

Abstract
Purpose:
We studied the phenotype and electroencephalographic (EEG) features, and therapeutic aspects of idiopathic generalized epilepsies (IGEs) in South Indian population.
Patients and Methods:
This prospective cross-sectional hospital-based study was carried out on non-consecutive 287 patients (age 22.2 ± 7.7 years; M:F = 139:148) with IGE syndrome. Their clinical and EEG observations were analyzed.
Results:
Majority of the patients had onset of seizures <20 years of age (n = 178; 62%). Thirty one patients (10.8%) had family history of epilepsy. Nearly half of them (49.9%) had <5 years of duration of seizures. The type of IGEs included Juvenile myoclonic epilepsy (JME): 115 (40.1%); IGE with generalized tonic-clonic seizures (GTCS) only: 102 (39.02%); childhood absence epilepsy (CAE): 35 (12.2%); GTCS on awakening: 15 (5.2%); Juvenile absence epilepsy (JAE): 11 (3.8%); and unclassified seizures: 9 (3.1%). The triggering factors noted in 45% were sleep deprivation (20%), non-compliance and stress in 5% each. The EEG (n = 280) showed epileptiform discharges in about 50% of patients. Epileptiform discharges during activation was observed in 40/249 patients (16.1%): Hyperventilation in 32 (12.8%) and photic stimulation in 19 (7.6%). The seizures were well controlled with anti-epileptic drugs (AEDs) in 232 (80.8%) patients and among them, 225 (78.4%) patients were on monotherapy. Valproate (n = 131) was the most frequently prescribed as monotherapy.
Conclusions:
This is one of the largest cohort of patients with IGE. This study reiterates the importance of segregating IGE syndrome and such analysis will aid to the current understanding and management.
Keywords: Electroencephalographic, idiopathic generalized epilepsy, epilepsy syndromes, seizure types
Introduction
Idiopathic generalized epilepsies (IGE) is a group of disorder with distinct clinical and electroencephalographic (EEG) features, and constitutes nearly one third of all epilepsies.[1,2] The IGEs are one of its four major groups emerging from a double dichotomy of generalized versus localization-related and idiopathic versus symptomatic. Generalized seizures occur with different and similar semiologies, frequencies, and patterns, ages at onset, and outcomes in different IGEs, suggesting common neuroanatomical pathways for seizure phenotypes. However, the same seizure phenotypes respond differently to the same treatments in different IGEs, suggesting different molecular defects across syndromes. Hence, incorrect and under diagnosis and inappropriate treatment and even usage of EEG is common, resulting in intractability and morbidity in IGE.[3,4]
Recognition of IGEs has important implications for management. Early diagnosis of IGEs can provide reassurance for adult patients and unnecessary repeated brain imaging can be avoided. Proper identification is also important for clinical genetic studies and regulatory trials of new anti-epileptic drugs (AEDs). However, the greatest value of accurate classification is in guiding the choice of treatment.[5] Studies on large cohort of IGE and its subtypes from developing country like India are few.[6–8]
Hence, the present study is planned to study the phenotype and EEG features and therapeutic aspects of IGEs in south Indian population.
Patients and Methods
This prospective cross-sectional hospital based study was carried out on non-consecutive recruitment of 287 patients (age 22.3 ± 10.7 years; M:F = 158:139) evaluated from 2001 to 2008 as IGE syndrome at the neurology out-patient services at our center, a university teaching hospital and major referral center for neuropsychiatric patients in South India. All the patients diagnosed to have IGE with or without family history of seizures were recruited. The definitions of seizure type and epilepsy syndrome were as per the International League Against Epilepsy (ILAE) (1981) and (1989) criteria,[9,10] and that proposed by Nordli (2005).[11] Patients with acute and remote symptomatic seizures; focal epilepsies with or without secondary generalization; evidence of abnormality in neuroimaging as the cause of seizures; neurodegenerative or metabolic disorders with epilepsy; non-epileptic attack disorders were excluded. Seizure control - were considered if patients were seizure free for at least 1 year while on AED; medically refractory seizures - those with seizure occurring with frequency of 1-2 per month while on regular ≥2 AEDs on standard dosages for the last 1 year.
