

Abstract
Ataxia, although rare, can be a symptom of many debilitating movement disorders. Hereditary ataxias are one subset of this condition and manifest when there is a genetic abnormality involved. Ataxia oculomotor apraxia type 1 (AOA1), an autosomal recessive ataxia, results from a mutation on the aprataxin gene (APTX). We characterized a novel homozygous deletion mutation (IVS4-12delT) on the APTX gene in a 14-year-old male born to consanguineous parents. This case report emphasizes the importance of investigating and increasing awareness of novel genetic mutations in order to help diagnose and further classify hereditary ataxias.
Keywords: AOA1, aprataxin gene, hereditary ataxia, mutation, oculomotor apraxia type 1
Introduction
Progressive ataxia is a neuromotor condition, the hallmark of degenerative ataxias. It is caused by neurodegeneration in the cerebellum and spinal cord, areas of the nervous system involved in regulating motor co-ordination and movement. Ataxia is characterized by symptoms such as loss of muscle co-ordination and balance as well as diminished fine motor skills. Ataxia is a rare condition that has been found to have a prevalence of 18.5 in 100,000.[1] There are many different types of degenerative ataxias, which are classified according to cause. One of these subgroups includes hereditary ataxias that are linked to genetic causes. These hereditary ataxias include Freidreich ataxia (FDRA), spinocerebellar ataxia, and ataxia with oculomotor apraxias types 1 (AOA1) and 2 (AOA2).[1]
FDRA is a recessive ataxia characterized by a mutation on chromosome 9q13.[2] It consists of a repeated trinucleotide sequence, GAA, in the intron of the frataxin gene.[2] This repetition is found in the normal gene however, in FDRA patients this same repetition is amplified. A lack of functioning frataxin leads to oxidative stress and an accumulation of iron concentration. This build-up of iron and free radicals may potentially be the cause of neurodegeneration in these patients. Another inherited ataxia is spinocerebellar ataxia, of which there are currently 25 known variants, each of which is caused by a different mutation.[3] Most of these mutations are also trinucleotide repeated sequences. AOA1 and AOA2 are caused by mutations on the aprataxin (APTX) and senataxin (SETX) gene, respectively.[1] APTX and SETX are genes, which encode for proteins involved in single strand DNA repair.[1]
Inherited ataxias are diagnosed mainly through DNA sequencing by the Sanger method. Other methods that can also be of diagnostic value are PCR analysis, MRI of the brain and spinal cord, motor and neurological examinations, and gait assessment. Currently, there is ongoing research into possible treatments for inherited ataxias although no viable treatments have yet been discovered. In this report, we present a case of a young male diagnosed with progressive AOA1, with a novel mutation on the APTX gene.
Case Report
This patient was a 17-year-old right-handed male of Afghani descent who developed progressive ataxia. His symptoms first started at age 10 when he developed dysarthria with scanning of speech, and he began to lose his balance as well as the ability to co-ordinate his movements. A former soccer player on his school team, he could no longer participate in the sport and in other athletic activities. He began to fall once or twice-a-day, and his typing and writing skills started to deteriorate due to lack of fine motor skills. In addition, he began to report difficulty in swallowing with frequent choking spells.
Born with no complications at birth, he started walking at 10 months, said his first words at 12 months, ran at age 2, and rode a bike at age 5. He has 3 sisters and a brother who are all healthy. The family is consanguineous as his parents are first cousins.
At age 13, he had his first neurological assessment where he was found to have slow speech with low volume. He had a moderate amplitude kinetic tremor of both upper extremities with target dysmetria on finger-nose-finger testing, as well as heal-knee-shin testing and dysdiadochokinesis. He had a wide-based, unsteady gait and was unable to stand with feet together for more than a few seconds. The rest of his neurological examination was unremarkable.
Investigations included complete blood count, serum electrolytes including calcium, magnesium, phosphorus, liver function tests, and a complete thyroid panel. An MRI of the brain revealed that he had an abnormal hypoplastic cerebellum as shown in Figure 1. Testing of somatosensory-evoked potentials and brainstem auditory response were both normal. Nerve conduction studies and electromyography were also both normal. However, his visual evoked potential was found to be abnormal upon binocular and monocular testing due to a low amplitude response.
