for 40 proteins.
Table 1.
Mean Percentage Volume for 40 Proteins in the Serum of Achalasia Patients and Controls
3. Spot identification by MALDI-TOF/MS
For matrix-assisted laser desorption ionization/time-of-flight mass spectrometry (MALDI-TOF/MS) analysis, peptides were concentrated using a POROS R2, Oligo R3 column (Applied Biosystems, Foster City, CA, USA). After washing the column sequentially with 70% acetonitrile, 100% acetonitrile, and 50 mM ammonium bicarbonate, samples were applied to the R2, R3 column and eluted with cyano-4-hydroxycinamic acid (CHCA) (Sigma-Aldrich, St. Louis, MO, USA) dissolved in 70% acetonitrile and 2% formic acid and spotted onto the MALDI plate (Opti-TOF™ 384-well Insert; Applied Biosystems). MALDI-TOF/MS was performed on 4800 MALDI-TOF/TOF™ Analyzer (Applied Biosystems, Carlsbad, CA, USA) equipped with a 355-nm Nd:YAG laser. The pressure in the TOF analyzer was approximately 7.6e-07 Torr. Mass spectra were obtained in the reflectron mode with an accelerating voltage of 20 kV and summed over 500 laser pulses. Calibration was performed using the 4700 calibration mixture (Applied Biosystems). Each sample spectrum was additionally calibrated using trypsin autolysis peaks. Data Explorer 4.4 (Applied Biosystems) was used for the data acquisition and extraction of monoisotopic masses. Mowse scores greater than 64 were considered to be significant.
4. Enzyme-linked immunosorbent assay
Serum samples of nine patients diagnosed with achalasia, in which five patients' serum samples used in the proteomic analysis were excluded, and 18 sex-matched and age-matched healthy controls were used for enzyme-linked immunosorbent assay (ELISA) tests. Blood was collected by vein puncture and the serum was separated from the coagulated or packed cells by centrifugation. Subsequently, specimens were stored at -20℃ until assayed. The serum level of complement C3 was measured using commercial ELISA kits (Young In Frontier Co., Seoul, Korea) according to the manufacturer's protocols. Incubation buffer was added to all wells and incubate the plate for 5 minutes at room temperature. For the standard curve, 100 µL of the standard was added to the appropriate microtiter wells. Serum require at least 100-fold dilution in phosphate buffered saline buffer (pH 7.4), and 100 µL of samples was added to each wells. The plate was covered with the plate cover and incubated for 2 hours at 37℃. Secondary antibody solutions were pipetted into each well. The plate was then incubated for 1 hour at 37℃. Avidin conjugated to horseradish peroxidase was added to each well. The plate was then incubated for 30 minutes at room temperature. Substrate was added to each well. The plate was then incubated at room temperature. The incubation time for chromogen substrate was determined by the microtiter plate reader (Bio-Rad) used. OD values were monitored. OD of the positive wells exceeded the limits of the instrument. Stop solution was added to each well, and the absorbance of each well at 450 nm was read within 20 minutes of adding the stop solution. The absorbance was measured in a microtiter plate reader at a wavelength of 450 nm and converted into units (units/mL) by plotting against the autoantibody titer of the calibrators/standards given by the manufacturer. In order to construct the standard curve, the absorbance of the standard was plotted on graph. The serum concentrations for the samples were measured, based on the standard curve.
5. Statistical analysis
The serum levels of C3 were compared by Student t-test. Statistical tests were performed using the SPSS system version 12.0 (SPSS Inc., Chicago, IL, USA). A p<0.05 was considered to be significant. The data are presented as mean±SE.