Patients who met the eligibility criteria were enrolled for the study and data regarding their demographic characteristics, pedigree, semiology of seizures, frequency, triggering factors, seizure control, etiology, general physical, and neurological examination was noted. Routine blood investigations (biochemical profile, urine analysis), and other specific tests like ultrasound of abdomen in patients on sodium valproate were carried out. All patients underwent brain imaging with computerized tomography (CT) scan of the brain. Magnetic resonance imaging (MRI) was carried out only in 10 patients based on clinical indications, feasibility, and affordability. Scalp EEGs were recorded from 2004 onwards on 16-channel “Galileo NT (EBN)” machine, employing International 10-20 system of electrode placement using standard parameters and procedures, e.g., high filter: 70 Hz; low filter: 0.1 Hz; recording time: 30 min; sensitivity: 7 μV/mm; sweep speed: 10 s/page; sampling rate: 256 Hz. Various referential and bipolar montages were used for the purpose. All patients underwent activation procedures in the form of hyperventilation for 2 min and photic stimulation (1-30 Hz for 5 s at a stretch with eyes open and closed). Additional Electrocardiograph (ECG) and electromyograph (EMG) electrodes were used whenever warranted. All the data, thus, obtained was entered onto a predesigned digital datasheet.
The data was incorporated into an MS excel spread sheet for analysis.
Results
The mean age at evaluation and onset of seizures were 22.2 ± 7.7 and 16.9 ± 6.8 years respectively. Majority of patients had seizure onset below 20 years of age (n = 178; 62.02%). Thirty-one patients (10.8%) had history of epilepsy in their family. The number of affected individuals in the family was: One: 16; two: 9; three: 4; and four: 2. The types of seizures in the family were generalized seizures (22), myoclonic seizures (7), and absence seizures (2). The age at onset of seizures in the family members were in the same decade compared to the patients in the study. Among the cohort of 287 patients with IGE syndrome in this study, history of febrile seizure was present in nine patients. Triggering factors noted in 128 patients were sleep deprivation (n = 56; 19.5%), non-compliance and stress in 14 each (4.8%). The details of clinical manifestations are mentioned in Table 1.
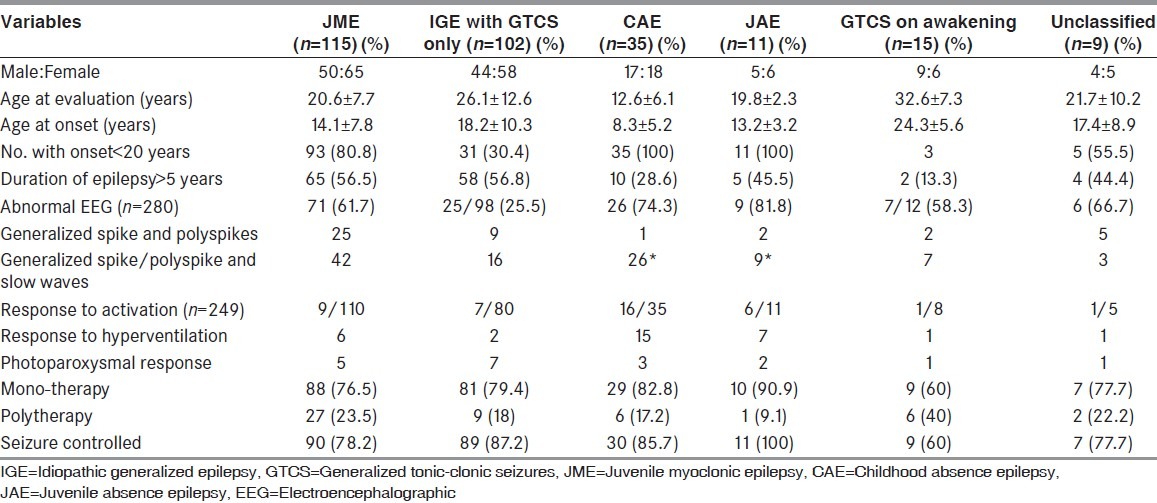
Table 1.