Figure 1.
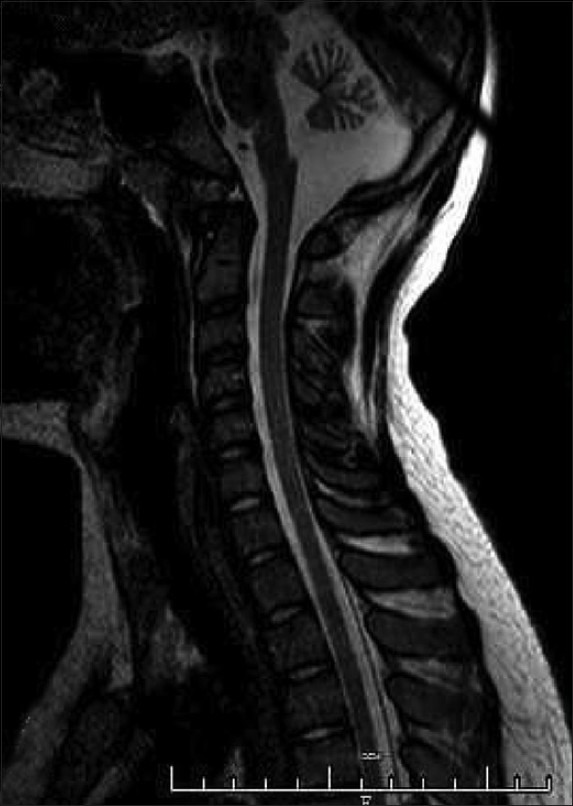
MRI Brainstem and C-Spine sagittal view T2 image showing moderate to severe cerebellar atrophy
Genetic testing for FDRA and spinocerebellar ataxia (SCA1, SCA2, SCA3, SCA6, SCA7, SCA8, SCA17) were found to be negative. However, DNA sequencing for AOA1 showed a mutation on the APTX gene. The mutation consists of a homozygous deletion of thymine at position 12 of IVS4 (IVS4-12delT). This specific mutation on the APTX gene has never been reported in the medical literature. Molecular testing of his parents showed that both of them were heterozygous for the same mutation and as a result, did not manifest any of the symptoms of ataxia at the time. His mother and father were 49 and 55 years of ages, respectively. His abnormal visual evoked potentials may be suggestive of ocular involvement, although it was not appreciated on history or physical examination.
Discussion
The APTX gene encodes for the single strand break DNA repair protein.[4,5] AOA1 is caused by a mutation on this gene.[6] The lack of functional APTX prevents the repair of breaks in DNA and has been shown to affect mitochondrial DNA to a great extent.[7] This mutation is located very close to the acceptor splice site of exon 5 and thus may result in the skipping of this exon during pre-mRNA processing. This leads to oculomotor apraxia, a condition that is characterized by the inability to make side-to-side eye movements.[8] The literature has described various types of genetic mutations associated with the APTX gene leading to AOA1 including substitution, missense and splicing mutations;[9–12] however, the IVS4-12delT mutation has never been specifically reported. This may play a major role in the observed oculomotor symptoms and may also be the cause of the abnormal visual evoked potentials seen in our patient.
Although this patient had phenotypic manifestations of the disease because of being homozygous for the mutation, his parents did not display any abnormalities due to their heterozygosity. Although not relevant to this case specifically, in trinucelotide repeat diseases, phenotypic appearance could manifest much earlier in the next generation due to a phenomenon of genetic anticipation. Genetic anticipation occurs when a disease caused by a mutation manifests earlier in the offspring than in the parents.[13] Progressive ataxias caused by trinucleotide repeat expansions are quite rare in the general population. Not only are these conditions rare, the manifestations of the disease as well as their contributing mutations vary to a great extent. This has led to an increase in the incidence of different types of ataxias. Therefore, clinicians must take an active role in reporting any new mutations to the scientific community, as this may lead to the discovery of a new inherited ataxia.