RESULTS
1. Proteomic analysis
Fig. 1 showed the representative result for protein spots separated by two-dimensional electrophoresis and a gradient polyacrylamide gel. The up-regulated protein spots were marked. Forty protein spots were up-regulated over two times in the serum samples from achalasia patients, compared with nonachalasia controls (Table 1). Of the 40 protein spots, 28 spots were identified by MALDI-TOF/MS (Table 2). Some of the up-regulated proteins suggested the involvement of immune-mediated response or neuronal degeneration in the pathogenesis of achalasia. These proteins included C4B5, C5, C3, cyclin-dependent kinase 5 (Cdk5), transthyretin (TTR), and A2M (Table 3).
Fig. 1.

Representative result for protein spots separated by by 2-dimensional gel electrophoresis and a gradient polyacrylamide gel. Protein spots up-regulated in achalasia patients were marked and excised from the gels, and identified using matrix-assisted laser desorption ionization/time-of-flight and a proteomics analyzer that operated in the mass spectrometry mode.
Table 2.
Up-Regulated Protein Spots in the Serum Samples from the Achalasia Group
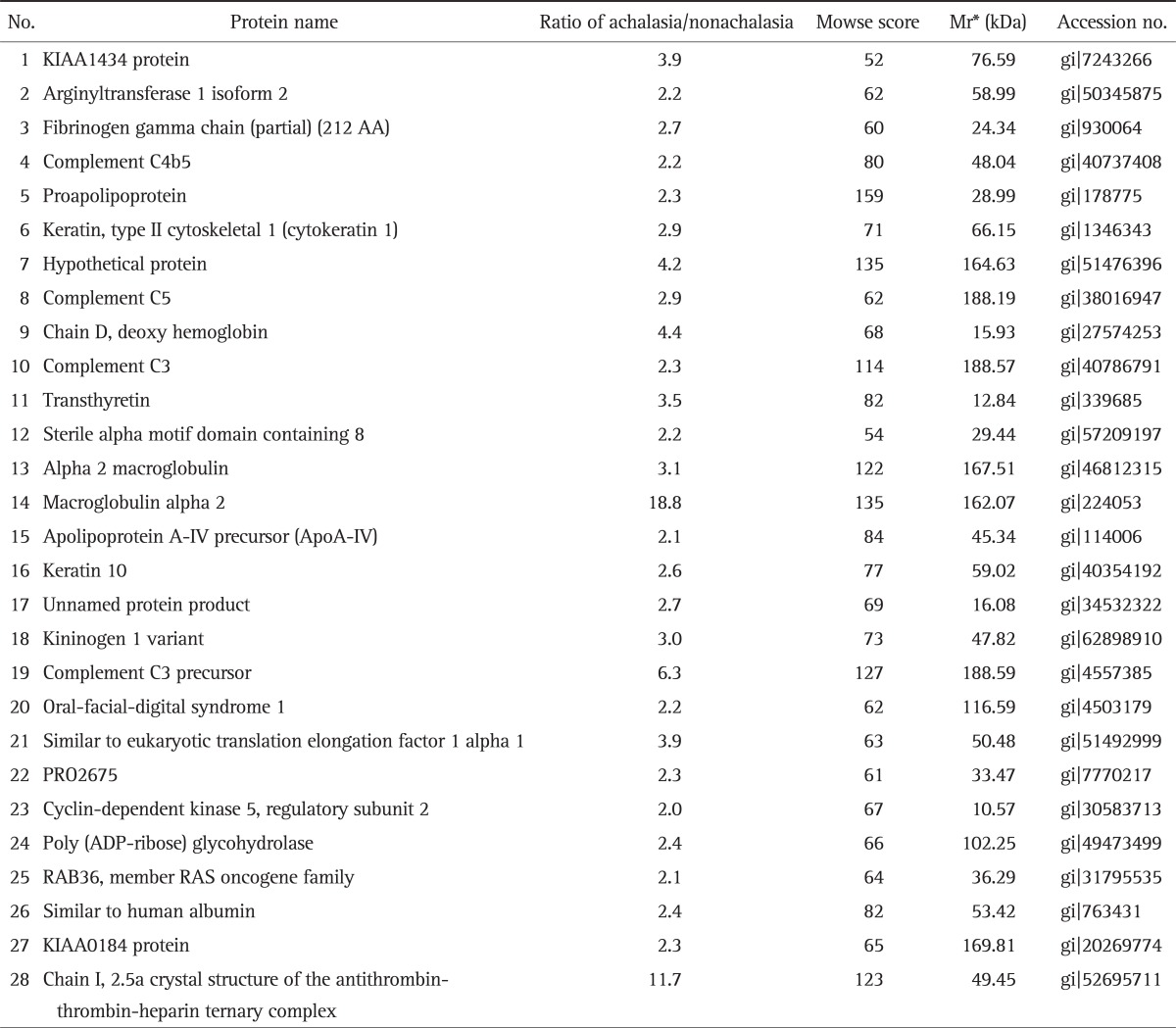
*Mr, relative mobility.
