

Abstract
Background:
Joubert Syndrome (JS) is a rare genetic developmental disorder, first identified in 1969. In patients with JS, certain regions of the brain (mainly cerebellar vermis and brainstem) are underdeveloped or malformed. This can lead to impaired attention, visual, spatial, motor, language and social functional skills. JS is characterized by a host of features, many of which do not occur in every patient.
Aim of the Study:
To spotlight and increase awareness of clinical profile and neuroimaging findings of children with Joubert syndrome.
Methods:
This is a retrospective case series study of patients with JS who attended the Pediatric Neurology Clinic in Aladan and Alfarawanya Hospitals in Kuwait, from September 2007 to September 2012. Clinical and radiological data were obtained from the patient medical records.
Results:
Cerebellar vermis hypoplasia/aplasia and apnea were present in all patients, polydactly in 3 of 16, renal problems with cysts in 5 patients and 11 of 16 had abnormal electroretinograms (ERGs). Blood investigations of organic acids, amino acids and very-long-chain fatty acid, were normal in the all the nine patients.
Conclusion:
JS is a rare genetic brain malformation with association of retinal dystrophy and renal abnormalities. The retinal dystrophy may be progressive. The prognosis of patients depends mainly on the degree of brain malformation.
Keywords: Cerebellar vermis hypoplasia, children, Joubert syndrome, renal anomalies, retinal dystrophy
Introduction
Joubert syndrome (JS) is a rare autosomal recessive disorder, first identified in 1969 by Marie Joubert,[1] with agenesis of the cerebellar vermis presenting episodic hyperpnoea, abnormal eye movements, ataxia and intellectual disability. Several years later, a pathognomonic midbrain-hindbrain malformation, the “molar tooth sign” (MTS) (distinctive cerebellar and brainstem malformation) on magnetic resonance imaging, was detected first in JS. The term Joubert syndrome and related disorders (JSRD) has been recently adopted to describe disorders presenting the MTS. JSRDs include JS, as well as other related conditions showing the MTS, such as the cerebello-oculo-renal syndrome, Dekaban-Arima syndrome, COACH syndrome (Cerebellar vermis hypoplasia/aplasia, Oligophrenia, Ataxia, Coloboma, and Hepatic fibrosis syndrome), Varadi-Papp syndrome and a minority of cases with Senior-Loken syndrome.[2–6]
Most cases of JS are sporadic; however in some families, JS appears to be inherited via a recessive gene. The specific gene was recently located on chromosome 6q23.2-q23.3.[7,8]
Other physical deformities that may be present in JS are polydactyly, cleft lip or palate, tongue abnormalities, hypotonia, encephalocele, meningocele, hydrocephalus, kidney problems, pituitary abnormality and autistic-like behavior. Seizures may also occur. Some children have a mild form of the disorder, with minimal motor disability and good mental development, whereas others may have severe motor disability and moderate mental retardation.[9–13] Treatment for JS is symptomatic and supportive. The prognosis depends on whether or not the cerebellar vermis is entirely absent or partially developed.
JS is often missed clinically and radiologically if enough attention is not paid to its subtle and variable clinical presentation. So the objective of this study is to clarify the clinical and radiological features of JS and to increase the awareness of this rare congenital malformation.
Subjects and Methods
The study was performed on the patients diagnosed as JS who attended the Pediatric Neurology clinic in Aladan and Alfarawanya Hospitals in Kuwait, from September 2009 to September 2012. Ethical approval was obtained from the hospital’s Ethics Committee, and informed consent was obtained from the parents of each patient.
Eleven patients were diagnosed as JS. The diagnosis of JS was based on history (abnormal neonatal breathing), physical and neurological examination (abnormal eye movements, developmental delay, and ataxia) and magnetic resonance imaging (MRI) findings (MTS).
Clinical, radiological and laboratory data were obtained from the patients’ medical records. The following data were extracted and reviewed: Perinatal history, age of onset of symptoms, presenting complaint (apnea, ataxia, visual symptoms, and seizures), laboratory investigation results, urinary tract investigations, and brain computed tomography (CT), brain MRI scans and Electroencephalography (EEG).