Footnotes
Source of Support: Nil
Conflict of Interest: Nil.
References
- 1.Klockgether T. Update on degeneration ataxias. Curr Opin Neurology. 2011;24:339–45. doi: 10.1097/WCO.0b013e32834875ba. [DOI] [PubMed] [Google Scholar]
- 2.Campuzano V, Montermini L, Moltò MD, Pianese L, Cossée M, Cavalcanti F, et al. Friedreich’s ataxia: Autosomal recessive disease caused by an intronic GAA triplet repeat expansion. Science. 1996;271:1423–7. doi: 10.1126/science.271.5254.1423. [DOI] [PubMed] [Google Scholar]
- 3.Pagon RA, Bird TD, Dolan CR. Heredity Ataxia Overview. Gene Reviews. 2012. [Last accessed on 05-07-2012]. Available from: http://www.ncbi.nlm.nih.gov/books/NBK1138/
- 4.Date H, Onodera O, Tanaka H, Iwabuchi K, Uekawa K, Igarashi S, et al. Early-onset ataxia with ocular motor apraxia and hypoalbuminemias caused by mutations in a new HIT superfamily gene. Nat Genet. 2001;29:184–8. doi: 10.1038/ng1001-184. [DOI] [PubMed] [Google Scholar]
- 5.Moreira MC, Barbot C, Tachi N, Kozuka N, Uchida E, Gibson T, et al. The gene mutated in ataxia-ocular apraxia 1 encodes the new HIT/Zn-finger protein APTX. Nat Genet. 2001;29:189–93. doi: 10.1038/ng1001-189. [DOI] [PubMed] [Google Scholar]
- 6.Tsao CY, Pauslson G. Type 1 ataxia with oculomotor apraxia with APTX gene mutations in two American Children. J Child Neurol. 2005;70:619–20. doi: 10.1177/08830738050200071701. [DOI] [PubMed] [Google Scholar]
- 7.Sykora P, Croteau DL, Bohr VA, Wilson DM. Aprataxin localizes to mitochondria and preserves mitochondrial function. PNAS. 2011;108:7437–42. doi: 10.1073/pnas.1100084108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.PeBenito R, Cracco JB. Congenital ocular motor apraxia: Case reports and literature review. Clin Pediatr. 1998;27:27–31. doi: 10.1177/000992288802700105. [DOI] [PubMed] [Google Scholar]
- 9.Amouri R, Moreira MC, Zouari M, El Euch G, Barhoumi C, Kefi M, et al. Aprataxin Gene Mutations in Tunisian Families. Neurology. 2004;63:928–9. doi: 10.1212/01.wnl.0000137044.06573.46. [DOI] [PubMed] [Google Scholar]
- 10.Ferrarini M, Squintani G, Cavallaro T, Ferrari S, Rizzuto N, Fabrizi GM. A novel mutation of aprataxin associated with ataxia ocular apraxia type 1: Phenotypical and genotypical characterization. J Neurol Sci. 2007;260:219–24. doi: 10.1016/j.jns.2007.05.015. [DOI] [PubMed] [Google Scholar]
- 11.Crimella C, Cantoni O, Guidarelli A, Vantaggiato C, Martinuzzi A, Fiorani M, et al. A novel nonsense mutation in the APTX gene associated with delayed DNA single-strand break removal fails to enhance sensitivity to different genotoxic agents. Hum Mutat. 2011;32:2118–33. doi: 10.1002/humu.21464. [DOI] [PubMed] [Google Scholar]
- 12.Le Ber I, Moreira MC, Rivaud-Pechoux S, Chamayou C, Ochsner F, Kuntzer T, et al. Cerebellar ataxia with oculomotor apraxia type 1: Clinical and genetic studies. Brain. 2003;126:2761–72. doi: 10.1093/brain/awg283. [DOI] [PubMed] [Google Scholar]
- 13.Wells RD. Molecular basis of genetic instability of triplet repeats. J Biol Chem. 1996;271:2875–8. doi: 10.1074/jbc.271.6.2875. [DOI] [PubMed] [Google Scholar]