Table 3.
Pathogenically Important Proteins Up-Regulated in the Serum of Achalasia Patients
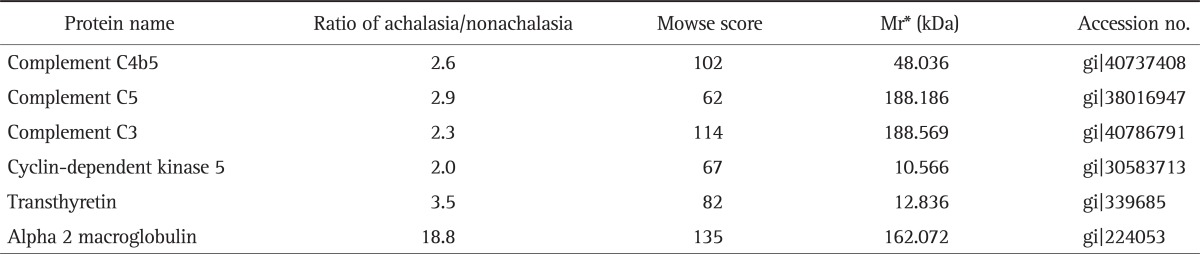
*Mr, relative mobility.
2. ELISA tests
The C3 levels of achalasia patients were significantly higher than those of nonachalasia controls (32.1±3.4 mg/mL vs 18.3±3.1 mg/mL; p<0.05).
DISCUSSION
Although many attempts have been made to determine the pathogenesis of achalasia, it remains to be unclear. It has been reported that the loss of normal inhibition in the esophagus is caused by inhibitory neuronal loss.2 The early disease stage such as vigorous achalasia is more likely to show myenteric inflammation with ganglionitis rather than classic achalasia. Progressive destruction of inhibitory neurons appears to lead to classic achalasia. All patients with achalasia in the present study presented classic achalasia, which was diagnosed by endoscopy and conventional esophageal manometry. Up-regulated proteins in the serum samples from our achalasia patients included C4B5, C5, C3, Cdk5, TTR, and alpha2 macroglobulin. These proteins may play a role in immune-mediated response or neuronal degeneration. The ELISA test demonstrated significantly higher serum level of C3 in achalasia patients, compared to nonachalasia controls. Therefore, our results of the present study may provide evidence for the involvement of immune-mediated response or neuronal degeneration in the pathogenesis of achalasia.
Available evidence has lead to several hypotheses regarding the etiology of achalasia. Inflammation of myenteric inhibitory ganglion cells has been suggested to be the primary mechanism underlying achalasia.2 Previous studies have demonstrated that loss of inhibitory neurons in the esophagus is induced by inflammation, leading to progressive neuronal destruction or fibrosis.6,7 Familial, infectious, and autoimmune factors may trigger the inflammatory process.2,8,9 Neuronal degeneration has also been proposed to be a possible etiologic factor.10-12 To date, studies on the pathophysiology and etiology of achalasia have been conducted using traditional laboratory tools. However, the number of target materials for the measurement is very limited in the traditional methodology. Proteomics is the large-scale analysis of proteins. Proteomics confirms the presence of the protein and provides a direct measure of the quantity present. Thus, in the present study, we tried to find the specific proteins involved in the pathogenesis of achalasia using a proteomic approach. Actually, proteomics has contributed to the elucidation of the mechanisms of diseases and to the identification of biomarkers for several neurodegenerative diseases.