Arrangements were made to recall the patients. Each recalled patient underwent a renal ultrasound and ophthalmological review including slit lamp microscopy of the anterior segment, fundus examination, studying of eye movement, electroretinograms (ERGs) and visual evoked potentials (VEPs). If any of the biochemical studies was inadequate, it was also performed. These included liver function tests, urea and electrolytes, very-long-chain fatty acids, serum amino acids and urine for amino acids and organic acids.
Statistical analysis
SPSS program version 18 was used to analyze the demographic data, neurological, ophthalmological, and renal manifestations, EEG, brain CT and MRI findings and results of renal ultrasound.
Results
The results are summarized in Table 1. In a total of 11 children (4 males and 7 females) were identified as having JS; as their final diagnosis. This included two pair of siblings from the same family. Two patients died; one from respiratory failure (at the age of 3 months) and the other one from aspiration pneumonia secondary to a cleft palate (at the age of 11 months). Therefore, 9 patients (3 males and 6 females) were reviewed in this study. Their age ranged from 6 months to 5 years (mean 2 years and 5 months). One child was delivered with meconium stained amniotic fluid. Consanguinity was observed in all the nine patients. The onset of symptoms which were usually in the form of respiratory symptoms or hypotonia was between 10 days and 5 months (mean in 40 days).
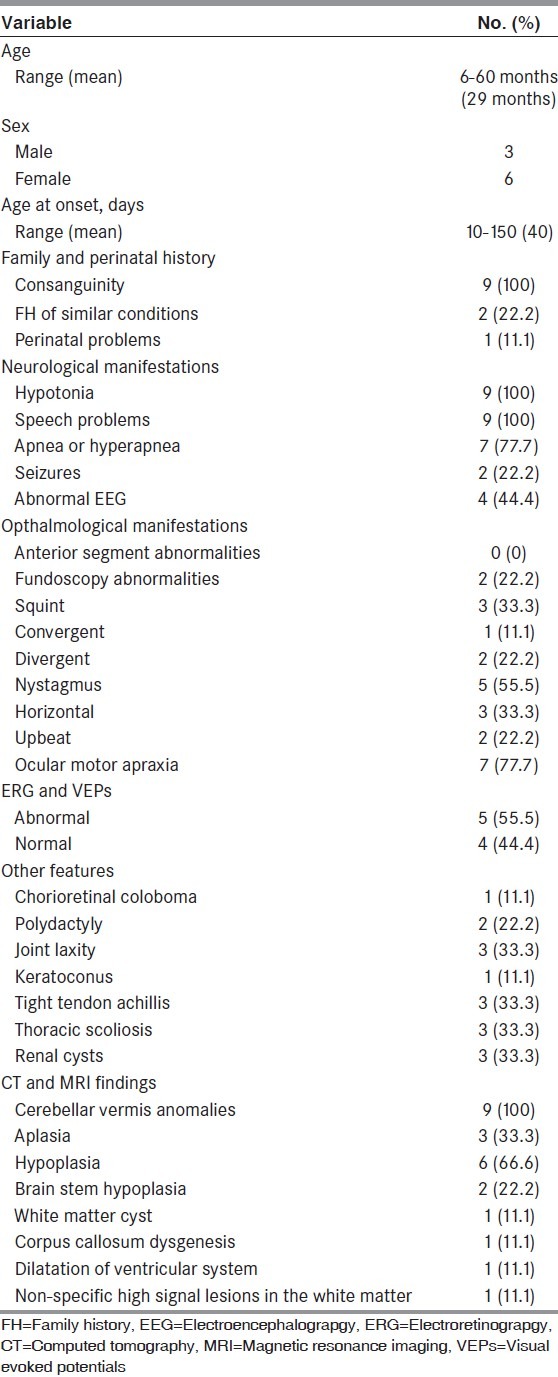
Table 1.