Details of various epilepsy syndromes in this cohort
All the 287 patients underwent CT scan of brain: Normal: 252 and in remaining 25 patients, it showed diffuse cerebral atrophy (11), cerebellar atrophy (10), incidental arachnoid cyst (2), and incidental calcified granuloma (2). The diffuse atrophy and cerebellar atrophy could be related to chronic epilepsy and long term AED usage. Twenty five patients underwent MRI scan of brain and all of them were normal.
EEG observations
The details of the EEG observations (n = 280) are mentioned in Table 2 and Figure 1. The background activity was predominantly alpha activity in 75% (n = 201) of patients. The EEG was normal in 143 (51.07%) patients and was abnormal in 137 (49.9%) patients. The abnormalities included generalized spike-wave and/or spike-polyspike morphology in majority. Forty patients out of 249 (16.1%) had increased epileptiform activity during activation procedures. Further, it is interesting to note that only young onset patients had response to activation procedures.
Table 2.
AEDs used by patients with each syndrome of idiopathic generalized epilepsy
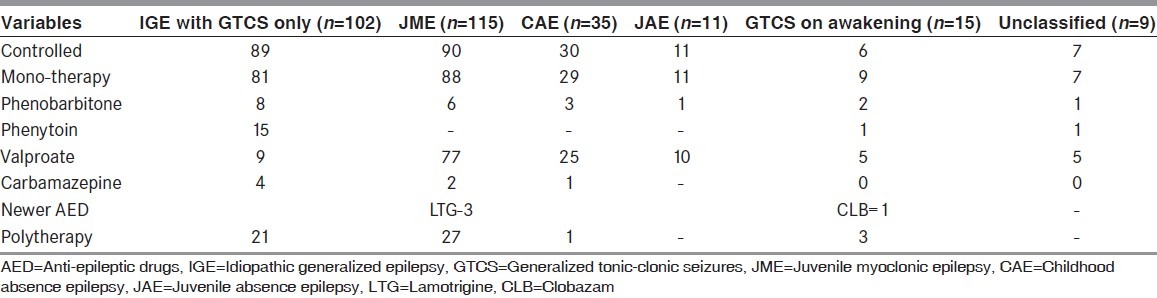
Figure 1.
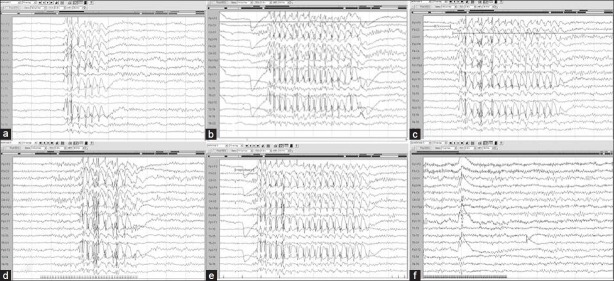
Mrs. K aged 25 years with uncontrolled seizures since 19 years of age on tablet phenobarbitone 60 mg/day. Initial EEG shows generalized bursts of high amplitude spikes and slow waves in (a) spontaneous record, (b) hyperventilation, (c) post-hyperventilation, (d) photic stimulation, (e) during eye closure; and (f) repeat EEG after 1 week of valproic acid therapy showed no epileptiform activity
Treatment details
Majority of the patients (n = 165) had consulted a physician for treatment within a month of having a seizure. One hundred ninety-one (66.5%) patients were compliant with the AED. Sixty three (21.9%) patients had documented adverse effects during the course of the AED therapy. The common adverse effects were hair loss (n = 17), tremor (n = 15), weight gain (n = 14), ataxia (n = 13), amenorrhoea (n = 5), and gum hyperplasia (n = 4). Two hundred sixteen patients were on monotherapy, while 71 patients (24.7%) were on polytherapy. Seizures were well controlled with AEDs in 232 (80.8%) patients and among them 210 (73.1%) were on monotherapy. About 40.06% had initiated AED within a month of onset of seizures.