Proteomic studies have been carried out on neurologic diseases, such as dementia, multiple sclerosis, amyotrophic lateral sclerosis, Huntington's disease, epilepsy, and gracile axonal dystrophy.13 Proteomic data for peripheral neuropathies, such as familial amyloid polyneuropathy (FAP), a rare autosomal dominant neuropathy characterized by a sensorimotor peripheral polyneuropathy and autonomic nervous system dysfunction, have been reported.14,15 FAP is known to be commonly caused by accumulation of deposits of TTR amyloid around peripheral nerves.15 Proteomic analysis in the present study showed that TTR was up-regulated in the serum samples from achalasia patients. Although there is no previous report that TTR is associated with esophageal achalasia, TTR might be involved in neuronal injury or autonomic dysfunction. Whether TTR is associated with the pathogenesis of esophageal achalasia needs further investigation.
The complement system is a major component of inflammatory responses.16 There is no direct evidence to show that the activated complimentary system causes the specific neuronal damage governing LES. However, when the complement system is activated at an inappropriate site and/or to an inappropriate extent, host tissues may be damaged, causing pathology as seen in neurodegenerative disorders of the central nervous system.17 In the present study, several complement proteins were found to be up-regulated in the serum proteomic analysis of achalasia patients. Moreover, significantly higher level of serum C3 in achalaisa patients, compared with normal controls, was reconfirmed by ELISA tests. C3 is cleaved into C3a and C3b. C3b is deposited on the surfaces of targeted cells. C3a and C5a function mediate inflammation.18 Therefore, it is conceivable that the complement system activation plays a role in inflammatory injury on myenteric neurons in achalasia patients, which warrants further investigation. In line with our findings, a previous study demonstrated that complement complex C5b-C9 and immunoglobulin M (IgM) were deposited within or at ganglion cells of Auerbach's plexus in achalasia patients.19 Antigenic stimuli provided by certain viruses and bacteria can induce the activation pathways of the complement system. Some neurotrophic viruses existing at neural structures of esophagus in achalasia patients may bind IgM to form immune complexes, which can activate the classical pathway of complements. C3, C4b5, and C5, which were significantly increased in the proteomic analysis of the serum samples from achalasia patients, are the main factors of the classical complement pathway. Thus, it is plausible that the classical complement pathway is activated by immune complexes and induces a complement-dependent autoimmune reaction against myenteric neurons in the esophagus.
Cdk5 is a member of the Cdk family that is involved in the regulation of the cell cycle. Cdk5 activity is restricted to the nervous system.20 Increased Cdk5 activity induces the hyperphosphorylation of tau protein, which inhibits the binding of tau protein to microtubules, and induces cytoskeletal disruption, morphological degeneration, and apoptosis.21 Studies have shown that Cdk5 has multiple activities in apoptosis, exocytosis, inflammation, gene transcription, and wound healing.22 To our knowledge, no study showing the relevance of Cdk5 to the pathogenesis of achalasia has been reported. Proteomic analysis in the present study showed that Cdk5 levels are significantly elevated in achalasia patients, compared with normal controls. Further study on the identification of Cdk5 in the esophageal tissues or larger serum samples of achalasia patients is required.
A2M is an inhibitor of matrix metalloproteases and forms A2M-proteinase complexes, removing proteolytic potential.23 High levels of A2M are associated with impaired immune efficiency. Increased levels of A2M is known to be associated with inflammation in some degenerative diseases.24 Furthermore, it has been reported that an excess of A2M has neurotoxic effects.25 The role of A2M in the pathogenesis of achalasia needs further study.