Clinical and radiological features of the studied population
Apneic episodes occurred in seven of nine patients, and six of them had transient phenomenon lasting up to 4 months and the fourth one continued to have attacks of hypercapnia with transient apneic episode required oxygen and apnea monitor at home up to the age of 10 months.
Neurologically, general hypotonia was an early observation in all nine patients. All patients also demonstrated some degree of motor and developmental delay, although this varied from mild to very severe. Two patients had walked unaided with a broad-based gait at the age of 3.5 years and 4 years. No intelligence quotient (IQ) assessment was performed during the study, but the speech was a problem in all our patients and seven of them were attending or had attended speech therapy. Of the seven children, one had developed intelligible speech. Those seven patients had mild to moderate disability although they had achieved toilet training and self-feeding. Two had severe disability, with failure to develop even those basic skills. Two patients had seizures and five patients had ataxia.
Other systemic features are also seen: Chorioretinal coloboma was seen in a patient, postaxial polydactyly was present in two patients (one of them was bilateral), general joint laxity in three patients, keratoconus in a patient, bilateral tight Achilles tendons in three patients, and thoracic scoliosis in four patients.
Three patients had ultrasound evidence of multiple renal cysts but they had normal renal function. Renal and liver function tests, urine for organic acids, serum, amino acids, very-long-chain fatty acids and routine karyotype were normal in all the patients. Nerve conduction velocity and electromyography were performed for all the seven patients and were normal. EEG was recorded in all our patients and was abnormal in four: Two had seizures; three had sharp discharges over focal areas and one had multifocal distribution consistent with the MRI of multifocal white matter intensities.
MRI and CT were reviewed in all patients. Cerebellar vermis aplasia/hypoplasia was present in all our patients [Figure 1]. The vermis was aplastic in three patients and hypoplastic in six patients, affecting mainly the postero-inferior part. In all patients, the midbrain and superior cerebellar peduncles displayed the MTS. Associated features were noted on the MRI and/or CT scans in five patients. These were brainstem hypoplasia in two patients, white matter cyst in a patient, corpus callosum dysgenesis in one, moderate dilation of the ventricular system in one, and non-specific high signal lesions in the white matter in a patient who also had seizures.
Figure 1.
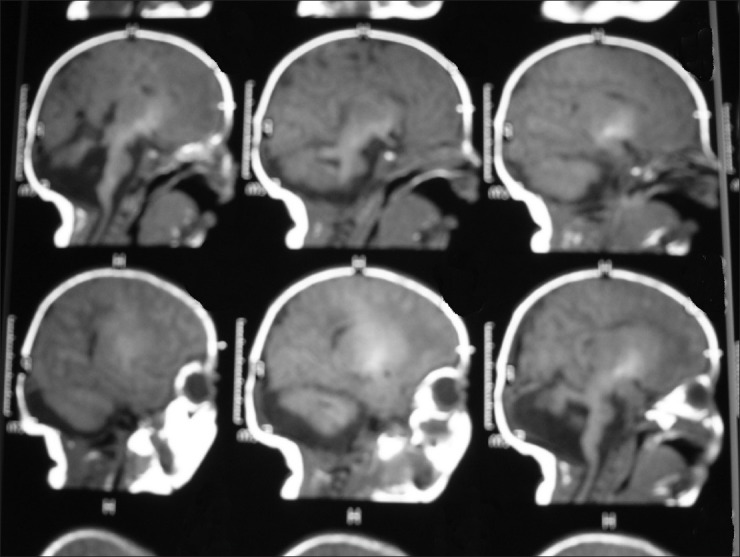
MRI of one patient with Joubert Syndrome
Ophthalmological examination
Anterior segment examinations were normal in all the nine patients but fundus examination revealed retinal pigment epithelium mottling in two patients who had abnormal ERG. There were three patients with squint (one with convergent squint and two with divergent squints). Five patients were associated with nystagmus (three with horizontal pendular nystagmus and two with upbeat nystagmus). In eye movement study; limited eye movement (ocular motor apraxia) was evident in seven of nine patients. ERG and VEPs were abnormal in five of our nine patients indicating that visual acuity was affected and at a rudimentary level.