Juvenile myoclonic epilepsy (n = 115)
Juvenile myoclonic epilepsy (JME) was the most common epilepsy syndrome seen in this cohort consisting of 115 (40.1%) patients. Among them, 65 (56.5%) were women. The mean age at evaluation was 20.6 ± 7.7 years (median 19, range 8-45). The mean age at onset of epilepsy was 14.1 ± 7.8 years (median 14, range 5-39). Fifty patients had duration <5 years. Seventy one (61.7%) had an abnormal EEG. The types of epileptiform discharges seen: Generalized spike/polyspikes and wave (42); generalized spike/polyspikes (25); focal discharges (4). Six patients had response to hyperventilation and 10 had paroxysmal photic response (PPR) to photic stimulation. Eighty eight (76.5%) patients were on monotherapy: Valproate (77); phenobarbitone (6); lamotrigine (3); and carbamazepine (2). Ninety (78.2%) patients had well controlled seizures.
IGE with generalized tonic-clonic seizures only (n = 102)
It was the next most common epilepsy syndrome seen in this cohort consisting of 102 (35.5%) patients. Among them, 44 (43.1%) patients were men and 58 (56.8%) were women. The mean age at evaluation was 26.1 ± 12.6 years (median 24 years, range 5-70 years). The mean age at onset of epilepsy was 18.2 ± 10.3 years (median 19 years, range 10-60 years). Fifty eight (56.8%) patient had duration of epilepsy >5 years. Twenty five among 98 patients (25.5%) had an abnormal EEG. Eighty one (79.4%) patients were on mono-therapy and eighty nine (87.2%) patients had good seizure control.
Childhood absence epilepsy (n = 35)
Among them, 17 patients were men. The mean age of at evaluation was 12.6 ± 6.1 years (median 11 years, range 3-24 years) and mean age at onset of epilepsy was 8.3 ± 5.2 years. Ten patients (28.6%) had duration of seizures >5 years. Twenty six (74.3%) had an abnormal EEG showing classical changes of 3-3.5 Hz spike-wave activity. Twenty nine (82.8%) patients were on mono-therapy. Thirty (85.7%) patients had a good seizure control.
Juvenile absence epilepsy (n = 11)
The Male:Female ratio was 5:6. The mean age at evaluation was 19.8 ± 2.26 years (median 19, range 18-23). The mean age at onset of epilepsy was 13.2 ± 3.2 years. Five patients had duration of seizures >5 years. Nine (81.8%) had an abnormal EEG. All patients with Juvenile absence epilepsy (JAE) had excellent seizure control.
GTCS on awakening
It was seen in 15 patients (Male: Female = 9:6). The mean age at evaluation was 32.6 ± 7.3 years (median 32 years, range 23-43 years). The mean age at onset was 24.3 ± 5.6 years. Thirteen (86.6%) had duration <5 years. Three out 12 patients (25%) had an abnormal EEG. Nine (60%) patients were on mono-therapy. Six (40%) had uncontrolled seizures.
Unclassified
Nine (3.1%; Male: Female = 4:5) patients could not be classified as per the ILAE. The mean age at evaluation and onset was 21.7 ± 10.2 and 17.4 ± 8.9 years, respectively. Seizures were controlled in majority (n = 7; 77.7%) while on mono-therapy (77.7%).
Discussion
Various studies have shown that IGE constitute 15-20% of all epilepsies.[12,13] There is a widespread assumption that IGE are rare beyond childhood and thus of little concern to adult neurologists. A tendency to over diagnose focal epilepsies with secondary generalization rather than IGE has its own consequences beyond the choice of AEDs. All the AEDs are not equally effective in IGE and moreover AEDs like carbamazepine and phenytoin might exacerbate absence and myoclonic seizures.[8] Genetic research in IGE depends largely on documentation of phenotype in patients and their relatives. There is a paucity of large series on IGE from India. Hence, this study was carried out on South Indian population with IGE syndrome for better phenotypic and EEG characterization.