Although the findings of the present study have important implications for research on the pathogenesis of esophageal achalasia, it has several limitations. First, and most obviously, the number of patients enrolled was small. This was unavoidable because of the low incidence of achalasia in Korea. Accordingly, a large scale multicenter study is required in the future. Second, proteomics experiments conducted in one laboratory are not easily reproduced in another. So, further validation tests using serum and tissues are necessary to confirm our proteomic results of the present study.
In conclusion, the serum proteomic analysis of achalasia patients shows a protein pattern suggesting immune-mediated response or neuronal degeneration. Further validation studies in larger samples and the esophageal tissue of achalasia patients are required.
Footnotes
No potential conflict of interest relevant to this article was reported.
References
- 1.Katz PO, Richter JE, Cowan R, Castell DO. Apparent complete lower esophageal sphincter relaxation in achalasia. Gastroenterology. 1986;90:978–983. doi: 10.1016/0016-5085(86)90876-0. [DOI] [PubMed] [Google Scholar]
- 2.Park W, Vaezi MF. Etiology and pathogenesis of achalasia: the current understanding. Am J Gastroenterol. 2005;100:1404–1414. doi: 10.1111/j.1572-0241.2005.41775.x. [DOI] [PubMed] [Google Scholar]
- 3.Wilkins MR, Pasquali C, Appel RD, et al. From proteins to proteomes: large scale protein identification by two-dimensional electrophoresis and amino acid analysis. Biotechnology (N Y) 1996;14:61–65. doi: 10.1038/nbt0196-61. [DOI] [PubMed] [Google Scholar]
- 4.Biomarkers Definitions Working Group. Biomarkers and surrogate endpoints: preferred definitions and conceptual framework. Clin Pharmacol Ther. 2001;69:89–95. doi: 10.1067/mcp.2001.113989. [DOI] [PubMed] [Google Scholar]
- 5.Sultana R, Boyd-Kimball D, Poon HF, et al. Redox proteomics identification of oxidized proteins in Alzheimer's disease hippocampus and cerebellum: an approach to understand pathological and biochemical alterations in AD. Neurobiol Aging. 2006;27:1564–1576. doi: 10.1016/j.neurobiolaging.2005.09.021. [DOI] [PubMed] [Google Scholar]
- 6.Goldblum JR, Whyte RI, Orringer MB, Appelman HD. Achalasia. A morphologic study of 42 resected specimens. Am J Surg Pathol. 1994;18:327–337. [PubMed] [Google Scholar]
- 7.Goldblum JR, Rice TW, Richter JE. Histopathologic features in esophagomyotomy specimens from patients with achalasia. Gastroenterology. 1996;111:648–654. doi: 10.1053/gast.1996.v111.pm8780569. [DOI] [PubMed] [Google Scholar]
- 8.Frieling T, Berges W, Borchard F, Lübke HJ, Enck P, Wienbeck M. Family occurrence of achalasia and diffuse spasm of the oesophagus. Gut. 1988;29:1595–1602. doi: 10.1136/gut.29.11.1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Robertson CS, Martin BA, Atkinson M. Varicella-zoster virus DNA in the oesophageal myenteric plexus in achalasia. Gut. 1993;34:299–302. doi: 10.1136/gut.34.3.299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Parrilla P, Aguayo JL, Martinez de Haro L, Ortiz A, Martinez DA, Morales G. Reversible achalasia-like motor pattern of esophageal body secondary to postoperative stricture of gastroesophageal junction. Dig Dis Sci. 1992;37:1781–1784. doi: 10.1007/BF01299876. [DOI] [PubMed] [Google Scholar]