Discussion
JS is a rare genetic developmental disorder, first identified in 1969. The diagnosis of JS as a definite diagnosis from other similar clinical conditions is difficult due to absence of a specific test or genetic marker.[14]
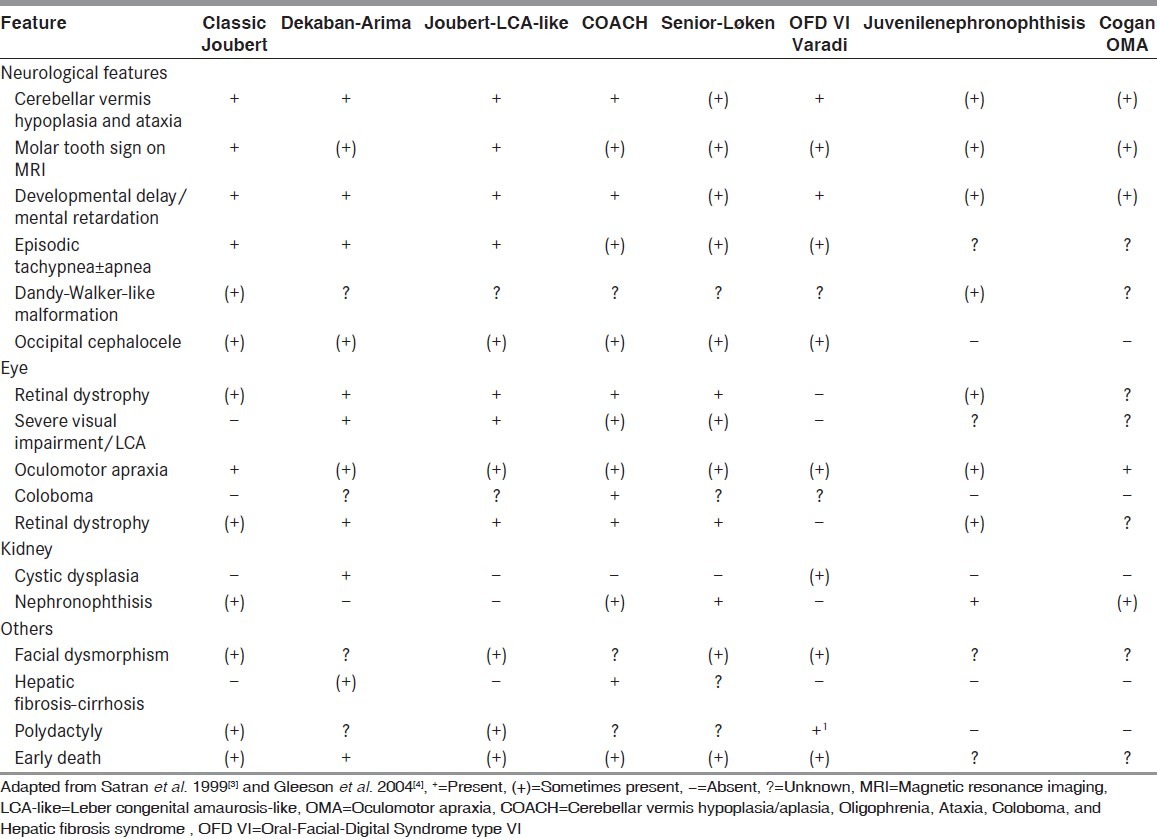
The term “JS” is reserved for those individuals fulfilling the diagnostic criteria that require the presence of developmental delay, abnormal ocular movements, and radiological evidence of marked cerebellar vermis abnormalities leading to the presence of the MTS on MRI.[15,16] This may also be termed classic JS. The term JSRD describes conditions that share the MTS and the some clinical features of JS, but that also have other manifestations that may represent a distinct syndrome. At least eight conditions in which a subset of affected individuals demonstrates the MTS have been identified[3,4] [Table 2]. There is a debate whether these represent subtypes of JS or distinct syndromes. In this study, only patients fulfilling the criteria mentioned before for diagnosis of JS were chosen and included in the study. Other features sometimes identified in JS include retinal dystrophy, renal disease, ocular colobomas, occipital encephalocele, hepatic fibrosis, polydactyly, oral hamartomas, and endocrine abnormalities.