Phenotypic observations
The phenotype and EEG parameters of 287 patients with IGE were studied. Some of the IGE subtypes like benign myoclonic epilepsy of infancy, Generalized epilepsy with febrile seizures (GEFS) plus and other unrecognized pediatric IGE syndromes were not observed possibly due to referral bias and inclusion of adult patients. About 11% of patients had a family history in the cohort. In a study by Valentin et al. (2007)[14] (IGE = 275) 14% had a family history of epilepsy. The most common type of epilepsy syndrome in this study were JME (40.06%) followed by “IGE with GTCS only” (35.5%), absence epilepsy (16.02%), and unclassified (3.1%). In a study (IGE = 58) by Benbadis et al.,[5] “IGE with GTCS only” (n = 36; 62%) comprised the largest group. Nicolson et al. (2004)[15,16] (n = 962) reported JME as commonest followed by “IGE with GTCS only” as in this study. Mohanraj and Brodie (2007)[17] (n = 103) confirmed the same findings in their study on IGE.
Michelucci et al. (1996)[18] defined adult onset IGE occurring in patients >20 years. We chose the age of 20 years as a pragmatic cut-off year for “adult” IGE. In this study, majority (n = 178, 62%) of the patients had onset of epilepsy <20 years. In the group with onset <20 years, 33.3% had GTCS, 12.9% had absence seizures, and 10.8% had myoclonic seizures as a single seizure type. In a study by Cutting et al. (2001)[19] (n = 313), 42 (13.4%) patients had adult onset IGE (mean: 23.8 years; range: 18-55 years). Marini et al.[20] (n = 121) found out that 34 (28%) had onset after 20 years. Gastaut (1981) reported that 35% of his IGE cases began >18 years and of these 95% began <50. In this cohort, among the 109 (38%) patients with adult onset IGE, the commonest type of epilepsy syndrome was GTCS on awakening (n = 12; 80%), epilepsy with GTCS only (n = 71; 65.1%), JME (n = 22; 20.2%), and unclassified (n = 4; 44.5%). In a study of 772 patients with seizure onset <20 years by Nicolson et al. (2004),[15,16] 93% had GTCS, 47% had myoclonic and 46.4% had absence seizures, but they did not subdivide patients with single or multiple types of seizures.[15,16]
JME was seen in 2/5th of patients in this study. The mean age at onset of JME was 14 years with slight female predominance. The clinical features were in accordance with the standard literature. An abnormal EEG was noted in 61.7% in this study similar to other studies. Studies had shown that EEG abnormalities are more often after a seizure and AEDs can reduce the abnormalities. Valproate as a mono-therapy was used by 67% of our patients. Jain et al. (2003)[7] and Panayiotopoulos et al. (1994)[21] reported its use in 74% and 44%, respectively. GTCS formed the 2nd common group in this cohort with abnormal EEG was seen in 1/4th of them.[20] This was in contrast to a study by Nicolson et al. (2004)[15,16] who observed EEG changes in 2/3rd of patients. Majority (79.4%) of patients with “IGE with GTCS” were on mono-therapy. Seizure control was higher in our study (87.2%) compared to other studies. Childhood absence epilepsy (CAE) was seen in 12.2% and valproate alone was used in 80% of patients. Polytherapy was used by 17.1% compared to 11% of patients in a study by Grosso (2005).[22] Seizure control was achieved in 85.7% patients, as in other studies. Juvenile absence epilepsy (JAE) was seen in 3.8% of patients with seizures control in all the JAE patients. Obeid (1994)[23] opined that polytherapy might not be essential if seizure control is achieved and not interfering activities of daily life. GTCS on awakening were observed in 5.2% of patients with mean age at onset of 24 years. Wolf (1992)[24] had described similar observations.
EEG findings
About half of patients had an abnormal EEG viz. spikes, polyspikes and slow waves. Stephan (2000)[25] had described similar observations in patients with JME. Betting et al. (2006)[26] (n = 180) observed that 1/3rd with IGE showed typical EEG abnormalities of IGE. Panayiotopoulos et al. (1994)[21] observed that 51.5% of patients had spike/multiple spike-slow wave discharges. In a study by Jain et al. (2003),[7] EEG was abnormal in 90% of drug-naïve JME. Forty out of 249 patients (16.1%) in this study had increased epileptiform activity during activation. Interestingly, response to hyperventilation was noted in 32 patients (12.8%) and PPR was observed in 19 (7.6%) of them. Photosensitivity in IGE is reported to range from 7.5% in JAE to 100% in pure photosensitive epilepsy.[27] below Panayiotopoulos et al. (1994)[21] found that 27.3% had PPR in 66 patients with JME.