- 11.Qualman SJ, Haupt HM, Yang P, Hamilton SR. Esophageal Lewy bodies associated with ganglion cell loss in achalasia. Similarity to Parkinson's disease. Gastroenterology. 1984;87:848–856. [PubMed] [Google Scholar]
- 12.Murphy MS, Gardner-Medwin D, Eastham EJ. Achalasia of the cardia associated with hereditary cerebellar ataxia. Am J Gastroenterol. 1989;84:1329–1330. [PubMed] [Google Scholar]
- 13.Tumani H, Lehmensiek V, Lehnert S, Otto M, Brettschneider J. 2D DIGE of the cerebrospinal fluid proteome in neurological diseases. Expert Rev Proteomics. 2010;7:29–38. doi: 10.1586/epr.09.99. [DOI] [PubMed] [Google Scholar]
- 14.Ando Y, Ueda M. Novel methods for detecting amyloidogenic proteins in transthyretin related amyloidosis. Front Biosci. 2008;13:5548–5558. doi: 10.2741/3098. [DOI] [PubMed] [Google Scholar]
- 15.Hou X, Aguilar MI, Small DH. Transthyretin and familial amyloidotic polyneuropathy. Recent progress in understanding the molecular mechanism of neurodegeneration. FEBS J. 2007;274:1637–1650. doi: 10.1111/j.1742-4658.2007.05712.x. [DOI] [PubMed] [Google Scholar]
- 16.Walport MJ. Complement. First of two parts. N Engl J Med. 2001;344:1058–1066. doi: 10.1056/NEJM200104053441406. [DOI] [PubMed] [Google Scholar]
- 17.Morgan BP, Gasque P. Expression of complement in the brain: role in health and disease. Immunol Today. 1996;17:461–466. doi: 10.1016/0167-5699(96)20028-f. [DOI] [PubMed] [Google Scholar]
- 18.Coleman RM, Lombard MF, Sicard RE, Rencricca NJ. Fundamental immunology. Dubuque: Brown Publishers; 1989. [Google Scholar]
- 19.Storch WB, Eckardt VF, Junginger T. Complement components and terminal complement complex in oesophageal smooth muscle of patients with achalasia. Cell Mol Biol (Noisy-le-grand) 2002;48:247–252. [PubMed] [Google Scholar]
- 20.Kesavapany S, Li BS, Amin N, Zheng YL, Grant P, Pant HC. Neuronal cyclin-dependent kinase 5: role in nervous system function and its specific inhibition by the Cdk5 inhibitory peptide. Biochim Biophys Acta. 2004;1697:143–153. doi: 10.1016/j.bbapap.2003.11.020. [DOI] [PubMed] [Google Scholar]
- 21.Maccioni RB, Otth C, Concha II, Muñoz JP. The protein kinase Cdk5. Structural aspects, roles in neurogenesis and involvement in Alzheimer's pathology. Eur J Biochem. 2001;268:1518–1527. doi: 10.1046/j.1432-1033.2001.02024.x. [DOI] [PubMed] [Google Scholar]
- 22.Honjyo Y, Kawamoto Y, Nakamura S, Nakano S, Akiguchi I. Immunohistochemical localization of CDK5 activator p39 in the rat brain. Neuroreport. 1999;10:3375–3379. doi: 10.1097/00001756-199911080-00022. [DOI] [PubMed] [Google Scholar]
- 23.Sottrup-Jensen L, Sand O, Kristensen L, Fey GH. The alpha-macroglobulin bait region. Sequence diversity and localization of cleavage sites for proteinases in five mammalian alpha-macroglobulins. J Biol Chem. 1989;264:15781–15789. [PubMed] [Google Scholar]
- 24.Mocchegiani E, Costarelli L, Giacconi R, Cipriano C, Muti E, Malavolta M. Zinc-binding proteins (metallothionein and alpha-2 macroglobulin) and immunosenescence. Exp Gerontol. 2006;41:1094–1107. doi: 10.1016/j.exger.2006.08.010. [DOI] [PubMed] [Google Scholar]
- 25.Kovacs DM. alpha2-macroglobulin in late-onset Alzheimer's disease. Exp Gerontol. 2000;35:473–479. doi: 10.1016/s0531-5565(00)00113-3. [DOI] [PubMed] [Google Scholar]