Table 2.
Clinical features of Joubert syndrome and Joubert syndrome and related disorders
The prevalence of JS has been estimated from to be 1:258,000.[17,18] In our study, 11 cases were reported in two general hospitals draining about 750.000 people within 2 years of study, so the prevalence is little higher. This can be explained by consanguineous marriage which is famous in the gulf area (multigenerational consanguinity) and this was present in all our patients.
A reported male: Female ratio of approximately 2:1[18] was not confirmed in other surveys.[19] In our study, male to female ratio was 4: 7.
Possible prenatal diagnosis of JS can be achieved by serial prenatal ultrasound imaging starting at 11-12 weeks’ gestation, with detailed evaluation of cerebellar and other fetal anatomy through 20 weeks’ gestation, followed by fetal MRI imaging at 20-22 weeks’ gestation.[20,21] However, in our study, no patient was diagnosed in-utero, inspire of good follow up for the mothers during pregnancy. Only one patient had a perinatal problem (meconium stained amniotic fluid).
Many of the clinical features of JS are evident in infancy.[1,22] In our study, the onset of symptoms (usually apnea or hyperapnea) occurs in the first 40 days (range: 10 days to 5 month).
Typical respiratory abnormalities in JS are represented by short alternate episodes of apnea and hyperpnoea or episodic hyperpnoea alone, which tend to occur shortly after birth, and progressively improve with age, usually disappearing around the 6th month of life. Their severity can range from occasional short-lasting episodes manifesting every few days to extremely frequent (up to several per day) and prolonged attacks of apnea.[5,23] In our study 7 out of 9 patients had respiratory symptoms occurred in early infancy, 6 of them improved within 3 months and the 4th one improved at the age of 10 months.
Early hypotonia is observed in nearly all JS patients and can be recognized in the neonatal period or in infancy.[5,11] This is also observed in all our nine patients. All patients also demonstrated some degree of motor and developmental delay, although this varied from mild to very severe. There are many reports about the developmental disabilities in JS, in particular language and motor skills, with variable degrees of severity.[11,24,25] However, it must be stressed that intellectual deficit is not a mandatory feature of JS and exceptional cases may have borderline or even normal intellect. One of our patients had developed intelligible speech. There is a strong relationship between articulatory deficits and abnormal eye movement, and this might be attributable to vermis malformation.[26]
Although ataxia and balance difficulties are non-specific findings IN JS but they represent a frequent finding. Seven of our patients (77.8%) had either ataxia or broad-based gait. Although epilepsy is a rare feature of JS, abnormal EEG was reported in four patients representing 44.4% of our patients but this can be explained by the associated central nervous system (CNS) malformations other than MTS as midbrain hypoplasia, white matter cyst, corpus callosum dysgenesis, dilation of the ventricular system, and high signal lesions in the white matter which have higher incidence of epilepsy. Small number of cases presents with occipital (meningo) encephalocele of variable severity[27,28] but this was not reported in our patients.