Treatment
Majority of patients (73%) in this cohort were on mono-therapy. Seizures were well controlled with AEDs in 232 (80.8%) patients. Valproate was most frequently prescribed (58.2%) in those on mono-therapy. In the polytherapy group, valproate was used in combination with either phenobarbitone or lamotrigine. Bourgeois et al. (1987)[28] also reported usefulness of valproate as the lone drug in JME. Glauser et al. (2006)[29] in ILAE task force laid down evidences for usage of AEDs as initial mono-therapy. Benbadis et al. (2003)[5] reported that 70% of IGE were receiving ill-advised AED, mostly phenytoin and carbamazepine and among them, only 22% were seizure-free. In this cohort, 1/4th of patients were receiving ill-advised AED, mostly phenytoin and carbamazepine. About 5.6% of patients were non-compliant. Non-compliance was reported in 5.1% of 155 patients by Gellisse et al. (2001).[30] He also observed that 74.8% of JME had seizure freedom. He reported that uncontrolled seizures seen in 3.2% were slightly more common in patients with seizure onset >20 years. Mohanraj and Brodie (2007)[17] (IGE = 107) reported seizure control in 74%. Among the various syndromes of IGE, absence epilepsies (CAE and JAE) had the best seizure control (93.3-100%) in this cohort. There are specific guidelines and practice parameters in women with IGE.[31,32]
Some of the limitations were retrospective study, non-consecutive recruitment, lack of follow-up data. Nevertheless, it highlights the importance of diagnosing the epilepsy type or syndrome so that the choice of AEDs is correct. A related issue is the widespread assumption that IGE are rare beyond childhood and thus of little concern to adult neurologists. As a result, GTC seizures (especially in adults) are often assumed as secondarily generalized, i.e., focal epilepsy. This might have serious consequences beyond the choice of medications. Clinicians should enquire about myoclonic jerks, absence seizures and certain precipitating factors such as hyperventilation and photosensitivity, normal imaging, and specific spike-wave patterns for clues toward making a diagnosis of IGE syndrome. Adequate steps to study newer AEDs in IGE syndrome might be required.
Footnotes
Source of Support: Nil
Conflict of Interest: Nil.
References
- 1.Panayiotopoulos CP, Koutroumanidis M, Giannakodimos S, Agathonikou A. Idiopathic generalised epilepsy in adults manifested by phantom absences, generalised tonic-clonic seizures, and frequent absence status. J Neurol Neurosurg Psychiatry. 1997;63:622–7. doi: 10.1136/jnnp.63.5.622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Panayiotopoulos CP. Idiopathic generalized epilepsies. In: Panayiotopoulos CP, editor. The Epilepsies: Seizures, Syndromes and Management. Oxford: Bladon Medical Publishing; 2005. pp. 271–348. [PubMed] [Google Scholar]
- 3.Durón RM, Medina MT, Martínez-Juárez IE, Bailey JN, Perez-Gosiengfiao KT, Ramos-Ramírez R, et al. Seizures of idiopathic generalized epilepsies. Epilepsia. 2005;46:34–47. doi: 10.1111/j.1528-1167.2005.00312.x. [DOI] [PubMed] [Google Scholar]
- 4.Koutroumanidis M, Smith S. Use and abuse of EEG in the diagnosis of idiopathic generalized epilepsies. Epilepsia. 2005;46:96–107. doi: 10.1111/j.1528-1167.2005.00320.x. [DOI] [PubMed] [Google Scholar]