Abnormal eye movements also represent a recurrent feature in JS. Oculomotor apraxia is one of the most characteristic and frequent abnormalities, that manifests with the inability to follow objects visually with compensatory head movements. Primary position nystagmus is also common. Five of our patients (55.6%) were associated with nystagmus. The nystagmus was horizontal pendular and upbeat nystagmus. These varieties of nystagmus were not typical of congenital sensory nystagmus even in those with an associated retinal dystrophy, and mostly can be attributed to a neurological cause probably resulting from brainstem malformation.[9,29] A range of eye movement abnormalities (ocular motor apraxia) were present, in seven of nine patients (77.8%) reviewed. There is a strong association of eye movement abnormalities with vermian malformations.[9,30,31]
Previous eye manifestations are present independently from the specific defects of the eyes and relate to the underlying midbrain-hindbrain malformation.[12,32] This was also observed in our patients who showed normal anterior segment by lit lamp examination. Dekaban (1969)[30] was the first to describe association of retinal problems with JS. Previously, Many studies have reported association of JS with very attenuated or undetectable rod-mediated ERGs.[9,29] Our study supports the reports in the literature through records of ERG and VEPs which showed retinal abnormalities in 55.6% of our patients. We found evidence of progressive retinal damage in one of the patients in VEPs compared with that in the old one done 2 years back. One of those patients had total visual impairment. Fundus examination of these patients revealed mottling of the retinal pigment epithelium, especially at the macula area in two patients with abnormal ERG.
Renal disease often occurs in 25% of individuals with JS. Two patients in our studies showed renal cysts, both of them had abnormal ERG. Many reports showed renal anomalies in JS. Other renal problems may be present in JS as shown in many reports such as renal dysplasia, and juvenile nephronophthisis, a form of chronic tubulointerstitial nephropathy.[5,33] Our patients showed normal renal function tests. Saraiva and Baraitser 1992 noted that retinal dystrophy was never absent when renal cysts were observed and can no longer be applied as a general rule.[19]
Some individuals with JS have congenital hepatic fibrosis as a result of anomalies of biliary structures and portal tracts during embryonic development.[5] Liver function tests with hepatic ultrasound are recommended biannually in children and at diagnosis in adults. None of our patients showed any hepatic fibrosis or abnormal hepatic function.
Other systemic features reported in our patients with JS as chorioretinal coloboma, postaxial polydactyly, general joint laxity, keratoconus, bilateral tight Achilles tendons, and thoracic scoliosis were also reported in other literatures.[5]
Conclusion
JS is a rare genetic brain malformation characterized by absence or underdevelopment of cerebellar vermis. Retinal dystrophy and renal abnormalities are common associations. The prognosis of patients depends mainly on the degree of brain malformation.
Footnotes
Source of Support: Nil
Conflict of Interest: Nil.
References
- 1.Joubert M, Eisenring JJ, Robb JP, Andermann F. Familial agenesis of the cerebellar vermis. A syndrome of episodic hyperpnea, abnormal eye movements, ataxia, and retardation. Neurology. 1969;19:813–25. doi: 10.1212/wnl.19.9.813. [DOI] [PubMed] [Google Scholar]