- 5.Benbadis SR, Tatum WO, 4th, Gieron M. Idiopathic generalized epilepsy and choice of antiepileptic drugs. Neurology. 2003;61:1793–5. doi: 10.1212/01.wnl.0000098891.76373.15. [DOI] [PubMed] [Google Scholar]
- 6.Jha S, Mathur VN, Mishra VN. Pitfalls in diagnosis of epilepsy of Janz and its implications. Neurol India. 2002;50:467–9. [PubMed] [Google Scholar]
- 7.Jain S, Tripathi M, Srivastava AK, Narula A. Phenotypic analysis of juvenile myoclonic epilepsy in Indian families. Acta Neurol Scand. 2003;107:356–62. doi: 10.1034/j.1600-0404.2003.00085.x. [DOI] [PubMed] [Google Scholar]
- 8.Chakravarty A, Mukherjee A, Roy D. Observations on juvenile myoclonic epilepsy amongst ethnic Bengalees in West Bengal: An Eastern Indian State. Seizure. 2007;16:134–41. doi: 10.1016/j.seizure.2006.10.012. [DOI] [PubMed] [Google Scholar]
- 9.Proposal for revised clinical and electroencephalographic classification of epileptic seizures. Commission on classification and terminology of the International League Against Epilepsy. Epilepsia. 1981;22:489–501. doi: 10.1111/j.1528-1157.1981.tb06159.x. [DOI] [PubMed] [Google Scholar]
- 10.Proposal for revised classification of epilepsies and epileptic syndromes. Commission on classification and terminology of the International League Against Epilepsy. Epilepsia. 1989;30:389–99. doi: 10.1111/j.1528-1157.1989.tb05316.x. [DOI] [PubMed] [Google Scholar]
- 11.Nordli DR., Jr Idiopathic generalized epilepsies recognized by the International League Against Epilepsy. Epilepsia. 2005;46:48–56. doi: 10.1111/j.1528-1167.2005.00313.x. [DOI] [PubMed] [Google Scholar]
- 12.Gastaut H, Gastaut JL, Gonçalves e Silva GE, Fernandez Sanchez GR. Relative frequency of different types of epilepsy: A study employing the classification of the International League Against Epilepsy. Epilepsia. 1975;16:457–61. doi: 10.1111/j.1528-1157.1975.tb06073.x. [DOI] [PubMed] [Google Scholar]
- 13.Jallon P, Latour P. Epidemiology of idiopathic generalized epilepsies. Epilepsia. 2005;46:10–4. doi: 10.1111/j.1528-1167.2005.00309.x. [DOI] [PubMed] [Google Scholar]
- 14.Valentin A, Hindocha N, Osei-Lah A, Fisniku L, McCormick D, Asherson P, et al. Idiopathic generalized epilepsy with absences: Syndrome classification. Epilepsia. 2007;48:2187–90. doi: 10.1111/j.1528-1167.2007.01226.x. [DOI] [PubMed] [Google Scholar]
- 15.Nicolson A, Chadwick DW, Smith DF. A comparison of adult onset and “classical” idiopathic generalised epilepsy. J Neurol Neurosurg Psychiatry. 2004;75:72–4. [PMC free article] [PubMed] [Google Scholar]
- 16.Nicolson A, Appleton RE, Chadwick DW, Smith DF. The relationship between treatment with valproate, lamotrigine, and topiramate and the prognosis of the idiopathic generalised epilepsies. J Neurol Neurosurg Psychiatry. 2004;75:75–9. [PMC free article] [PubMed] [Google Scholar]
- 17.Mohanraj R, Brodie MJ. Outcomes of newly diagnosed idiopathic generalized epilepsy syndromes in a non-pediatric setting. Acta Neurol Scand. 2007;115:204–8. doi: 10.1111/j.1600-0404.2006.00791.x. [DOI] [PubMed] [Google Scholar]