- 2.Maria BL, Hoang KB, Tusa RJ, Mancuso AA, Hamed LM, Quisling RG, et al. Joubert syndrome revisited: Key ocular motor signs with magnetic resonance imaging correlation. J Child Neurol. 1997;12:423–30. doi: 10.1177/088307389701200703. [DOI] [PubMed] [Google Scholar]
- 3.Satran D, Pierpont ME, Dobyns WB. Cerebello-oculo-renal syndromes including Arima, Senior-Löken and COACH syndromes: More than just variants of Joubert syndrome. Am J Med Genet. 1999;86:459–69. [PubMed] [Google Scholar]
- 4.Gleeson JG, Keeler LC, Parisi MA, Marsh SE, Chance PF, Glass IA, et al. Molar tooth sign of the midbrain-hindbrain junction: Occurrence in multiple distinct syndromes. Am J Med Genet A. 2004;125A:125–34. doi: 10.1002/ajmg.a.20437. [DOI] [PubMed] [Google Scholar]
- 5.Brancati F, Dallapiccola B, Valente EM. Joubert Syndrome and related disorders. Orphanet J Rare Dis. 2010;5:20. doi: 10.1186/1750-1172-5-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sattar S, Gleeson JG. The ciliopathies in neuronal development: A clinical approach to investigation of Joubert syndrome and Joubert syndrome-related disorders. Dev Med Child Neurol. 2011;53:793–8. doi: 10.1111/j.1469-8749.2011.04021.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ferland RJ, Eyaid W, Collura RV, Tully LD, Hill RS, Al-Nouri D, et al. Abnormal cerebellar development and axonal decussation due to mutations in AHI1 in Joubert syndrome. Nat Genet. 2004;36:1008–13. doi: 10.1038/ng1419. [DOI] [PubMed] [Google Scholar]
- 8.Valente EM, Brancati F, Dallapiccola B. Genotypes and phenotypes of joubert syndrome and related disorders. Eur J Med Genet. 2008;51:1–23. doi: 10.1016/j.ejmg.2007.11.003. [DOI] [PubMed] [Google Scholar]
- 9.Khan AO, Oystreck DT, Seidahmed MZ, AlDrees A, Elmalik SA, Alorainy IA, et al. Ophthalmic features of Joubert syndrome. Ophthalmology. 2008;115:2286–9. doi: 10.1016/j.ophtha.2008.08.005. [DOI] [PubMed] [Google Scholar]
- 10.Merritt L. Recognition of the clinical signs and symptoms of Joubert syndrome. Adv Neonatal Care. 2003;3:178–86. doi: 10.1016/s1536-0903(03)00137-1. [DOI] [PubMed] [Google Scholar]
- 11.Braddock BA, Farmer JE, Deidrick KM, Iverson JM, Maria BL. Oromotor and communication findings in joubert syndrome: Further evidence of multisystem apraxia. J Child Neurol. 2006;21:160–3. doi: 10.1177/08830738060210020501. [DOI] [PubMed] [Google Scholar]
- 12.Weiss AH, Doherty D, Parisi M, Shaw D, Glass I, Phillips JO. Eye movement abnormalities in Joubert syndrome. Invest Ophthalmol Vis Sci. 2009;50:4669–77. doi: 10.1167/iovs.08-3299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Maria BL, Boltshauser E, Palmer SC, Tran Tx. Clinical features and revised diagnostic criteria in Joubert syndrome. J Child Neurol. 1999;14:583–90. doi: 10.1177/088307389901400906. [DOI] [PubMed] [Google Scholar]
- 14.Blair IP, Gibson RR, Bennett CL, Chance PF. Search for genes involved in Joubert syndrome: Evidence that one or more major loci are yet to be identified and exclusion of candidate genes EN1, EN2, FGF8, and BARHL1. Am J Med Genet. 2002;107:190–6. [PubMed] [Google Scholar]
- 15.Maria BL, Quisling RG, Rosainz LC, Yachnis AT, Gitten J, Dede D, et al. Molar tooth sign in Joubert syndrome: Clinical, radiologic, and pathologic significance. J Child Neurol. 1999;14:368–76. doi: 10.1177/088307389901400605. [DOI] [PubMed] [Google Scholar]
- 16.McGraw P. The molar tooth sign. Radiology. 2003;229:671–2. doi: 10.1148/radiol.2293020764. [DOI] [PubMed] [Google Scholar]