- 18.Michelucci R, Rubboli G, Passarelli D, Riguzzi P, Volpi L, Parmeggiani L, et al. Electroclinical features of idiopathic generalised epilepsy with persisting absences in adult life. J Neurol Neurosurg Psychiatry. 1996;61:471–7. doi: 10.1136/jnnp.61.5.471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cutting S, Lauchheimer A, Barr W, Devinsky O. Adult-onset idiopathic generalized epilepsy: Clinical and behavioral features. Epilepsia. 2001;42:1395–8. doi: 10.1046/j.1528-1157.2001.14901.x. [DOI] [PubMed] [Google Scholar]
- 20.Marini C, King MA, Archer JS, Newton MR, Berkovic SF. Idiopathic generalised epilepsy of adult onset: Clinical syndromes and genetics. J Neurol Neurosurg Psychiatry. 2003;74:192–6. doi: 10.1136/jnnp.74.2.192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Panayiotopoulos CP, Obeid T, Tahan AR. Juvenile myoclonic epilepsy: A 5-year prospective study. Epilepsia. 1994;35:285–96. doi: 10.1111/j.1528-1157.1994.tb02432.x. [DOI] [PubMed] [Google Scholar]
- 22.Grosso S, Galimberti D, Vezzosi P, Farnetani M, Di Bartolo RM, Bazzotti S, et al. Childhood absence epilepsy: Evolution and prognostic factors. Epilepsia. 2005;46:1796–801. doi: 10.1111/j.1528-1167.2005.00277.x. [DOI] [PubMed] [Google Scholar]
- 23.Obeid T. Clinical and genetic aspects of juvenile absence epilepsy. J Neurol. 1994;241:487–91. doi: 10.1007/BF00919710. [DOI] [PubMed] [Google Scholar]
- 24.Wolf P. Epilepsy with grand mal on awakening. In: Roger J, Bureau M, Dravet Ch, Dreifuss FE, Ferret A, Wolf P, editors. Epileptic Syndromes in Infancy, Childhood and Adolescence. 2nd ed. London: John Libbey; 1992. pp. 329–41. [Google Scholar]
- 25.Stephan W. The EEG in juvenile myoclonic epilepsy. In: Schmitz B, Sander T, editors. Juvenile Myoclonic Epilepsy: The Janz Syndrome. Philadelphia: Wrightson Biomedical Publishing; 2000. pp. 41–55. [Google Scholar]
- 26.Betting LE, Mory SB, Lopes-Cendes I, Li LM, Guerreiro MM, Guerreiro CA, et al. EEG features in idiopathic generalized epilepsy: Clues to diagnosis. Epilepsia. 2006;47:523–8. doi: 10.1111/j.1528-1167.2006.00462.x. [DOI] [PubMed] [Google Scholar]
- 27.Covanis A. Photosensitivity in idiopathic generalized epilepsies. Epilepsia. 2005;46:67–73. doi: 10.1111/j.1528-1167.2005.00315.x. [DOI] [PubMed] [Google Scholar]
- 28.Bourgeois B, Beaumanoir A, Blajev B, de la Cruz N, Despland PA, Egli M, et al. Monotherapy with valproate in primary generalized epilepsies. Epilepsia. 1987;28:S8–11. doi: 10.1111/j.1528-1157.1987.tb05769.x. [DOI] [PubMed] [Google Scholar]
- 29.Glauser T, Ben-Menachem E, Bourgeois B, Cnaan A, Chadwick D, Guerreiro C, et al. ILAE treatment guidelines: Evidence-based analysis of antiepileptic drug efficacy and effectiveness as initial monotherapy for epileptic seizures and syndromes. Epilepsia. 2006;47:1094–120. doi: 10.1111/j.1528-1167.2006.00585.x. [DOI] [PubMed] [Google Scholar]
- 30.Gelisse P, Genton P, Thomas P, Rey M, Samuelian JC, Dravet C. Clinical factors of drug resistance in juvenile myoclonic epilepsy. J Neurol Neurosurg Psychiatry. 2001;70:240–3. doi: 10.1136/jnnp.70.2.240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Crawford P. Best practice guidelines for the management of women with epilepsy. Epilepsia. 2005;46:117–24. doi: 10.1111/j.1528-1167.2005.00323.x. [DOI] [PubMed] [Google Scholar]
- 32.Thomas SV. Management of epilepsy and pregnancy. J Postgrad Med. 2006;52:57–64. [PubMed] [Google Scholar]