- 17.Flannery DB, Hudson JG. A survey of Joubert syndrome. David W Smith workshop. Proc Greenwood Genet Ctr. 1994;13:130. [Google Scholar]
- 18.Badhwar A, Andermann F, Valerio RM, Andermann E. Founder effect in Joubert Syndrome. Ann Neurol. 2000;48:435–6. [Google Scholar]
- 19.Saraiva JM, Baraitser M. Joubert syndrome: A review. Am J Med Genet. 1992;43:726–31. doi: 10.1002/ajmg.1320430415. [DOI] [PubMed] [Google Scholar]
- 20.Doherty D, Glass IA, Siebert JR, Strouse PJ, Parisi MA, Shaw DW, et al. Prenatal diagnosis in pregnancies at risk for Joubert syndrome by ultrasound and MRI. Prenat Diagn. 2005;25:442–7. doi: 10.1002/pd.1145. [DOI] [PubMed] [Google Scholar]
- 21.Aslan H, Ulker V, Gulcan EM, Numanoglu C, Gul A, Agar M, et al. Prenatal diagnosis of Joubert syndrome: A case report. Prenat Diagn. 2002;22:13–6. doi: 10.1002/pd.220. [DOI] [PubMed] [Google Scholar]
- 22.Boltshauser E, Isler W. Joubert syndrome: Episodic hyperpnea, abnormal eye movements, retardation and ataxia, associated with dysplasia of the cerebellar vermis. Neuropadiatrie. 1977;8:57–66. doi: 10.1055/s-0028-1091505. [DOI] [PubMed] [Google Scholar]
- 23.Boltshauser E, Herdan M, Dumermuth G, Isler W. Joubert syndrome: Clinical and polygraphic observations in a further case. Neuropediatrics. 1981;12:181–91. doi: 10.1055/s-2008-1059650. [DOI] [PubMed] [Google Scholar]
- 24.Fennell EB, Gitten JC, Dede DE, Maria BL. Cognition, behavior, and development in Joubert syndrome. J Child Neurol. 1999;14:592–6. doi: 10.1177/088307389901400907. [DOI] [PubMed] [Google Scholar]
- 25.Gitten J, Dede D, Fennell E, Quisling R, Maria BL. Neurobehavioral development in Joubert syndrome. J Child Neurol. 1998;13:391–7. doi: 10.1177/088307389801300806. [DOI] [PubMed] [Google Scholar]
- 26.Hodgkins PR, Harris CM, Shawkat FS, Thompson DA, Chong K, Timms C, et al. Joubert syndrome: Long-term follow-up. Dev Med Child Neurol. 2004;46:694–9. doi: 10.1017/s0012162204001161. [DOI] [PubMed] [Google Scholar]
- 27.Shian WJ, Chi CS, Mak SC, Chen CH. Joubert syndrome in Chinese infants and children: A report of four cases. Zhonghua Yi Xue Za Zhi (Taipei) 1993;52:342–5. [PubMed] [Google Scholar]
- 28.Wang P, Chang FM, Chang CH, Yu CH, Jung YC, Huang CC. Prenatal diagnosis of Joubert syndrome complicated with encephalocele using two-dimensional and three-dimensional ultrasound. Ultrasound Obstet Gynecol. 1999;14:360–2. doi: 10.1046/j.1469-0705.1999.14050360.x. [DOI] [PubMed] [Google Scholar]
- 29.Schild AM, Fricke J, Herkenrath P, Bolz H, Neugebauer A. Neuro-ophthalmological and ophthalmological findings in Joubert syndrome. Klin Monbl Augenheilkd. 2010;227:786–91. doi: 10.1055/s-0029-1245735. [DOI] [PubMed] [Google Scholar]
- 30.Dekaban AS. Hereditary syndrome of congenital retinal blindness (Leber), polycystic kidneys and maldevelopment of the brain. Am J Ophthalmol. 1969;68:1029–37. doi: 10.1016/0002-9394(69)93443-6. [DOI] [PubMed] [Google Scholar]
- 31.Malaki M, Nemati M, Shoaran M. Joubert syndrome presenting as unilateral dysplastic kidney, hypotonia, and respiratory problem. Saudi J Kidney Dis Transpl. 2012;23:325–9. [PubMed] [Google Scholar]
- 32.Dekaban AS. Hereditary syndrome of congenital retinal blindness (Leber), polycystic kidneys and maldevelopment of the brain. Am J Ophthalmol. 1969;68:1029–1037. doi: 10.1016/0002-9394(69)93443-6. [DOI] [PubMed] [Google Scholar]
- 33.Sturm V, Leiba H, Menke MN, Valente EM, Poretti A, Landau K, et al. Ophthalmological findings in Joubert syndrome. Eye (Lond) 2010;24:222–5. doi: 10.1038/eye.2009.116. [DOI] [PubMed] [Google Scholar]